

Гибкость,аморфное фаз н.pptx
- Количество слайдов: 58
 Химия и физика полимеров основной курс Физика полимеров
Химия и физика полимеров основной курс Физика полимеров
 Гибкость цепи полимера Гибкость – это способность полимерной цепи изгибаться в пространстве и принимать различные конформации. Различают два вида гибкости: термодинамическая и кинетическая. Термодинамическая гибкость характеризует способность полимера изгибаться в пространстве, что предопределяется химическим строением макромолекул. По способности изгибаться в пространстве полимеры делятся на две группы: гибкоцепные и жесткоцепные. К гибкоцепным относятся большинство полимеров винилового и диенового рядов, ряд гетероцепных полимеров. К жесткоцепным относятся полимеры, например, с наличием сопряжения в основной цепи (поли-п-бензамид, поли-н-бутилизоцианат). -----------------------------------------------Конформация – это способ расположения различных частей молекулы в пространстве без разрыва химических связей. В результате теплового движения или внешнего воздействия для каждой макромолекулы обычно реализуется большое количество конформаций (за исключением предельно жестких полимеров): от предельно вытянутой до плотной глобулы.
Гибкость цепи полимера Гибкость – это способность полимерной цепи изгибаться в пространстве и принимать различные конформации. Различают два вида гибкости: термодинамическая и кинетическая. Термодинамическая гибкость характеризует способность полимера изгибаться в пространстве, что предопределяется химическим строением макромолекул. По способности изгибаться в пространстве полимеры делятся на две группы: гибкоцепные и жесткоцепные. К гибкоцепным относятся большинство полимеров винилового и диенового рядов, ряд гетероцепных полимеров. К жесткоцепным относятся полимеры, например, с наличием сопряжения в основной цепи (поли-п-бензамид, поли-н-бутилизоцианат). -----------------------------------------------Конформация – это способ расположения различных частей молекулы в пространстве без разрыва химических связей. В результате теплового движения или внешнего воздействия для каждой макромолекулы обычно реализуется большое количество конформаций (за исключением предельно жестких полимеров): от предельно вытянутой до плотной глобулы.
 Поворотно-изомерный механизм гибкости Существует два механизма гибкости: поворотно-изомерный, характерный для гибкоцепных полимеров, и персистентный, характерный для жесткоцепных. Поворотно-изомерный механизм гибкости заключается во внутреннем вращении атомов или групп атомов относительно простых -связей, лежащих в основной цепи макромолекул. Внутреннее вращение предполагает наличие большого количества поворотных (конформационных) изомеров у макромолекулы. Внутреннее вращение в НМС, например метильных групп в этане приводит к существованию большого количества поворотных изомеров (конформеров), различающихся расположением Н в соседних метильных группах и, соответственно значением потенциальной энергии. Два граничных поворотных изомера с “заслоненной” (цис) и “заторможенной” (транс) конформацией характеризуются максимальной и минимальной потенциальной энергией соответственно.
Поворотно-изомерный механизм гибкости Существует два механизма гибкости: поворотно-изомерный, характерный для гибкоцепных полимеров, и персистентный, характерный для жесткоцепных. Поворотно-изомерный механизм гибкости заключается во внутреннем вращении атомов или групп атомов относительно простых -связей, лежащих в основной цепи макромолекул. Внутреннее вращение предполагает наличие большого количества поворотных (конформационных) изомеров у макромолекулы. Внутреннее вращение в НМС, например метильных групп в этане приводит к существованию большого количества поворотных изомеров (конформеров), различающихся расположением Н в соседних метильных группах и, соответственно значением потенциальной энергии. Два граничных поворотных изомера с “заслоненной” (цис) и “заторможенной” (транс) конформацией характеризуются максимальной и минимальной потенциальной энергией соответственно.
 Поворотно-изомерный механизм гибкости На проекциях Ньюмена хорошо видно, что два крайних положения сдвинуты относительно друга на 60 . Изменение потенциальной энергии при последовательном повороте одной метильной группы из заторможенной конформации на угол от 0 до 360 отразится изменением потенциальной энергии, которая есть функция угла поворота U = f( )
Поворотно-изомерный механизм гибкости На проекциях Ньюмена хорошо видно, что два крайних положения сдвинуты относительно друга на 60 . Изменение потенциальной энергии при последовательном повороте одной метильной группы из заторможенной конформации на угол от 0 до 360 отразится изменением потенциальной энергии, которая есть функция угла поворота U = f( )
 Поворотно-изомерный механизм гибкости 1, 2 дихлорэтан – наиболее термодинамически выгодная конформация, когда атомы хлора и водорода максимально удалены (транс); наиболее не выгодной является та, где атомы хлора в заслоненном положении (цис); промежуточная по значению потенциальной энергии – скошенная или гош конформация. Зависимость потенциальной энергии поворотных изомеров 1, 2 дихлорэтана от угла поворота группы СН 2 Сl относительно связи С-С
Поворотно-изомерный механизм гибкости 1, 2 дихлорэтан – наиболее термодинамически выгодная конформация, когда атомы хлора и водорода максимально удалены (транс); наиболее не выгодной является та, где атомы хлора в заслоненном положении (цис); промежуточная по значению потенциальной энергии – скошенная или гош конформация. Зависимость потенциальной энергии поворотных изомеров 1, 2 дихлорэтана от угла поворота группы СН 2 Сl относительно связи С-С
 Поворотно-изомерный механизм гибкости Энергия, необходимая для перехода молекулы из одного положения в другое, т. е. энергия для совершения конформационного перехода называется потенциальным или активационным барьером вращения U 0. Для молекулы этана высота такого барьера = 12, 1 к. Дж/моль. В таблице представлены величины потенциального барьера вращения U 0 относительно различных связей. Наибольшее из них соответствует вращению относительно С-С связи. Тип связи Величина потенциального барьера вращения, к. Дж/моль С-С 18, 85 С-S 4, 44 C-O 4, 48 C-N 7, 90 – 8, 21 C-Si 7, 12 В НМС переход от одной конформации к другой осуществляется очень быстро, со скоростью 1010 раз в секунду, поэтому выделить химическим или физическим способом поворотные изомеры не удается.
Поворотно-изомерный механизм гибкости Энергия, необходимая для перехода молекулы из одного положения в другое, т. е. энергия для совершения конформационного перехода называется потенциальным или активационным барьером вращения U 0. Для молекулы этана высота такого барьера = 12, 1 к. Дж/моль. В таблице представлены величины потенциального барьера вращения U 0 относительно различных связей. Наибольшее из них соответствует вращению относительно С-С связи. Тип связи Величина потенциального барьера вращения, к. Дж/моль С-С 18, 85 С-S 4, 44 C-O 4, 48 C-N 7, 90 – 8, 21 C-Si 7, 12 В НМС переход от одной конформации к другой осуществляется очень быстро, со скоростью 1010 раз в секунду, поэтому выделить химическим или физическим способом поворотные изомеры не удается.
 Поворотно-изомерный механизм гибкости В полимерной цепи ПВХ, где мономерные звенья соединены “х к х”, участок макромолекулы можно рассматривать как замещенную молекулу 1, 2 дихлорэтана. Относительно С-С связи возможно вращение хлорметиновых СНСl групп, при этом энергетически выгодными способами в расположении заместителей являются одна транс и две гош-конформации:
Поворотно-изомерный механизм гибкости В полимерной цепи ПВХ, где мономерные звенья соединены “х к х”, участок макромолекулы можно рассматривать как замещенную молекулу 1, 2 дихлорэтана. Относительно С-С связи возможно вращение хлорметиновых СНСl групп, при этом энергетически выгодными способами в расположении заместителей являются одна транс и две гош-конформации:
 Поворотно-изомерный механизм гибкости Внутреннее вращение в полиэтилене Кривая потенциальной энергии конформационных изомеров в полиэтилене
Поворотно-изомерный механизм гибкости Внутреннее вращение в полиэтилене Кривая потенциальной энергии конформационных изомеров в полиэтилене
 Поворотно-изомерный механизм гибкости Согласно поворотно-изомерной модели в ПВХ вращение вокруг связей основной цепи осуществляется дискретно, в результате чего фиксируются транс- и две гош- конформации. Если угол поворота =0, (транс-конформация), то основная цепь представляет собой плоскую ленту (плоский зигзаг). Вращение вокруг любой из С-С связей приводит к излому плоской ленты в том месте, где 0. Совокупность изломов приводит к тому, что макромолекула в пространстве располагается не прямолинейно, как жесткий стержень, а криволинейно, причем искривление может происходить в различных направлениях и изменяется во времени.
