лекция 3 мит.pptx
- Количество слайдов: 45



ЗАБОЛЕВАНИЯ, СВЯЗАННЫЕ С ТОЧЕЧНЫМИ МУТАЦИЯМИ В МИТОХОНДРИАЛЬНЫХ ГЕНАХ СУБЪЕДИНИЦ ДЫХАТЕЛЬНОЙ ЦЕПИ

Фактором, который усложняет картину наследования митохондриевых болезней, является существование такого явления, как гетероплазмия — генетическая гетерогенность популяции митохондрий у некоторых индивидуумов. Когда мутация возникает в митохондриевой ДНК, она воспроизводится во всех копиях этой мт. ДНК, но при этом нет механизма, посредством которого данная мутация распространилась бы на все другие молекулы мт. ДНК в той же клетке. гетероплазмия в каждой новой клетке может быть большей или меньшей, чем она была в родительской клетке

Примером синдромов, вызванных нуклеотидными заменами в генах мт. ДНК, кодирующих белковые цепи, является LHON (Leber's hereditary optic neuropathy) наследственная зрительная (оптическая) нейропатия [невропатия] Лебера — наиболее распространенное митохондриальное заболевание с двусторонней атрофией зрительного нерва. Среди болезней OXPHOS (болезни окислительного фосфорилирования) это заболевание было одним из первых, для которого удалось найти мутационные повреждения митохондриального генома.

LHON имеет исключительно нейроофтальмологические симптомы. В основном, болезнь случается у мужчин.
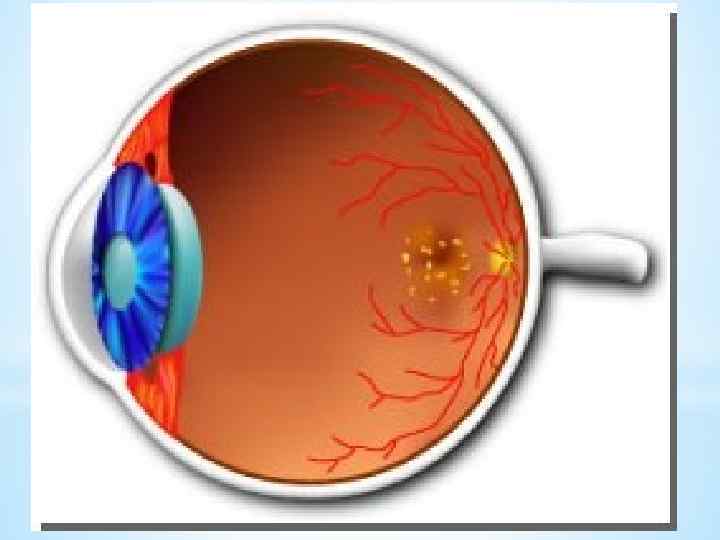

- Всегда наследуемая по материнской линии - болезнь начинается, как правило, в молодом возрасте — чаще всего между 15 и 35 годами (описаны случаи появления симптомов в возрасте от 1 года до 80) - Потеря зрения не сопровождается болевыми ощущениями и обычно начинается с одного глаза - Ухудшение зрения во втором глазу начинается через несколько недель или месяцев - При обследовании отмечаются центральные или цекоцентральные дефекты полей зрения. - Иногда попутно обнаруживаются тремор, дистония, изменения на ЭКГ - Позднее присоединяются нарастающие по тяжести неврологические и психические расстройства и выраженная мышечная слабость
")
При LHON описано несколько различных мутаций мт. ДНК Наиболее часто (69% всех случаев LHON) – замена G 11778 А, приводящая к замене аргинина в 340 -й позиции полипептидной цепи на гистидин. Этот нуклеотид входит в ген, кодирующий четвертую субъединицу НАДНдегидрогеназы (ND 4). две другие мутации — в позициях 3460 (ген ND 1) и 14484 (ген ND 6) встречаются примерно с одинаковой частотой — 13% и 14%, соответственно. Это «основные мутации LHON» . более 90% случаев заболевания во всем мире вызвано какой-либо из них

Другие точечные мутации обнаруживаются у больных LHON с большей частотой, нежели в контроле, но причинно-следственная связь между этими второстепенными мутациями и возникновением заболевания не всегда очевидна. Некоторые из них могут играть роль первопричины заболевания, но их распространенность сводится к нескольким родословным на всем Земном шаре

