a87e767b512d0cbc768e2eeb611184bc.ppt
- Количество слайдов: 61



β-Thalassemia and G 6 PD deficiency as Public Health Problems in Gaza Strip, Palestine Mahmoud Sirdah, Ph. D Associate Professor of Blood Pathophysiology Al-Azhar University Gaza

Why Genetics is important in Gaza? Overall Consanguinity is high 39. 9 % Consanguineous marriages are significantly higher in semi -urban areas (41. 6%) than in urban areas (39. 1%). Compound consanguinity (two generation) is 20. 7%. The average of those with: first-cousin marriages 22. 4 ± 4. 4 years) second-cousin marriages (24. 3 ± 6. 1 years) non-consanguineous (26. 5 ± 8. 2 years).


The Thalassemias • Thalassemias reflect quantitative abnormalities in Hb production. • The term thalassemia is derived from the Greek words: thalassa (sea) and eima (blood), so it means sea in the blood. • Also known as Cooley's Anemia, Mediterranean Anemia

Human Hemoglobins • Heterogeneous proteins packed inside the RBCs. • About 270 millions of molecules of hemoglobin/ RBC. • Tetrameric in structure, made up of two different pairs of globin polypeptide chains: • 2 like (141 a. a), controlled by genes on chrs 16 • 2 like (146 a. a), controlled by genes on chrs 11. • This heterogeneity is expressed at all stages of development, during which different hemoglobin forms are synthesized during human life.

Heterogeneity of Human Hemoglobin Type Structure Developmental Stage Hb Gower 1 2 2 embryo Hb Gower 2 2 2 embryo Hb Portland 2 2 embryo Hb. F 2 2 fetal and adult Hb. A 2 2 2 adult Hb. A 2 2 adult

Genetics of Human Hemoglobin

Fetal to Adult Hemoglobin Switch • After birth, the adult and globin chains begin gradually to replace the -globin chain. • This results in a major switch from Hb. F ( 2 2) to the adult hemoglobin Hb. A ( 2 2) synthesis. • This switch occurs about the time of birth and ends 6 -9 months later. • After the Hb. F ( 2 2) to Hb. A ( 2 2) switch, 97 -98% of the hemoglobin is Hb. A, while Hb. A 2 ( 2 2) accounts for approximately 2%. • Small amounts ( 1%) of Hb. F are also found in adults blood.
 to Hb A ( 2 2) Switch")
Hb F ( 2 2) to Hb A ( 2 2) Switch
 • The inherited disorders of hemoglobin are known")
The Inherited Disorders of Hemoglobin (Hemoglobinopathies) • The inherited disorders of hemoglobin are known as hemoglobinopathies: • Quantitative: defect in the rate of production of Quantitative one or more of the globin chains, known as thalassemias. • Qualitative: structural change in the globin chain, Qualitative known as Hb variants which alter the Hb stability or function of Hb, e. g. Hb. S, Hb. C, Hb. D, Hb. E, etc. .

The Thalassemias • A group of recessively autosomal inherited conditions, leads to hereditary anemias. • Characterized by decreased or absence of synthesis of one or more polypeptide chains (α-like or β-like ) that form Hb. • The commonest genetic diseases of mankind. • WHO estimates : 1. 5 % of the world’s population might be carriers of βthalassaemia, 60, 000 thalassemic births /year • Affect men and women equally.

Geographic Distribution • Thalassemias have been encountered practically in every racial group and geographic location of the world. • Most common in the Mediterranean region and equatorial or near equatorial regions of Africa and Asia. • The high frequencies of thalassemia are found in those areas historically afflicted with endemic malaria

Worldwide Distribution of Thalassemias
 is")
Classification of Thalassemias • Thalassemias are classified according to which particular globin chain(s) is produced in a reduced amount. • The main types which have now been defined with certainty are the , , , and -thalassemias. • and -thalassemias are the most common classes

-Thalassemia • The most important type of thalassemias. • So common, widely distributed and result in severe anemia in the homozygous and compound heterozygous states. • High prevalence in a broad belt extending from the Mediterranean basin and parts of north and west Africa through the Middle East and Far East. • In some regions of Italy, Greece, Cyprus, Sardinia and South Eastern Asia carriers frequencies ranges between 1520 %