Поворотно-изомерный механизм гибкости Согласно поворотно-изомерной модели в ПВХ вращение вокруг связей основной цепи осуществляется дискретно, в результате чего фиксируются транс- и две гош- конформации. Если угол поворота =0, (транс-конформация), то основная цепь представляет собой плоскую ленту (плоский зигзаг). Вращение вокруг любой из С-С связей приводит к излому плоской ленты в том месте, где 0. Совокупность изломов приводит к тому, что макромолекула в пространстве располагается не прямолинейно, как жесткий стержень, а криволинейно, причем искривление может происходить в различных направлениях и изменяется во времени.
 приводит к изменению формы") Поворотно-изомерный механизм гибкости Внутреннее вращение в виниловом полимере (выделенная триада) приводит к изменению формы небольшого участка макромолекулы, но не целиком макромолекулы. Положение триады до поворота характеризуется потенциальной энергией U 1, а после поворота - энергией U 2. Разность ΔU= U 1–U 2 характеризует вероятность конформационного перехода. Чем меньше ΔU, тем выше термодинамическая гибкость. Однако, чтобы реализовался конформационный переход необходимо наличие энергии U 0 для преодоления потенциального барьера. Если запаса кинетической энергии атомов недостаточно для преодоления потенциального барьера вращения, то вращения не происходит. Наблюдается только колебательное движение элементов.
Поворотно-изомерный механизм гибкости Внутреннее вращение в виниловом полимере (выделенная триада) приводит к изменению формы небольшого участка макромолекулы, но не целиком макромолекулы. Положение триады до поворота характеризуется потенциальной энергией U 1, а после поворота - энергией U 2. Разность ΔU= U 1–U 2 характеризует вероятность конформационного перехода. Чем меньше ΔU, тем выше термодинамическая гибкость. Однако, чтобы реализовался конформационный переход необходимо наличие энергии U 0 для преодоления потенциального барьера. Если запаса кинетической энергии атомов недостаточно для преодоления потенциального барьера вращения, то вращения не происходит. Наблюдается только колебательное движение элементов.
 Факторы, влияющие на внутреннее вращение в гибкоцепных полимерах Вращение в макромолекулах может тормозиться: - внутри- и межмолекулярным взаимодействием; - наличием объемных боковых заместителей. Среди сил физического молекулярного взаимодействия различают: ионное, водородное взаимодействие и силы Ван-дер-Ваальса. Из них самое сильное водородное связывание, а самое слабое – дисперсионное взаимодействие. Особенности теплового движения в полимерах – макромолекулы целиком не способны участвовать в тепловом движении подобно молекулам НМС. Чтобы переместить макромолекулу целиком необходимо разорвать все физические связи, для этого необходимо затратить большое количество энергии. В тепловом движении участвуют атомы и группы атомов, совершающие колебательные движения относительно положения равновесия, а также независимые участки полимерной цепи (сегменты), совершающие поступательные и вращательные движения.
Факторы, влияющие на внутреннее вращение в гибкоцепных полимерах Вращение в макромолекулах может тормозиться: - внутри- и межмолекулярным взаимодействием; - наличием объемных боковых заместителей. Среди сил физического молекулярного взаимодействия различают: ионное, водородное взаимодействие и силы Ван-дер-Ваальса. Из них самое сильное водородное связывание, а самое слабое – дисперсионное взаимодействие. Особенности теплового движения в полимерах – макромолекулы целиком не способны участвовать в тепловом движении подобно молекулам НМС. Чтобы переместить макромолекулу целиком необходимо разорвать все физические связи, для этого необходимо затратить большое количество энергии. В тепловом движении участвуют атомы и группы атомов, совершающие колебательные движения относительно положения равновесия, а также независимые участки полимерной цепи (сегменты), совершающие поступательные и вращательные движения.
 Особенности теплового движения в полимерах Характеристика сегментов: - сегменты – участи полимерной цепи, состоящие из нескольких звеньев; - положение сегментов в пространстве не зависит от положения соседних участков макромолекулы; - движение сегментов изменяет конформации цепей, которые могут принимать более и менее развернутую или свернутую форму; - чем гибче цепь, тем меньше длина сегмента и наоборот. На рисунке показана полимерная цепь с выделенными сегментами, которые перемещаются как единое целое под действием теплового (механического или электрического импульса), при этом остальная часть молекулы остается неподвижной. Длина сегмента (сегмента Куна по имени автора, который впервые произвел расчеты длины сегмента), а также количество звеньев в сегменте являются количественными характеристиками термодинамической гибкости (табл. ).
Особенности теплового движения в полимерах Характеристика сегментов: - сегменты – участи полимерной цепи, состоящие из нескольких звеньев; - положение сегментов в пространстве не зависит от положения соседних участков макромолекулы; - движение сегментов изменяет конформации цепей, которые могут принимать более и менее развернутую или свернутую форму; - чем гибче цепь, тем меньше длина сегмента и наоборот. На рисунке показана полимерная цепь с выделенными сегментами, которые перемещаются как единое целое под действием теплового (механического или электрического импульса), при этом остальная часть молекулы остается неподвижной. Длина сегмента (сегмента Куна по имени автора, который впервые произвел расчеты длины сегмента), а также количество звеньев в сегменте являются количественными характеристиками термодинамической гибкости (табл. ).
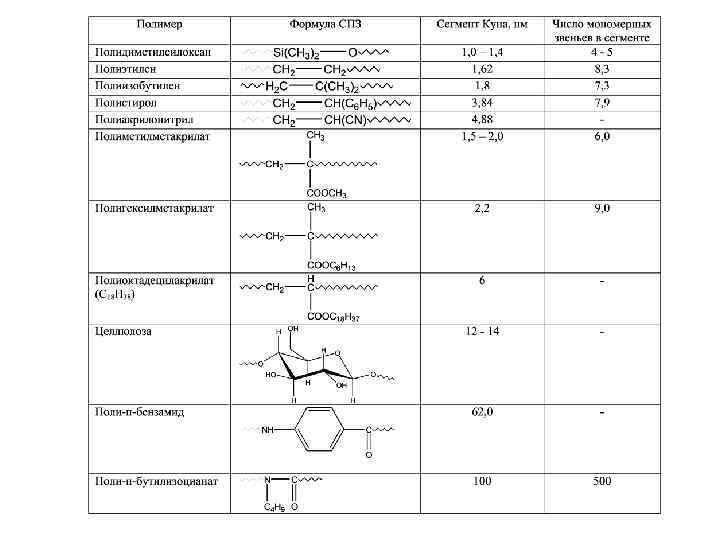
 Персистентный механизм гибкости Для большинства гибкоцепных полимеров значение сегмента Куна находится в пределах 2 – 3 нм. Если длина сегмента Куна не превышает 10 нм, то полимер относится к гибкоцепным. Персистентный механизм гибкости характерен для жесткоцепных полимеров. Жесткость цепи увеличивается: 1. При наличии объемных боковых заместителей; 2. При наличии циклов в основной цепи; 3. При наличии сопряжении в цепи. При увеличении размера заместителей в боковых цепях гибкость сначала изменяется незначительно, в дальнейшем, когда число атомов углерода в боковом радикале более 8, гибкость уменьшается (растет U 0) или увеличивается жесткость (сравнение ПММА, полигексилметакрилата и полиоктадецилакрилата, табл. ). Наличие циклов в цепи резко уменьшает или даже полностью исключает возможность внутримолекулярных вращений вокруг валентных связей, образующих главную цепь. Например, в двутяжевых полиорганосилоксанах величина сегмента Куна 20 – 30.