Во многих случаях LHON развивалась у пациентов с гетероплазмией, имеющей тенденцию к гомоплазмии по мутантной мт. ДНК (с заменой G>A 11778). При других missense-мутациях, также приводящих к замене высоко- или умеренно-консервативных аминокислот, отмечался аналогичный феномен. Критерии, позволяющие считать основные мутации LHON непосредственно связанными с возникновением клинического фенотипа: 1) гетероплазмия с нарастанием доли мутантной мт. ДНК; 2) существенно более высокая доля мутантной мт. ДНК у пациентов, чем в митохондриях здоровых индивидуумов; 3) замена эволюционно консервативной аминокислоты (постоянство ее присутствия в нормальном белке, т. е. «консервативность» , свидетельствует о биохимической значимости).

- Вероятность возникновения LHON существенно возрастает с приобретением каждой новой мутации. - чаще всего поражаются мутациями гены субъединиц НАДН-дегидрогеназы - Прогноз заболевания более благоприятен у пациентов с мутацией в позиции 14484. До 72% пациентов с этой мутацией могут рассчитывать на умеренное улучшение зрения по сравнению лишь с 5% больных с мутацией в позиции 11778

- Присутствие мутации в мт. ДНК необходимо для получения клинического фенотипа, - Необходимо также достижение определенного порога концентрации мутантной мт. ДНК, в развитии митохондриальной болезни (гетероплазмия). - Однако, описаны случаи, когда индивидуум со 100% мутантной мт. ДНК не имел никаких признаков ухудшения зрения. В некоторых родословных обнаружено по две основных мутации, связанных с возникновением LHON, но у членов этих семейств не отмечалось никаких признаков усугубления тяжести течения болезни. - Отсутствие каких-либо особенностей при ухудшении зрения касалось и пенетрантности заболевания, и гетероплазмии, и даже возможности улучшения состояния пациентов
 и внешней среды Например,")
Имеет место взаимодействия генетических факторов (как митохондриальных, так и ядерных) и внешней среды Например, сцепление с Х-хромосомой какого-либо признака, связанного со зрительной функцией, может серьезно изменить фенотипические проявления LHON Патофизиология LHON остается неизвестной или очень слабо изученной. Предполагается, что звеньями патогенеза могут быть нарушение OXPHOS и недостаточная продукция АТФ, что прямо или косвенно связано с выбросом свободных кислородных радикалов, вызывающих необратимые повреждения клеток ганглиев и аксонов
 должно вызвать три «биохимических» осложнения. Во-первых, в дыхательной цепи")
угнетение функций комплекса I (НАДН-дегидрогеназы) должно вызвать три «биохимических» осложнения. Во-первых, в дыхательной цепи митохондрий не будет происходить окисления НАДН до НАД. Во-вторых, комплекс I не будет перекачивать протоны, что снизит общий уровень продукции АТФ в митохондриях. В-третьих, усилится продукция супероксидных радикалов, вызывающих повреждение мт. ДНК, перекисное окисление липидов в мембранах митохондрий и денатурацию митохондриальных белков.

непонятно, почему эти процессы общего характера избирательно поражают зрительный нерв Интерес вызывает сообщение о генетическом исследовании другого типа атрофии зрительного нерва — так называемой атрофии 1 -го типа, наиболее распространенной формы наследственных атрофий, передающейся как аутосомно-доминантный признак. Оказалось, что мутации в ядерном гене ОРА-1 способны изменить нормальную функцию белка с тем же названием, связанного с группой белков-динаминов, импортируемого в митохондрии и играющего важную роль в их подвижности и формировании митохондриальной сети

атрофия зрительного нерва может встречаться как непостоянный или вторичный симптом и в других заболеваниях, вызванных нарушениями OXPHOS зрительную нейропатию практически у любого пациента следует рассматривать как маркер нарушенных функций митохондрий.