Types of -Thalassemia • -thalassemia: defect in -chain production alone, is a due mainly to point mutations, and it is the most common type. • -thalassemia: reduced/ absence of and chain synthesis resulting usually from deletions in the and globin genes, , it is a less common type. • -thalassemia and -thalassemias : are rare forms of thalassemia which results from a long deletion in the -like globin cluster, and so removing the, , and globin gene
 in subjects with")
Clinical Classification of -Thalassemia The three main clinical phenotypes (Severity Classification) in subjects with β thalassaemia are major, intermedia, and minor • -Thalassemia major, Cooley’s anemia or homozygous -thalassemia, double heterozygous thalassemia is a severe, transfusion dependent disorder • -thalassemia intermedia: is applied to a less severe clinical phenotype in which significant anemia occurs but regular transfusion therapy is not required • -thalassemia minor or trait or heterozygous thalassemia is the symptomless carrier state.

Molecular pathology of -Thalassemia • -Thalassemia is very heterogeneous at the molecular level, with more than 400 different mutations (most of which are point mutations, large deletion mutations are found in rare cases) identified that causes the disease. • The disease arise from mutations that affect every step in the pathway of -globin gene expression: transcription, m. RNA processing or m. RNA translation • The basic molecular defect in -thalassemia (HBB gene) results in either absence ( 0) or reduced ( +) beta chain production.
 or ( +) beta chain production.")
Pathophysiology of -Thalassemia • Consequences of ( 0) or ( +) beta chain production. : • Reduced production of Hb A ( 2 2). • Imbalanced globin chain synthesis : . • The excess chains are unstable and precipitate in BM RBC precursors, giving rise to a large intracellular and subsequently intramedullarly (ineffective erythropoiesis) and extramedullarly destructions of RBC become the norm. • Anemia of -thalassemia results: • • • Hb production Ineffective Erythropoiesis Shortened RBC survival

Clinical Features of -Thalassemia Major • Without treatment/ management, β-thalassemia major is lethal in early childhood, • Treatment/ management with a regular transfusion program and iron chelation allows for normal growth and development and extends life expectancy into the 3 rd to 5 th decade. • Most deaths are caused by the cardiac complications of iron overload

-Thalassemia In Gaza Strip, Palestine Before 1997 • About 300 -Thalassemia major patients • All efforts were directed toward providing the patients with blood transfusion and iron chelating agents. • -Thalassemia new births 15 -25/ year. • No estimation of the prevalence of the gene in the population. • Knowledge and awareness is very limited or lacking among the population

-Thalassemia In Gaza Strip, Palestine • 1997 Establishment of the first official Palestinian Thalassemia foundation that take care of all social and medical aspects of the thalassemic patients and their families. • 1997 -1998 first screening study that estimated the prevalence (4. 3%, range 3. 37. 9%) of the -Thalassemia in Gaza Strip.


-Thalassemia In Gaza Strip, Palestine • 1999 -2000, a series of nationwide activities that included: • Public lectures that covered people at urban, rural, refugee camps as well as other remote communities. • Media awareness programs and brochures • Scientific lectures to students at high School and universities • Discussions and meetings with national policy makers. • TV interviews with geneticist, hematologist together with affected families and patients.

The obligatory premarital tests for thalassemia • September 2000, the superior legitimate judge adopted the implementation of the premarital tests for -thalassemia as an obligatory step before any proposed couple can be issued with a marriage certificate.


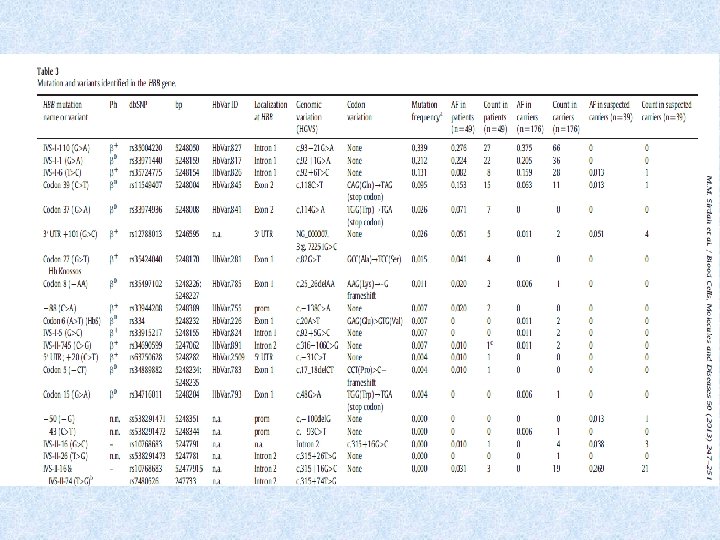
2010. Molecular characterization of thalassemia in Gaza Strip We screened patients and carriers IVS-I-110 G>A IVS-I-1 G>A IVS-I-6 T>C Cd 39 C>T Cd 37 G>A Three novel HBB variants were discovered by direct sequencing of the gene: 5′ UTR-50 (−/G) 5′ UTR-43 C>T IVS-II-26 T>G.