Персистентный механизм гибкости Для большинства гибкоцепных полимеров значение сегмента Куна находится в пределах 2 – 3 нм. Если длина сегмента Куна не превышает 10 нм, то полимер относится к гибкоцепным. Персистентный механизм гибкости характерен для жесткоцепных полимеров. Жесткость цепи увеличивается: 1. При наличии объемных боковых заместителей; 2. При наличии циклов в основной цепи; 3. При наличии сопряжении в цепи. При увеличении размера заместителей в боковых цепях гибкость сначала изменяется незначительно, в дальнейшем, когда число атомов углерода в боковом радикале более 8, гибкость уменьшается (растет U 0) или увеличивается жесткость (сравнение ПММА, полигексилметакрилата и полиоктадецилакрилата, табл. ). Наличие циклов в цепи резко уменьшает или даже полностью исключает возможность внутримолекулярных вращений вокруг валентных связей, образующих главную цепь. Например, в двутяжевых полиорганосилоксанах величина сегмента Куна 20 – 30.
 Персистентный механизм гибкости Особенно сильно жесткость цепи увеличивается при наличии в ней сопряжения атомов. Например, в полиамидах существует р- сопряжение в амидной группе, выраженное с помощью граничных структур. Алифатические полиамиды, где HNCO группы разделены гибкими метиленовыми мостиками (СН 2)n проявляют достаточно высокую гибкость. Ароматические полиамиды, где HNCO группы разделены бензольным ядром – жесткие полимеры. Полиалкилизоцианаты (поли-н-бутилизоцианат), где цепь построена только из амидных групп предельно жесткие (длина сегмента составляет до 100 нм, что соответствует ~500 звеньям); вращение в них не реализуется, так как нарушение сопряжения крайне энергетически не выгодно. Это можно выразить с помощью резонансных структур:
Персистентный механизм гибкости Особенно сильно жесткость цепи увеличивается при наличии в ней сопряжения атомов. Например, в полиамидах существует р- сопряжение в амидной группе, выраженное с помощью граничных структур. Алифатические полиамиды, где HNCO группы разделены гибкими метиленовыми мостиками (СН 2)n проявляют достаточно высокую гибкость. Ароматические полиамиды, где HNCO группы разделены бензольным ядром – жесткие полимеры. Полиалкилизоцианаты (поли-н-бутилизоцианат), где цепь построена только из амидных групп предельно жесткие (длина сегмента составляет до 100 нм, что соответствует ~500 звеньям); вращение в них не реализуется, так как нарушение сопряжения крайне энергетически не выгодно. Это можно выразить с помощью резонансных структур:
 Персистентный механизм гибкости При небольшой длине макромолекулы жесткоцепные полимеры находятся в конформации вытянутого стержня и “коленчатого вала”. В случае большой длины цепей изгибание возможно за счет деформации валентных углов, а также малых (до 3 %) колебаний длин связей – сущность персистентного механизма гибкости. Эта гибкость не велика, но благодаря ей, достаточно удаленные отрезки цепи могут ориентироваться независимо. Количественной характеристикой персистентной гибкости является персистентная длина l. На рис. S – контурная длина отрезка аб, - угол между касательными, проведенными к концам отрезка, характеризует величину изгибания отрезка цепи; соs - средний косинус угла изгибания (закручивания цепи).
Персистентный механизм гибкости При небольшой длине макромолекулы жесткоцепные полимеры находятся в конформации вытянутого стержня и “коленчатого вала”. В случае большой длины цепей изгибание возможно за счет деформации валентных углов, а также малых (до 3 %) колебаний длин связей – сущность персистентного механизма гибкости. Эта гибкость не велика, но благодаря ей, достаточно удаленные отрезки цепи могут ориентироваться независимо. Количественной характеристикой персистентной гибкости является персистентная длина l. На рис. S – контурная длина отрезка аб, - угол между касательными, проведенными к концам отрезка, характеризует величину изгибания отрезка цепи; соs - средний косинус угла изгибания (закручивания цепи).
 Персистентный механизм гибкости Значение косинуса угла изгибания изменяется по уравнению соs = е –S/l В зависимости от жесткости цепи возможны случаи: 1) Если l >> S, то соs → 1; это значит, что → 0° и данный отрезок близок к форме стержня; 2) Если l << S, то соs → 0; это значит, что → 90 ° и данный отрезок сильно искривлен; 3) Если S = l , то = 67 °. Таким образом, персистентная длина – это длина отрезка полимерной цепи, где угол между касательными, проведенными из концов отрезка, составляет 67 °. Чем больше l, тем выше жесткость полимера
Персистентный механизм гибкости Значение косинуса угла изгибания изменяется по уравнению соs = е –S/l В зависимости от жесткости цепи возможны случаи: 1) Если l >> S, то соs → 1; это значит, что → 0° и данный отрезок близок к форме стержня; 2) Если l << S, то соs → 0; это значит, что → 90 ° и данный отрезок сильно искривлен; 3) Если S = l , то = 67 °. Таким образом, персистентная длина – это длина отрезка полимерной цепи, где угол между касательными, проведенными из концов отрезка, составляет 67 °. Чем больше l, тем выше жесткость полимера
 Кинетическая гибкость Термодинамическая гибкость характеризует способность полимера изгибаться в пространстве, что предопределяется химическим строением макромолекул. Если в основной цепи для положения атомов водорода или заместителей существует хотя бы два термодинамически выгодных положения, обуславливающие возможность перехода из одного положения в другое в результате внутреннего вращения, то такой полимер потенциально способен изгибаться в пространстве и принимать большое количество конформаций (в случае ПВХ это транс и гош конформации). Однако реализация конформационных переходов зависит от величины потенциального барьера внутреннего вращения U 0 и наличия кинетической энергии Екин (k. T), необходимой для преодоления этого барьера. Кинетическая гибкость характеризует скорость перехода макромолекулы из одной конформации в другую, определяется соотношением между U 0 и Екин. Количественной мерой кинетической гибкости полимеров служит среднее время , необходимое для изменения конформации. Чем больше , тем ниже кинетическая гибкость цепи.
Кинетическая гибкость Термодинамическая гибкость характеризует способность полимера изгибаться в пространстве, что предопределяется химическим строением макромолекул. Если в основной цепи для положения атомов водорода или заместителей существует хотя бы два термодинамически выгодных положения, обуславливающие возможность перехода из одного положения в другое в результате внутреннего вращения, то такой полимер потенциально способен изгибаться в пространстве и принимать большое количество конформаций (в случае ПВХ это транс и гош конформации). Однако реализация конформационных переходов зависит от величины потенциального барьера внутреннего вращения U 0 и наличия кинетической энергии Екин (k. T), необходимой для преодоления этого барьера. Кинетическая гибкость характеризует скорость перехода макромолекулы из одной конформации в другую, определяется соотношением между U 0 и Екин. Количественной мерой кинетической гибкости полимеров служит среднее время , необходимое для изменения конформации. Чем больше , тем ниже кинетическая гибкость цепи.
 Поворотно-изомерный механизм гибкости Внутреннее вращение в полиэтилене Кривая потенциальной энергии конформационных изомеров в полиэтилене
Поворотно-изомерный механизм гибкости Внутреннее вращение в полиэтилене Кривая потенциальной энергии конформационных изомеров в полиэтилене
 Факторы, определяющие гибкость цепи полимера Схема, отражающая влияние строения и внешних условий на гибкость цепи полимера.
Факторы, определяющие гибкость цепи полимера Схема, отражающая влияние строения и внешних условий на гибкость цепи полимера.
 Факторы, определяющие гибкость цепи полимера Термодинамическая гибкость цепи сильно зависит от наличия кратных связей, циклов, сопряжения в основной цепи. Т. к. вращение относительно атомов, связанных кратными связями не возможно, то появление двойных или тройных связей в цепи ограничивает ее гибкость. Например, такие полимеры, как и -карбины, полиацетилен являются жесткими полимерами, имеют форму стержня (при небольшой ММ). При нагревании они не размягчаются, а при высоких температурах разлагаются. Однако, если кратные связи разделены гибкими фрагментами (метиленовыми цепочками), то такие полимеры проявляют гибкость. Полиизопрен, полибутадиен, полихлоропрен характеризуются высокой гибкостью, так как вращение относительно простой С-С связи, расположенной рядом с двойной ( С-С=С) осуществляется легче, чем С-С-С (влияние природы связи на U 0).