ЗАБОЛЕВАНИЯ, СВЯЗАННЫЕ С ТОЧЕЧНЫМИ МУТАЦИЯМИ В ГЕНАХ МИТОХОНДРИАЛЬНЫХ т. РНК

Примером заболеваний OXPHOS, вызванных нуклеотидными заменами в генах, кодирующих т. РНК, являются MERRF и MELAS Синдром MERRF (англ. Myoclonic Epilepsy with Ragged Red Fibers, миоклоническая эпилепсия с рваными мышечными волокнами) — редкое митохондриальное заболевание, вызываемое мутациями в следующих генах: MTTK, MTTL 1, MTTH, MTTS 1, MTTS 2, MTTF. Симптомы MERRF также проявляются при мутациях гена MTND 5

Синдром MELAS (англ. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes — «митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные эпизоды» ) — прогрессирующее нейродегенеративное заболевание, сопровождающееся полиморфной симптоматикой — диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями. В каждом конкретном случае набор симптомов и их тяжесть может сильно отличаться, поскольку синдром связан с мутациями во многих генах: MTTL 1, MTTQ, MTTH, MTTK, MTTS 1, MTND 1, MTND 5, MTND 6, MTTS 2.
 достигает гетероплазмия по")
MERRF возникает, когда некоего порогового уровня (для каждой ткани — своего) достигает гетероплазмия по мт. ДНК с мутацией в гене, кодирующем т. РНКЛиз (транзиция A>G в 8344 -й позиции). MELAS вызывается гетероплазмией также по транзиции A>G в 3243 -й позиции, что соответствует гену т. РНКЛей. Эти две мутации наиболее типичны для каждого из заболеваний, соответственно, однако и для MERRF, и для MELAS описаны и другие нуклеотидные замены.

Синдром MERRF

Синдром MELAS Двусторонняя кальцификация базальных ганглий, легкая атрофия мозжечка, ишемические изменения, повышение лактата.

Оба заболевания были известны как неврологические расстройства еще до обнаружения причинно-следственной связи между мутациями в мт. ДНК и появлением клинического фенотипа. Для обоих было отмечено разнообразие клинических проявлений, которое лишь увеличилось, когда, благодаря генетическому обследованию, нуклеотидные замены A 8344 G и A 3243 G были выявлены в больших группах пациентов, чей клинический фенотип не вполне соответствовал условным стандартам.

MERRF наследуется по материнской линии в тяжелых случаях у пациентов отмечаются миоклоническая эпилепсия (неконтролируемые подергивания тела), энцефаломиопатия, судороги, мозжечковая атаксия, нейросенсорные слуховые расстройства, дилатационная кардиомиопатия, почечная недостаточность, респираторные нарушения, слабоумие. Все эти тяжелые расстройства являются проявлением недостаточности дыхательных комплексов I и IV, вызванной, в свою очередь, нарушенным биосинтезом их компонентов

С возрастом состояние пациентов ухудшается При окрашивании срезов из биоптатов мышечной ткани Гомори трехцветным у пациентов обнаруживаются рванокрасные волокна, что свидетельствует о присутствии большого числа митохондрий. Специфична и гистохимическая картина, наблюдаемая при параллельном окрашивании срезов: Активность сукцинатдегидрогеназы в рвано-красных волокнах повышена, цитохромоксидазы — практически сведена к нулю.

Пациенты с MELAS страдают от периодически возникающих инсультоподобных состояний, оставляющих в коре и белом веществе головного мозга следы, выявляемые с помощью ЯМР или компьютерной томографии. Нередко к этому присоединяются симптомы корковой слепоты, молочнокислого ацидоза, неврологические расстройства, а общая тяжесть заболевания также нарастает с возрастом. Иногда у пациентов обнаруживается гипертрофия левого желудочка с нарушениями ритма в виде аритмий, реже — экстрасистолий У некоторых больных отмечалось явное снижение остроты слуха
 2 х синдромов 1. оба заболевания затрагивают не одну, а несколько систем")
Обобщение (сравнение) 2 х синдромов 1. оба заболевания затрагивают не одну, а несколько систем органов. У пациентов можно обнаружить как довольно мягкое течение болезни, так и крайне тяжелые формы, приводящие к ранней гибели. 2. некоторые клинические проявления характерны для одного из заболеваний, но крайне редко встречаются в другом. Из типичных для MERRF симптомов (но редких для MELAS) можно отметить нейропатию, атаксию, миоклонические судороги, возникновение липом и атрофию зрительного нерва. Для клинической картины MELAS типичными симптомами являются повторные инсульты или инсультоподобные состояния, сахарный диабет, пигментная ретинопатия, нередко — СРЕО

3. некоторые симптомы, такие, как эпилепсия, миопатия, глухота, с одинаковой частотой встречаются в клинической картине каждого из двух заболеваний. Характерно, что пациенты и с MERRF, и с MELAS, как правило, имеют небольшой рост. Помимо наиболее распространенной замены A 3243 G, при синдроме MELAS было описано не менее 13 других точечных мутаций, а также гигантские делеции мт. ДНК В связи с этим большой интерес представляют исследования вопроса о том, как мутационная нагрузка влияет на частоту возникновения основных клинических симптомов данного заболевания.