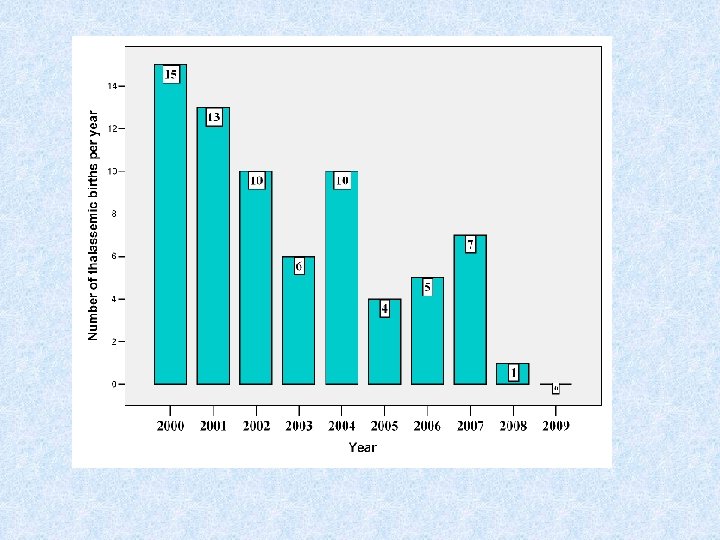
Outcomes 13 -year experience in running premarital tests • The first year of the implementation of obligatory tests was considered to be the most problematic. • After that, this process is easier and the obligatory tests procedure is going very smoothly. • About 10, 000 couples are screened /year. • Most ( 95 %) couples at risk (carries for thalassemia) separated and selected another partner (not carrier). • The relative success of the Palestinian thalassemia prevention program is clear through the actual and significant chronological reduction in the number of -thalassaemia major births to almost zero in the last years.
 deficiency")
Glucose-6 -phosphate dehydrogenase (G 6 PD) deficiency

RBC metabolic pathways • Mature RBC has no mitochondria. • ATP is generated by anaerobic glycolysis. • Glucose is anaerobically catabolize to lactic acid. • This pathway is known as (Embden-Meyerhof pathway). • Other shunts or pathways are also directly connected to Embden. Meyerhof pathway: • Hexose-Monophosphate Shunt (pentose phosphate pathway. • Rapoport-Luebering shunt

Embden-Meyerhof pathway

Function of Embden-Meyerhof pathway • Production of ATP molecules (ATP is needed for Na+/K+ ATPase pump, maintain cell shape and flexibility (deformability) • NADPH production through hexose monophosphate shunt (NADPH used to prevent accumulation of H 2 O 2, this shunt is dependent on G 6 PD enzyme). • 2, 3 diphosphoglycerate (2, 3 DPG) production (generated in Rapoport -Luebering shunt). • NADH production. ( Used by methemoglobin reductase to reduce Met. Hb to Hb)
 • Under normal conditions, about 10%")
Importance of the Hexose-Monophosphate Shunt (pentose phosphate pathway) • Under normal conditions, about 10% of the glucose utilized by RBC is catabolized via the hexose-monophosphate shunt. • This shunt provides a critical protection against oxidative damage through generation of the NADPH which enables cells to counterbalance oxidative stress. • Since RBCs do not contain mitochondria, the HMS is their only source of NADPH; therefore, defense against oxidative damage is dependent on G 6 PD. • G 6 PD deficient red cells are highly susceptible to oxidative damage.
 • G 6 PD is the")
Importance of the Hexose-Monophosphate Shunt (pentose phosphate pathway) • G 6 PD is the first and rate-controlling enzyme in the Hexose-Monophosphate Shunt. • G 6 PD oxidizes glucose-6 -phosphate to 6 -phosphogluconate. NADP+, which serves as a cofactor in this reaction, is reduced to NADPH • NADPH is a cofactor for glutathione reductase, which then reduces oxidized glutathione NADPH (GSSG) to the reduced form (GSH) of glutathione. • GSH serves as a cofactor for the enzyme glutathione peroxidase, which reduces various peroxides to water.