Факторы, определяющие гибкость цепи полимера Термодинамическая гибкость цепи сильно зависит от наличия кратных связей, циклов, сопряжения в основной цепи. Т. к. вращение относительно атомов, связанных кратными связями не возможно, то появление двойных или тройных связей в цепи ограничивает ее гибкость. Например, такие полимеры, как и -карбины, полиацетилен являются жесткими полимерами, имеют форму стержня (при небольшой ММ). При нагревании они не размягчаются, а при высоких температурах разлагаются. Однако, если кратные связи разделены гибкими фрагментами (метиленовыми цепочками), то такие полимеры проявляют гибкость. Полиизопрен, полибутадиен, полихлоропрен характеризуются высокой гибкостью, так как вращение относительно простой С-С связи, расположенной рядом с двойной ( С-С=С) осуществляется легче, чем С-С-С (влияние природы связи на U 0).
 Факторы, определяющие гибкость цепи полимера Наличие циклических фрагментов (фенильных, моносахаридных, имидных и т. п. ) сильно снижает гибкость цепи, так как вращение внутри цикла не возможно. Такие полимеры, как полифенилен, целлюлоза и ее производные, лестничные полиарилены, полиимиды, полимеры фталиевой кислоты, поли-п-бензамид являются достаточно жесткими полимерами, как правило не размягчающимися при нагревании или размягчающимися при очень высоких температурах: В сложных полиэфирах, полиамидах и т. д. термодинамическая гибкость дополнительно снижается из-за р- сопряжения в основной цепи. При этом алифатические сложные полиэфиры и полиамиды более гибкие, чем ароматические.
Факторы, определяющие гибкость цепи полимера Наличие циклических фрагментов (фенильных, моносахаридных, имидных и т. п. ) сильно снижает гибкость цепи, так как вращение внутри цикла не возможно. Такие полимеры, как полифенилен, целлюлоза и ее производные, лестничные полиарилены, полиимиды, полимеры фталиевой кислоты, поли-п-бензамид являются достаточно жесткими полимерами, как правило не размягчающимися при нагревании или размягчающимися при очень высоких температурах: В сложных полиэфирах, полиамидах и т. д. термодинамическая гибкость дополнительно снижается из-за р- сопряжения в основной цепи. При этом алифатические сложные полиэфиры и полиамиды более гибкие, чем ароматические.
 Факторы, определяющие гибкость цепи полимера Кинетическая гибкость цепи определяется: величиной U 0 , ММ полимера, частотой пространственной сетки, температурой и скоростью внешнего воздействия. Потенциальный барьер внутреннего вращения U 0 зависит от природы связи относительно которой осуществляется вращение, интенсивности внутри- и межмолекулярного взаимодействия, размера, количества и расположения заместителей. Наиболее низкие значения U 0 при вращении относительно связей С-О, С-S, С-N, C-Si, Si-O, Р-О, Р-N поэтому полимеры с такими связями, при отсутствии полярных заместителей, обладают высокой термодинамической и кинетической гибкостью: алифатические полиэфиры, полисилоксаны, полисульфиды и т. д. Карбоцепные полимеры, не содержащие полярных заместителей можно разделить на ненасыщенные и насыщенные. Вращение в элементе C-C=C судя по величине U 0 осуществляется гораздо легче: СН 3 -СН=СН 2 (8, 2 к. Дж/моль); СН 3 -СН 3 (18 к. Дж/моль) поэтому СКИ, СКД - полимеры, проявляющие свою гибкость при комнатной температуре, в отличие от ПЭ, ПП и т. д. кинетическая гибкость которых проявляется только при повышенных температурах (при высокой т/д гибкости).
Факторы, определяющие гибкость цепи полимера Кинетическая гибкость цепи определяется: величиной U 0 , ММ полимера, частотой пространственной сетки, температурой и скоростью внешнего воздействия. Потенциальный барьер внутреннего вращения U 0 зависит от природы связи относительно которой осуществляется вращение, интенсивности внутри- и межмолекулярного взаимодействия, размера, количества и расположения заместителей. Наиболее низкие значения U 0 при вращении относительно связей С-О, С-S, С-N, C-Si, Si-O, Р-О, Р-N поэтому полимеры с такими связями, при отсутствии полярных заместителей, обладают высокой термодинамической и кинетической гибкостью: алифатические полиэфиры, полисилоксаны, полисульфиды и т. д. Карбоцепные полимеры, не содержащие полярных заместителей можно разделить на ненасыщенные и насыщенные. Вращение в элементе C-C=C судя по величине U 0 осуществляется гораздо легче: СН 3 -СН=СН 2 (8, 2 к. Дж/моль); СН 3 -СН 3 (18 к. Дж/моль) поэтому СКИ, СКД - полимеры, проявляющие свою гибкость при комнатной температуре, в отличие от ПЭ, ПП и т. д. кинетическая гибкость которых проявляется только при повышенных температурах (при высокой т/д гибкости).
 Факторы, определяющие гибкость цепи полимера Полярные заместители увеличивают внутри- и межмолекулярное взаимодействие (а, следовательно, U 0 ), интенсивность которого зависит от степени полярности функциональных групп, расстояния между ними и симметричности расположения в цепи. Наиболее полярные группы: С N, NO 2 ; менее полярные: ОН, атомы Hal. Если полярные группы расположены редко, взаимодействие между ними практически не проявляется; величины U 0 в таких полимерах невелики и макромолекулы обладают высокой термодинамической и кинетической гибкостью (СКН-18). Близко расположенные полярные группы взаимодействуют между собой поэтому ПАН, ПВС, ПВХ менее гибкие (кинетическая гибкость проявляется при повышенных температурах).
Факторы, определяющие гибкость цепи полимера Полярные заместители увеличивают внутри- и межмолекулярное взаимодействие (а, следовательно, U 0 ), интенсивность которого зависит от степени полярности функциональных групп, расстояния между ними и симметричности расположения в цепи. Наиболее полярные группы: С N, NO 2 ; менее полярные: ОН, атомы Hal. Если полярные группы расположены редко, взаимодействие между ними практически не проявляется; величины U 0 в таких полимерах невелики и макромолекулы обладают высокой термодинамической и кинетической гибкостью (СКН-18). Близко расположенные полярные группы взаимодействуют между собой поэтому ПАН, ПВС, ПВХ менее гибкие (кинетическая гибкость проявляется при повышенных температурах).
 Факторы, определяющие гибкость цепи полимера Если полярные заместители расположены симметрично относительно какоголибо атома С, то суммарный дипольный момент снижается (или равен нулю), гибкость увеличивается – ПТФЭ, поливинилиденхлорид, политрифторхлорэтилен. В гетероцепных полимерах, где вращение происходит вокруг связей С-О, С-N, обладающих низкими значениями U 0 (полиамиды, сложные полиэфиры, полиуретаны) кинетическая гибкость цепи ограничена сильным межмолекулярным взаимодействием близко расположенных участков цепи, где проявляются различные виды взаимодействия, в частности водродное связывание:
Факторы, определяющие гибкость цепи полимера Если полярные заместители расположены симметрично относительно какоголибо атома С, то суммарный дипольный момент снижается (или равен нулю), гибкость увеличивается – ПТФЭ, поливинилиденхлорид, политрифторхлорэтилен. В гетероцепных полимерах, где вращение происходит вокруг связей С-О, С-N, обладающих низкими значениями U 0 (полиамиды, сложные полиэфиры, полиуретаны) кинетическая гибкость цепи ограничена сильным межмолекулярным взаимодействием близко расположенных участков цепи, где проявляются различные виды взаимодействия, в частности водродное связывание:
 Факторы, определяющие гибкость цепи полимера Большие по объему и массе заместители снижают гибкость цепи. При наличии у одного и того же атома углерода двух заместителей, гибкость, напротив увеличивается из-за того, что их влияние на барьер внутреннего вращения частично компенсируется. Например, макромолекулы ПС, содержащие объемный заместитель обладают меньшей кинетической гибкостью по сравнению с ПЭ, ПП; ПММА более гибкий, чем ПМА; это отражается на величинах температуры стеклования полимеров. У ПММА Тс на 10 С ниже, чем у ПМА, а у поливинилиденхлорида Тс ниже на 40 С по сравнению с поливинилхлоридом. С увеличением ММ возрастает число возможных конформаций, которые может принимать молекула. Так, n сегментам цепи соответствует 2 n+1 конформаций. Поэтому полимеры с высокой ММ более гибкие.