мутационная нагрузка (на практике — степень гетероплазмии по одной или нескольким мутациям в мт. ДНК) в миоцитах прямо соответствует частоте клинических проявлений MELAS и MERRF динамика развития гетероплазмии в дальнейшем остается неизвестной, так что сопоставить ее с клиническим течением болезни не представляется возможным. Возможно, не только относительная величина мутационной нагрузки в пределах какой-либо ткани, но и гетерогенность мутаций может влиять на появление клинических симптомов заболевания.

не только у пациентов с MERRF, но и в случае MELAS микроскопия обнаруживает в мышцах рвано-красные волокна — феномен, вызванный компенсаторным увеличением числа и размеров митохондрий в ответ на неэффективный процесс OXPHOS В семьях с мутацией MELAS A 3243 G нередко наблюдается сочетание сахарного диабета с нейросенсорной глухотой.

не все нуклеотидные замены в генах митохондриальных т. РНК связаны с патологией При обнаружении новой мутации в мт. ДНК полезно применить известные критерии ее патогенности: 1) Мутация не должна обнаруживаться в репрезентативных когортах здоровых людей, и, помимо проверяемой нуклеотидной замены, в исследуемой мт. ДНК не должно быть других замен, потенциально связанных с митохондриальной патологией. 2) У пациента должна обнаруживаться гетероплазмия в отношении проверяемой мутации.
 Мутация должна находиться в высоко консервативном участке гена т. РНК. 4) Должны присутствовать")
3) Мутация должна находиться в высоко консервативном участке гена т. РНК. 4) Должны присутствовать проявления нарушений в дыхательной цепи митохондрий (обычно проверяют наличие рвано-красных волокон и отсутствие активности цитохром с оксидазы в мышечных биоптатах). 5) Необходимо наличие родственников пациента по материнской линии, у которых также обнаруживается проверяемая мутация. 6) Следует показать связь между мутационной нагрузкой (на практике — выраженностью гетероплазмии в пораженной ткани или в референтных клетках) и тяжестью заболевания.

Патогенез MERRF и MELAS: молекулярные исследования и невропатология Неврологическая картина MERRF, в основном, обусловлена дегенерацией нейронов в зубчатом и нижнем оливном ядрах головного мозга. Отмечены случаи нейродегенерации также в коре больших полушарий и в мозжечке. Именно патологическими изменениями в коре объясняют появление у пациентов с MERRF деменции, миоклонуса и эпилепсии

при высоком содержании мутации MERRF 8344 в митохондриях снижены уровни синтеза белка, потребления кислорода, а также отмечается недостаточная активность цитохром с оксидазы. При этом особенно угнетен был синтез кодируемых митохондриальным геномом белковых цепей с относительно более высоким содержание лизина. было обнаружено, что при мутации MERRF 8344 нарушен синтез нормальной т. РНКЛиз

при иммуногистохимическом исследовании разных отделов мозга пациента с MERRF для оценки уровня экспрессии субъединиц дыхательных комплексов, кодируемых как в митохондриальном, так и в ядерном геноме, было обнаружено специфическое снижение содержания II субъединицы цитохром с оксидазы. При этом содержание VIII субъединицы АТФазы и I субъединицы НАДН-дегидрогеназы, кодируемых в митохондриях, оставалось нормальным. субъединица АТФазы имеет наивысшее содержание лизина - противоречие. высказано предположение, что при MERRF происходит избирательное повреждение цитохром с оксидазы.

При MELAS чаще всего встречаются множественные некротические очаги в разных отделах головного мозга (затронуты бывают кора больших полушарий, базальные ганглии, таламус, мозжечок, ствол мозга) и нейродегенеративные изменения в коре мозжечка и зубчатом ядре. Нередко наблюдаются минеральные отложения в перечисленных структурах. Отмечается пролиферация митохондрий, чаще имеющих аномальную морфологию, в эндотелии и гладкой мускулатуре мозговых сосудов.