Hexose-Monophosphate Shunt

The G 6 PD gene, is located near the telomeric region of the long arm of the X chromosome (Xq 28)

The G 6 PD gene • Consists of 13 Exons and 12 Introns, Exon 1 is noncoding • Encodes 515 amino acids. • About 18. 5 kilobases (kb) in length. • Intron 2 constitutes about 12 kb. • The significance of this large Intron 2 is unknown • Important for efficient transcription or for processing? ? ?

The G 6 PD enzyme • The enzymatically active form is either a dimer or a tetramer of a single polypeptide subunit of about 59 kd.

G 6 PD Dimer

G 6 PD Tetramer

Genetics of G 6 PD • Because it is X-linked, • Males can be only: • normal hemizygotes • or deficient hemizygotes. • Females can be: • normal homozygotes, • deficient homozygotes • heterozygotes

Consequences of X-linkage • G 6 PD Mutations display the typical pattern of Mendelian X-linked recessive inheritance (crisscross pattern). • Phenotype pedigree

Consequences of X-linkage • Severe G 6 PD deficiency is much more common in males than in females. • Because of X-chromosome inactivation, G 6 PD heterozygous females exhibit mosaic somatic cells.

WHO Classification of G 6 PD deficiency Class I Severely deficient, associated with chronic non-spherocytic hemolytic anemia Class II Severely deficient (1– 10% residual activity), associated with acute hemolytic anemia Class III Moderately deficient (10– 60% residual activity) Class IV Normal activity (60– 150%) Class V Increased activity (>150%)

Molecular Basis of G 6 PD Deficiency • Almost all variants of the G 6 PD gene that result in enzyme deficiency affect the coding sequence. • About 186 variants have been reported, most of which are singlebase substitutions leading to amino acid replacements. • In a few cases, two amino acid replacements are found. • a few cases (8) of small in-frame deletions have been discovered. • Only one mutation affecting splicing site has been discovered. • No mutations have yet been reported in the promoter region

Most common mutations along coding sequence of G 6 PD gene
 deficiency is the")
Global distribution of G 6 PD deficiency • G 6 PD) deficiency is the most common human enzyme defect, being present in more than 500 million people worldwide. • The global distribution of G 6 PD deficiency is remarkably similar to that of malaria, providing a support to the so-called malaria protection hypothesis. • G 6 PD deficient RBCs are more resistant to Malaria parasite Plasmodium falciparum malaria.

World map distribution of G 6 PD deficiency

World distribution of G 6 PD deficiency and mutation spectrum

Molecular Heterogeneity of G 6 PD deficiency in Gaza Strip Palestinians • Mutations within G 6 PD were identified and characterized by Sanger sequencing of the 13 exons using 3100 Genetic Analyser.

 hemolytic anemic")
Results • Following biochemical evaluation: • 65 out 80 ( 81. 3%) hemolytic anemic children (60 males and 5 females, were found to be G 6 PD deficient. • Hemolytic crisis in all G 6 PD deficient children was due to the ingestion of fava beans (either green or dried).

Results • Genetic analysis of G 6 PD deficient Palestinian children revealed three predominant and two less frequent mutations within the Gaza Strip population.

G 6 PD Gaza c. 536 G>A A novel missense mutation G 6 PD Gaza is characterized by a G > A transition at nucleotide c. 536, which changes serine p. 179 to asparagine

Molecular modeling and dynamics studies suggest that Gaza mutation affects key protein conformation regions, leading to decreased binding affinity of the enzyme for NADP+ due to general displacement of active site residues within the binding site

Conclusions about G 6 PD in Gaza • we characterize the molecular heterogeneity of G 6 PD variants among Gaza Strip Palestinians, which shows a wide molecular diversity of G 6 PD deficient variants. • Favism was the predominant cause of hemolytic anaemia in Palestinian children at Gaza Strip, representing 81% of patients admitted with hemolytic crisis. • The association of these G 6 PD mutants with clinically significant hemolysis requiring hospital admission justifies nationwide programs of newborn screening for G 6 PD deficiencies

Thank you so much for your attendance
a87e767b512d0cbc768e2eeb611184bc.ppt