Факторы, определяющие гибкость цепи полимера Большие по объему и массе заместители снижают гибкость цепи. При наличии у одного и того же атома углерода двух заместителей, гибкость, напротив увеличивается из-за того, что их влияние на барьер внутреннего вращения частично компенсируется. Например, макромолекулы ПС, содержащие объемный заместитель обладают меньшей кинетической гибкостью по сравнению с ПЭ, ПП; ПММА более гибкий, чем ПМА; это отражается на величинах температуры стеклования полимеров. У ПММА Тс на 10 С ниже, чем у ПМА, а у поливинилиденхлорида Тс ниже на 40 С по сравнению с поливинилхлоридом. С увеличением ММ возрастает число возможных конформаций, которые может принимать молекула. Так, n сегментам цепи соответствует 2 n+1 конформаций. Поэтому полимеры с высокой ММ более гибкие.
 Факторы, определяющие гибкость цепи полимера Сшивание существенно ограничивает подвижность участка цепи. В ряду: каучук (не сшитый), резина (редко сшитый), эбонит (густо сшитый) по мере роста степени сшивания снижается гибкость и нарастает жесткость материала. Густо сшитые полимеры утрачивают способность к обратимой деформации, набуханию в растворителях. Скорость внешнего воздействия оказывает влияние на кинетическую гибкость полимеров, т. к. для перехода из одной конформации в другую необходимо определенное количество времени. При больших скоростях воздействия на образец полимера конформации не успевают изменяться, поэтому образец, не смотря на потенциальную термодинамическую гибкость ведет себя как жесткий полимер. С повышением температуры U 0 не изменяется, но возрастает энергия теплового движения. Однако, если k. T U 0, то даже термодинамически гибкие полимеры не могут проявить свою кинетическую гибкость.
Факторы, определяющие гибкость цепи полимера Сшивание существенно ограничивает подвижность участка цепи. В ряду: каучук (не сшитый), резина (редко сшитый), эбонит (густо сшитый) по мере роста степени сшивания снижается гибкость и нарастает жесткость материала. Густо сшитые полимеры утрачивают способность к обратимой деформации, набуханию в растворителях. Скорость внешнего воздействия оказывает влияние на кинетическую гибкость полимеров, т. к. для перехода из одной конформации в другую необходимо определенное количество времени. При больших скоростях воздействия на образец полимера конформации не успевают изменяться, поэтому образец, не смотря на потенциальную термодинамическую гибкость ведет себя как жесткий полимер. С повышением температуры U 0 не изменяется, но возрастает энергия теплового движения. Однако, если k. T U 0, то даже термодинамически гибкие полимеры не могут проявить свою кинетическую гибкость.
 Агрегатные и фазовые состояния полимеров Особенности для полимеров: 1. У полимеров отсутствует газообразное агрегатное состояние, т. к. суммарная энергия физических связей намного превышает энергию химической связи в основной цепи. Попытки испарения полимеров нагреванием приводят к их термической деструкции. 2. В полимерах в тепловом движении участвуют структурные единицы макромолекул, при этом основными кинетическими единицами являются: атомы и группы атомов, совершающие колебательные движения; сегменты, совершающие поступательные, вращательные движения.
Агрегатные и фазовые состояния полимеров Особенности для полимеров: 1. У полимеров отсутствует газообразное агрегатное состояние, т. к. суммарная энергия физических связей намного превышает энергию химической связи в основной цепи. Попытки испарения полимеров нагреванием приводят к их термической деструкции. 2. В полимерах в тепловом движении участвуют структурные единицы макромолекул, при этом основными кинетическими единицами являются: атомы и группы атомов, совершающие колебательные движения; сегменты, совершающие поступательные, вращательные движения.
 Агрегатные и фазовые состояния полимеров 1. Газообразное фазовое состояние для полимеров отсутствует. 2. Кристаллическое фазовое состояние полимеров - трехмерный дальний порядок в расположении сегментов и/или макромолекул. Жидкое (аморфное) фазовое состояние - ближний порядок в расположении сегментов. 3. Полимеры определенного строения могут существовать в мезоморфном (промежуточном) фазовом состоянии – жидкие кристаллы, где существует одно - или двумерный дальний порядок в расположении макромолекул.
Агрегатные и фазовые состояния полимеров 1. Газообразное фазовое состояние для полимеров отсутствует. 2. Кристаллическое фазовое состояние полимеров - трехмерный дальний порядок в расположении сегментов и/или макромолекул. Жидкое (аморфное) фазовое состояние - ближний порядок в расположении сегментов. 3. Полимеры определенного строения могут существовать в мезоморфном (промежуточном) фазовом состоянии – жидкие кристаллы, где существует одно - или двумерный дальний порядок в расположении макромолекул.
 Агрегатные, фазовые и физические состояния полимеров 4. По агрегатному состоянию аморфные полимеры могут быть: - твердыми (стеклообразные полимеры); - жидкими (выше температуры плавления). По агрегатному состоянию трехмерные кристаллические полимеры – твердые. Для описания деформационных свойств полимеров понятий об агрегатном и фазовом состояниях недостаточно; вводится понятие о физических состояниях полимера, которые различаются природой структурных элементов, участвующих в тепловом движении.
Агрегатные, фазовые и физические состояния полимеров 4. По агрегатному состоянию аморфные полимеры могут быть: - твердыми (стеклообразные полимеры); - жидкими (выше температуры плавления). По агрегатному состоянию трехмерные кристаллические полимеры – твердые. Для описания деформационных свойств полимеров понятий об агрегатном и фазовом состояниях недостаточно; вводится понятие о физических состояниях полимера, которые различаются природой структурных элементов, участвующих в тепловом движении.
 Термомеханический метод исследования полимеров Температурные интервалы существования полимера в том или ином физическом состоянии можно определить термомеханическим методом, суть которого - измерение величины деформации образца полимера под действием силы F в условиях непрерывного нагрева. Результат - зависимость = f(Т), которая называется термомеханической кривой (ТМК). Для аморфного линейного гибкоцепного полимера: на ТМК четыре участка, разделенные температурой стеклования Тс и температурой текучести Тт. Каждому из состояний соответствует определенный тип деформаций: упругая в стеклообразном, высокоэластическая в высокоэластическом, вязкого течения (или пластическая) в вязкотекучем состоянии.
Термомеханический метод исследования полимеров Температурные интервалы существования полимера в том или ином физическом состоянии можно определить термомеханическим методом, суть которого - измерение величины деформации образца полимера под действием силы F в условиях непрерывного нагрева. Результат - зависимость = f(Т), которая называется термомеханической кривой (ТМК). Для аморфного линейного гибкоцепного полимера: на ТМК четыре участка, разделенные температурой стеклования Тс и температурой текучести Тт. Каждому из состояний соответствует определенный тип деформаций: упругая в стеклообразном, высокоэластическая в высокоэластическом, вязкого течения (или пластическая) в вязкотекучем состоянии.
; в тепловом движении участвуют атомы или группы атомов,") I участок - стеклообразное состояние (СТС); в тепловом движении участвуют атомы или группы атомов, совершающие колебательные движения относительно положения равновесия. Реализуется упругая деформация, связанная с незначительным изменением валентных углов и длин связей; по величине - мала (не более 1 %), обратима; практически не зависит от температуры. II участок (переходный) – появляется подвижность сегментов, полимер “расстекловывается”; величина деформации возрастает с ростом температуры; полимер уже не ведет себя как твердое, жесткое тело, но и не является пока истинным эластомером. III участок – высокоэластическое состояние (ВЭС); в тепловом движении участвуют атомы и группы атомов, колеблющиеся относительно положения равновесия, сегменты, которые перемещаются в поле действия механических сил, что сопровождается изменением формы макромолекул. ВЭД обратима; величина достигает нескольких сотен процентов, практически не зависит от температуры. IV участок - вязкотекучее состояние (ВТС), когда движение сегментов в поле действия механических сил приводит к смещению центра тяжести макромолекул, перемещению макромолекул относительно друга – необратимая деформация течения, величина которой увеличивается с ростом температуры. Если при снятии ТМК не происходит деструкция, то при медленном охлаждении образец последовательно переходит из ВТС в ВЭС и СТС соответственно.