при изучении гетероплазмии по мутации MELAS 3243 в цибридах, где реципиентами выступали клетки остеосаркомы, было показано, что уже по достижении 50% мутантной мт. ДНК снижалась эффективность комплекса I (НАДН-дегидрогеназы) измерение содержания продуктов трансляции митохондриально кодируемых субъединиц комплексов дыхательной цепи обнаружило обратную зависимость между содержанием мутантной мт. ДНК и уровнем синтеза этих субъединиц

Группа наблюдений говорит в пользу патогенетического механизма MELAS, основанного на нарушении не транскрипции, а трансляции в митохондриях с мутантным геномом. В модельных экспериментах на лимфобластоидных клетках, трансформированных вирусом Эпштейн-Барра и содержавших мутацию MELAS 3243, получили клеточные линии с разной мутационной нагрузкой — 30% и 70%. В клетках, содержавших 30% мутантной мт. ДНК, аномалий в дыхательной цепи митохондрий обнаружено не было, тогда как при 70% мутантных копий митохондриального генома отмечалось существенное снижение активности комплекса IV и небольшое уменьшение активности всех остальных комплексов, кроме НАДН-дегидрогеназы.

Пептидные карты транслируемых белковых цепей выявили нарушение их структуры. Возможным механизмом в этом случае может являться нарушение т. РНК-опосредованной доставки аминокислот или сбой в узнавании рибосомами т. РНК не только мутации в мт. ДНК, но и многие другие события, приводящие к нарушениям структуры и функций одного или нескольких комплексов дыхательной цепи митохондрий, могут вызывать почти однотипные симптомы

ЗАБОЛЕВАНИЯ, СВЯЗАННЫЕ С ОБШИРНЫМИ ДЕЛЕЦИЯМИ В МИТОХОНДРИАЛЬНОЙ ДНК
 и KSS —")
К болезням, вызванным делениями мт. ДНК, относятся СРЕО, синдром Пирсона (PS) и KSS — синдром Кернса-Сейра; развивается в молодом возрасте; проявляется офтальмоплегией, пигментной дегенерацией сетчатки, гиперпаратиреоидизмом; иногда проявлениям KSS предшествуют проявления синдрома де Тони-Дебре-Фанкони — глюкозурия, генерализованная гипераминоацидурия, фосфатурия, тубулярная протеинурия, гипокалиемия, гипоуремия; СРЕО — хроническая прогрессирующая офтальмоплегия; симптоматика сходна с симптоматикой KSS, со значительными дисфункциями центральной нервной системы;

синдром Пирсона - развивается в детстве; проявляется системными нарушениями окислительного фосфорилирования, поражением центральной нервной системы, костного мозга и поджелудочной железы; как и в случае KSS проявлениям синдрома Пирсона могут предшествовать проявления синдрома де Тони-Дебре. Фанкони;

При всех трех описываемых заболеваниях в мт. ДНК обнаруживаются одинаковые гигантские делении. в отдельных случаях KSS были обнаружены протяженные вставки (инсерции) в мт. ДНК, также нарушающие функции генома. синдромы, вызванные гигантскими делециями, являются исключением из правила о материнском наследовании болезней OXPHOS. Почти всегда они возникают спорадически и не имеют семейной истории, т. е. обследование родственников пациента не обнаруживает аномальной мт. ДНК и указывает на возникновение мутации de novo

Гигантские делеции единичны, в пределах одной молекулы мт. ДНК чаще встречается только одна делеция таких размеров, хотя иногда обнаруживаются и димеры Делеции захватывают как гены, кодирующие т. РНК, так и гены субъединиц дыхательных комплексов. Почти у половины всех пациентов была обнаружена так называемая «обычная делеция» протяженностью 4977 п. н. — от гена 8 -й субъединицы АТФазы до гена, кодирующего ND 5, хотя встречаются и делеции больших размеров — длиной примерно в 7, 4 и 10, 4 тысяч нуклеотидных пар.

Считается, что «обычная делеция» возникает в той области мт. ДНК, где чаще случаются сбои в механизме репликации, например, аномальная рекомбинация или соскальзывание реплицируемой цепи у многих пациентов с KSS и/или СРЕО в клетках наряду с мт. ДНК дикого типа (нормальной) присутствуют разные формы мт. ДНК, подвергшиеся той или иной структурной перестройке

При измерении доли полностью синтезированных за определенное время молекул мт. ДНК в культуре фибробластов оказалось, что время, затрачиваемое на репликацию полноразмерных копий и мт. ДНК с гигантской делецией, одинаково Вероятно, делеции в мт. ДНК возникают в оогенезе или в раннем эмбриогенезе.
лекция 3 мит.pptx