I участок - стеклообразное состояние (СТС); в тепловом движении участвуют атомы или группы атомов, совершающие колебательные движения относительно положения равновесия. Реализуется упругая деформация, связанная с незначительным изменением валентных углов и длин связей; по величине - мала (не более 1 %), обратима; практически не зависит от температуры. II участок (переходный) – появляется подвижность сегментов, полимер “расстекловывается”; величина деформации возрастает с ростом температуры; полимер уже не ведет себя как твердое, жесткое тело, но и не является пока истинным эластомером. III участок – высокоэластическое состояние (ВЭС); в тепловом движении участвуют атомы и группы атомов, колеблющиеся относительно положения равновесия, сегменты, которые перемещаются в поле действия механических сил, что сопровождается изменением формы макромолекул. ВЭД обратима; величина достигает нескольких сотен процентов, практически не зависит от температуры. IV участок - вязкотекучее состояние (ВТС), когда движение сегментов в поле действия механических сил приводит к смещению центра тяжести макромолекул, перемещению макромолекул относительно друга – необратимая деформация течения, величина которой увеличивается с ростом температуры. Если при снятии ТМК не происходит деструкция, то при медленном охлаждении образец последовательно переходит из ВТС в ВЭС и СТС соответственно.
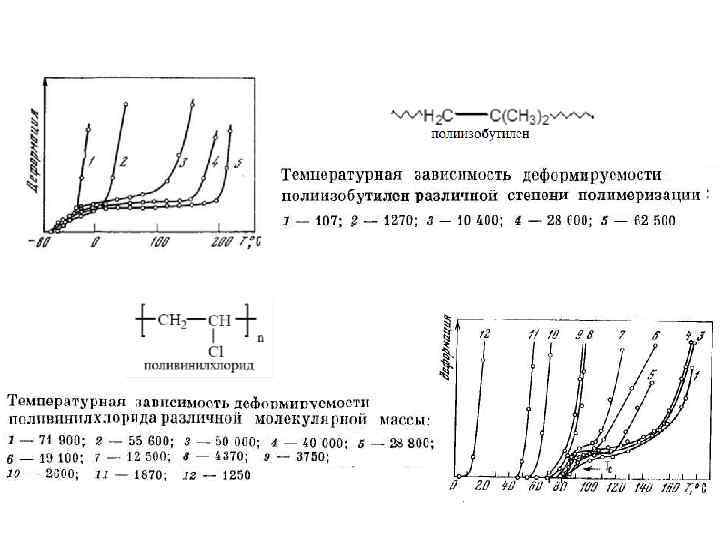
 Стеклообразное состояние ограничено температурами хрупкости Тхр и Тс. Высокоэластическое - температурами стеклования Тс и текучести Тт; Вязкотекучее – температурами текучести Тт и начала деструкции Тр. Эластомеры перерабатываются в области Тт – Тр, а эксплуатируются Тс – Тт. Большинство пластомеров перерабатываются Тт – Тр, а эксплуатируются Тхр – Тс. Вид ТМК для аморфных линейных полимеров может изменяться в зависимости от гибкости макромолекул (а), ММ полимера (б). Так как для ВЭС характерна ВЭД, связанная с раскручиванием молекулярных клубков, то с увеличением жесткости цепей интервал ВЭС смещается в область более высоких температур (необходимо применить более высокие температуры для преодоления U 0 , увеличивающегося с ростом полярности, количества и объема заместителей, циклических фрагментов и т. д. ). Для очень жестких полимеров область ВЭС “вырождается”. ТМА- кривые полимеров: 1 - линейного гибкоцепного с высокой ММ; 2 – линейного с умеренной жесткостью; 3 - линейного с очень жесткими цепями.
Стеклообразное состояние ограничено температурами хрупкости Тхр и Тс. Высокоэластическое - температурами стеклования Тс и текучести Тт; Вязкотекучее – температурами текучести Тт и начала деструкции Тр. Эластомеры перерабатываются в области Тт – Тр, а эксплуатируются Тс – Тт. Большинство пластомеров перерабатываются Тт – Тр, а эксплуатируются Тхр – Тс. Вид ТМК для аморфных линейных полимеров может изменяться в зависимости от гибкости макромолекул (а), ММ полимера (б). Так как для ВЭС характерна ВЭД, связанная с раскручиванием молекулярных клубков, то с увеличением жесткости цепей интервал ВЭС смещается в область более высоких температур (необходимо применить более высокие температуры для преодоления U 0 , увеличивающегося с ростом полярности, количества и объема заместителей, циклических фрагментов и т. д. ). Для очень жестких полимеров область ВЭС “вырождается”. ТМА- кривые полимеров: 1 - линейного гибкоцепного с высокой ММ; 2 – линейного с умеренной жесткостью; 3 - линейного с очень жесткими цепями.
 Тст растет с ростом ММ, область ВЭС") В случае олигомеров (образцы 1 – 4) Тст растет с ростом ММ, область ВЭС состояния отсутствует. Зависимость Тст от ММ прекращается, когда молекула приобретает гибкость (длина молекулы заметно больше длины сегмента). С ростом ММ макромолекул (образцы 5 – 9) Тт растет, температурный интервал ВЭС расширяется.
В случае олигомеров (образцы 1 – 4) Тст растет с ростом ММ, область ВЭС состояния отсутствует. Зависимость Тст от ММ прекращается, когда молекула приобретает гибкость (длина молекулы заметно больше длины сегмента). С ростом ММ макромолекул (образцы 5 – 9) Тт растет, температурный интервал ВЭС расширяется.
, однако область высокоэластического состояния ограничена") Вид ТМК для аморфного слабо сшитого полимера аналогичный (а), однако область высокоэластического состояния ограничена температурой начала химического разложения (термодеструкции). Выше Тр пластическая деформация осуществляется за счет перемещения осколков макромолекул – продуктов деструкции. Вид ТМК для кристаллического полимера зависит от содержания кристаллической фазы (степени кристалличности) и температуры ее плавления (разупорядочивания). Если Тпл кристаллической фазы ниже температуры текучести Тт, т. е. Тс Тпл Тт, то полимер после разупорядочивания кристаллической фазы переходит в ВЭС, а затем в вязкотекучее (б). Если Тт Тпл, то полимер сразу переходит в вязкотекучее состояние, вид ТМК иной (в).
Вид ТМК для аморфного слабо сшитого полимера аналогичный (а), однако область высокоэластического состояния ограничена температурой начала химического разложения (термодеструкции). Выше Тр пластическая деформация осуществляется за счет перемещения осколков макромолекул – продуктов деструкции. Вид ТМК для кристаллического полимера зависит от содержания кристаллической фазы (степени кристалличности) и температуры ее плавления (разупорядочивания). Если Тпл кристаллической фазы ниже температуры текучести Тт, т. е. Тс Тпл Тт, то полимер после разупорядочивания кристаллической фазы переходит в ВЭС, а затем в вязкотекучее (б). Если Тт Тпл, то полимер сразу переходит в вязкотекучее состояние, вид ТМК иной (в).
 Деформационные свойства аморфных полимеров. стеклообразное состояние, сущность Стеклование – переход полимера из ВЭС или ВТС в СТС. Различают структурное (истинное) стеклование и механическое. Механизм структурного стеклования (переход полимера из ВЭС или ВТС в СТС при охлаждении). Надмолекулярная структура аморфного полимера флуктуационная сетка. Под действием теплового движения или теплового движения и Емех одновременно, узлы сетки распадаются в одном месте и возникают в другом. С понижением Т уменьшается энергия теплового движения сегментов и увеличивается время жизни узлов сетки; при некоторых значениях Т энергии теплового движения недостаточно для преодоления межмолекулярных взаимодействий в узлах флуктуационной сетки; одна из случайных структур ближнего порядка фиксируется; полимер застекловывается, ведет себя как твердое, жесткое тело, не способное к проявлению ВЭД (отсутствует сегментальная подвижность). Прекращению сегментальной подвижности способствует уменьшение свободного объема в образце полимера с понижением температуры. Коэффициент теплового сжатия для эластомеров составляет 6 -7 10 -4 1/град. Если свободный объем становится менее 2, 5 % от общего объема полимера, то тепловое движение сегментов прекращается. Температура, при которой это происходит - Тс.
Деформационные свойства аморфных полимеров. стеклообразное состояние, сущность Стеклование – переход полимера из ВЭС или ВТС в СТС. Различают структурное (истинное) стеклование и механическое. Механизм структурного стеклования (переход полимера из ВЭС или ВТС в СТС при охлаждении). Надмолекулярная структура аморфного полимера флуктуационная сетка. Под действием теплового движения или теплового движения и Емех одновременно, узлы сетки распадаются в одном месте и возникают в другом. С понижением Т уменьшается энергия теплового движения сегментов и увеличивается время жизни узлов сетки; при некоторых значениях Т энергии теплового движения недостаточно для преодоления межмолекулярных взаимодействий в узлах флуктуационной сетки; одна из случайных структур ближнего порядка фиксируется; полимер застекловывается, ведет себя как твердое, жесткое тело, не способное к проявлению ВЭД (отсутствует сегментальная подвижность). Прекращению сегментальной подвижности способствует уменьшение свободного объема в образце полимера с понижением температуры. Коэффициент теплового сжатия для эластомеров составляет 6 -7 10 -4 1/град. Если свободный объем становится менее 2, 5 % от общего объема полимера, то тепловое движение сегментов прекращается. Температура, при которой это происходит - Тс.
 Стеклообразное состояние полимеров Механическое стеклование характерно для полимеров, находящихся в ВЭС; заключается в потере способности полимера к ВЭД при увеличении скорости деформации. Когда скорость действия силы велика и превышает скорость теплового движения сегментов, то в нем не успевает развиваться ВЭД и полимер ведет себя как застеклованный, твердый полимер. Механическое стеклование отличается от структурного тем, что тепловое движение сегментов не прекращается, структура полимера не фиксируется. При прочих равных условиях, чем больше скорость действия силы, тем выше Тс. Факторы, влияющие на температуру стеклования: - Скорость охлаждения; - Гибкость цепи полимера (интенсивность внутри- и межмолекулярного взаимодействия, объем и расположение заместителей); - Молекулярная масса полимера.
Стеклообразное состояние полимеров Механическое стеклование характерно для полимеров, находящихся в ВЭС; заключается в потере способности полимера к ВЭД при увеличении скорости деформации. Когда скорость действия силы велика и превышает скорость теплового движения сегментов, то в нем не успевает развиваться ВЭД и полимер ведет себя как застеклованный, твердый полимер. Механическое стеклование отличается от структурного тем, что тепловое движение сегментов не прекращается, структура полимера не фиксируется. При прочих равных условиях, чем больше скорость действия силы, тем выше Тс. Факторы, влияющие на температуру стеклования: - Скорость охлаждения; - Гибкость цепи полимера (интенсивность внутри- и межмолекулярного взаимодействия, объем и расположение заместителей); - Молекулярная масса полимера.
 Факторы, влияющие на температуру стеклования аморфных полимеров Скорость охлаждения. При медленном охлаждении сегменты успевают перемещаться даже при температурах близких к Тс, поэтому требуется сильнее охладить полимер, для прекращения любой перестройки. Например, если охлаждение ПВА проводить так, что при каждой температуре выдерживать в одном случае 0, 02 ч, а в другом - 100 ч, то значения Тс составляют 32 и 23 С. Т. о. чем выше скорость охлаждения, тем выше измеренная Тс. Гибкость цепи полимера. Неполярные полимеры с высокой гибкостью характеризуются низкими значениями Тс, например: НК -70; ПИБ -74, СКД -105 С соответственно. Наличие полярных групп увеличивает межмолекулярное взаимодействие и повышает Тс, например, полихлоропрен -40 С. Увеличение количества полярных групп повышает Тс, например в ряду СКН-18, СКН-26, СКН-40 изменяется -50; -30; -20 С соответственно.
Факторы, влияющие на температуру стеклования аморфных полимеров Скорость охлаждения. При медленном охлаждении сегменты успевают перемещаться даже при температурах близких к Тс, поэтому требуется сильнее охладить полимер, для прекращения любой перестройки. Например, если охлаждение ПВА проводить так, что при каждой температуре выдерживать в одном случае 0, 02 ч, а в другом - 100 ч, то значения Тс составляют 32 и 23 С. Т. о. чем выше скорость охлаждения, тем выше измеренная Тс. Гибкость цепи полимера. Неполярные полимеры с высокой гибкостью характеризуются низкими значениями Тс, например: НК -70; ПИБ -74, СКД -105 С соответственно. Наличие полярных групп увеличивает межмолекулярное взаимодействие и повышает Тс, например, полихлоропрен -40 С. Увеличение количества полярных групп повышает Тс, например в ряду СКН-18, СКН-26, СКН-40 изменяется -50; -30; -20 С соответственно.
 Факторы, влияющие на температуру стеклования аморфных полимеров На Тс оказывает влияние расположение полярных групп: полярные связи С-Сl в поливинилиденхлориде расположены симметрично, электрические полярных связей взаимно компенсируются, Тс -17 С, для ПВХ Тс составляет 80 С, цепи макромолекул при комнатной температуре жесткие. Наличие объемных заместителей ведет к росту Тс , например, в сополимерах стирола и бутадиена в ряду: СКС-10, СКС-30, СКС-60 -80 -72 -24 С соответственно. Молекулярная масса полимера влияет на Тс: при переходе от олигомера к полимеру Тс возрастает, достигая постоянного значения; с дальнейшим ростом ММ полимера практически не изменяется. Влияние молекулярной массы полимера на величины Тхр, Тс, Тт
Факторы, влияющие на температуру стеклования аморфных полимеров На Тс оказывает влияние расположение полярных групп: полярные связи С-Сl в поливинилиденхлориде расположены симметрично, электрические полярных связей взаимно компенсируются, Тс -17 С, для ПВХ Тс составляет 80 С, цепи макромолекул при комнатной температуре жесткие. Наличие объемных заместителей ведет к росту Тс , например, в сополимерах стирола и бутадиена в ряду: СКС-10, СКС-30, СКС-60 -80 -72 -24 С соответственно. Молекулярная масса полимера влияет на Тс: при переходе от олигомера к полимеру Тс возрастает, достигая постоянного значения; с дальнейшим ростом ММ полимера практически не изменяется. Влияние молекулярной массы полимера на величины Тхр, Тс, Тт
 Деформационные свойства аморфных полимеров Деформационные свойства характеризуют способность полимеров деформироваться под действием нагрузки (внешних мех. сил) без разрушения. Величина деформирующей силы характеризуется напряжением, т. е. силой, приходящейся на единицу площади поперечного сечения образца. Различают условное f и истинное напряжение при деформировании: f = Р/S 0; = Р/S , где Р–деформирующая сила, Н (кгс); S 0 -площадь поперечного сечения образца до нагружения, м 2; S –площадь поперечного сечения образца в процессе деформирования , м 2. Условное напряжение f - сила, приходящаяся на единицу площади поперечного сечения исходного образца (до деформирования), Н/м 2 или Па. Истинное напряжение – сила, приходящаяся на единицу площади поперечного сечения образца в процессе деформирования (например, при удлинениях 50, 100, 200 % в процессе растяжения, когда площадь поперечного сечения образца изменяется). Различают три вида механического воздействия: простой сдвиг, одноосное растяжение, всестороннее сжатие (или растяжение). Деформационные свойства полимеров оцениваются зависимостью = f ( ) – деформационная (механическая) кривая.
Деформационные свойства аморфных полимеров Деформационные свойства характеризуют способность полимеров деформироваться под действием нагрузки (внешних мех. сил) без разрушения. Величина деформирующей силы характеризуется напряжением, т. е. силой, приходящейся на единицу площади поперечного сечения образца. Различают условное f и истинное напряжение при деформировании: f = Р/S 0; = Р/S , где Р–деформирующая сила, Н (кгс); S 0 -площадь поперечного сечения образца до нагружения, м 2; S –площадь поперечного сечения образца в процессе деформирования , м 2. Условное напряжение f - сила, приходящаяся на единицу площади поперечного сечения исходного образца (до деформирования), Н/м 2 или Па. Истинное напряжение – сила, приходящаяся на единицу площади поперечного сечения образца в процессе деформирования (например, при удлинениях 50, 100, 200 % в процессе растяжения, когда площадь поперечного сечения образца изменяется). Различают три вида механического воздействия: простой сдвиг, одноосное растяжение, всестороннее сжатие (или растяжение). Деформационные свойства полимеров оцениваются зависимостью = f ( ) – деформационная (механическая) кривая.
 Аморфные полимеры при температурах Т < Тхр ведут себя как жесткие, хрупкие материалы (рис. а), способные только к упругой деформации, обратимой и малой по величине (единицы %), подчиняющейся закону Гука: = Е* , где Е – модуль упругости, МПа; - относительная деформация равная (l - l 0 )/l, %; - истинное напряжение, МПа. Рис. Механические кривые аморфного стеклообразного полимера: а – жесткого и хрупкого (ниже Тхр); б – жесткого, но пластичного (в интервале Тхр < Тс); I и III – область упругой деформации; II – область вынужденной высокоэластической деформации; в – предел вынужденной эластичности В температурном интервале Тхр < Тс аморфные полимеры способны вести себя как жесткие, но пластичные материалы, способные существенно деформироваться (десятки, сотню %) приложении нагрузки (рис. б). Этот вид деформации называется вынужденной эластической деформацией (вынужденной-высокоэластической). На кривой - три участка.
Аморфные полимеры при температурах Т < Тхр ведут себя как жесткие, хрупкие материалы (рис. а), способные только к упругой деформации, обратимой и малой по величине (единицы %), подчиняющейся закону Гука: = Е* , где Е – модуль упругости, МПа; - относительная деформация равная (l - l 0 )/l, %; - истинное напряжение, МПа. Рис. Механические кривые аморфного стеклообразного полимера: а – жесткого и хрупкого (ниже Тхр); б – жесткого, но пластичного (в интервале Тхр < Тс); I и III – область упругой деформации; II – область вынужденной высокоэластической деформации; в – предел вынужденной эластичности В температурном интервале Тхр < Тс аморфные полимеры способны вести себя как жесткие, но пластичные материалы, способные существенно деформироваться (десятки, сотню %) приложении нагрузки (рис. б). Этот вид деформации называется вынужденной эластической деформацией (вынужденной-высокоэластической). На кривой - три участка.
 I участок. Прямолинейный отрезок оа соответствует упругой деформации, угол наклона прямой к оси абсцисс велик. В области ав прямолинейная зависимость нарушается. Это связано с концентрацией напряжения в месте дефекта в образце полимера (микротрещина). Напряжение в вершине микротрещины в несколько раз превышает действующее в образце. В результате в области вершины микротрещины под действием перенапряжения происходит вынужденное перемещение сегментов: раскручивание части молекулярных клубков и ориентация сегментов в направлении действия силы. Ориентация сегментов в области микродефекта приводит к упрочнению, трещина дальше не растет. В том месте, где произошла ориентация, возникает шейка; ее возникновение соответствует максимуму на кривой. Напряжение, при котором в стеклообразном полимере начинает развиваться вынужденная эластическая деформация, называется пределом вынужденной эластичности в. Тхр – это температура, при которой полимер разрушается, не достигая предела вынужденной эластичности.
I участок. Прямолинейный отрезок оа соответствует упругой деформации, угол наклона прямой к оси абсцисс велик. В области ав прямолинейная зависимость нарушается. Это связано с концентрацией напряжения в месте дефекта в образце полимера (микротрещина). Напряжение в вершине микротрещины в несколько раз превышает действующее в образце. В результате в области вершины микротрещины под действием перенапряжения происходит вынужденное перемещение сегментов: раскручивание части молекулярных клубков и ориентация сегментов в направлении действия силы. Ориентация сегментов в области микродефекта приводит к упрочнению, трещина дальше не растет. В том месте, где произошла ориентация, возникает шейка; ее возникновение соответствует максимуму на кривой. Напряжение, при котором в стеклообразном полимере начинает развиваться вынужденная эластическая деформация, называется пределом вынужденной эластичности в. Тхр – это температура, при которой полимер разрушается, не достигая предела вынужденной эластичности.
 II участок. Продолжающееся растяжение приводит к постепенному увеличению области ориентации за счет раскручивания молекулярных клубков; область шейки растет постепенно, пока весь образец не перейдет в шейку (кроме зажимов); при этом в образце остается практически постоянным; на кривой горизонтальный участок, где величина деформации может достигать сотню процентов. Если образец освободить из зажимов, то он, будучи, застеклованным, не сократится, но при нагревании выше Тс - сократится до размеров близких к исходному. Т. о. , при растяжении молекулярные клубки разворачиваются в направлении действия силы, принимая развернутые конформации, а после нагревания – сворачиваются под действием теплового движения. Так как разворачивание клубков происходит вынужденно под действием возникших в образце перенапряжений, то деформация называется вынужденно-эластической. Изменение формы образца при растяжении: а – исходный образец до деформирования, б – появление шейки, в – переход всего образца в шейку, г – разрыв образца III участок. При дальнейшем растяжении, вплоть до разрушения, образец растягивается как единое целое, перестройки структуры не происходит; ориентированный образец деформируется за счет изменения валентных углов, длин связей; величина деформации - единицы процентов.
II участок. Продолжающееся растяжение приводит к постепенному увеличению области ориентации за счет раскручивания молекулярных клубков; область шейки растет постепенно, пока весь образец не перейдет в шейку (кроме зажимов); при этом в образце остается практически постоянным; на кривой горизонтальный участок, где величина деформации может достигать сотню процентов. Если образец освободить из зажимов, то он, будучи, застеклованным, не сократится, но при нагревании выше Тс - сократится до размеров близких к исходному. Т. о. , при растяжении молекулярные клубки разворачиваются в направлении действия силы, принимая развернутые конформации, а после нагревания – сворачиваются под действием теплового движения. Так как разворачивание клубков происходит вынужденно под действием возникших в образце перенапряжений, то деформация называется вынужденно-эластической. Изменение формы образца при растяжении: а – исходный образец до деформирования, б – появление шейки, в – переход всего образца в шейку, г – разрыв образца III участок. При дальнейшем растяжении, вплоть до разрушения, образец растягивается как единое целое, перестройки структуры не происходит; ориентированный образец деформируется за счет изменения валентных углов, длин связей; величина деформации - единицы процентов.
 Изделия из пластомеров эксплуатируются в температурном интервале вынужденной эластичности. На температурный интервал вынужденной эластичности влияет: 1. величина межмолекулярного взаимодействия. С ее ростом Тхр понижается, благодаря образованию полярными группами межмолекулярных связей; 2. плотность упаковки макромолекул (величина свободного объема в полимере). При стекловании менее гибкие макромолекулы не успевают плотно упаковываться, величина свободного объема несколько выше; конформационных возможностей при вынужденно-эластической деформации больше; шире температурный интервал вынужденной эластичности. 3. ММ полимера. У олигомеров значения Тс и Тхр совпадают. Когда молекулы становятся достаточно длинными и появляется гибкость, Тс растет быстрее, чем Тхр и возникает температурный интервал вынужденной эластичности. При дальнейшем росте ММ Тхр даже несколько понижается, что приводит к увеличению интервала вынужденной эластичности. Зависимость температур стеклования и хрупкости от ММ полимера
Изделия из пластомеров эксплуатируются в температурном интервале вынужденной эластичности. На температурный интервал вынужденной эластичности влияет: 1. величина межмолекулярного взаимодействия. С ее ростом Тхр понижается, благодаря образованию полярными группами межмолекулярных связей; 2. плотность упаковки макромолекул (величина свободного объема в полимере). При стекловании менее гибкие макромолекулы не успевают плотно упаковываться, величина свободного объема несколько выше; конформационных возможностей при вынужденно-эластической деформации больше; шире температурный интервал вынужденной эластичности. 3. ММ полимера. У олигомеров значения Тс и Тхр совпадают. Когда молекулы становятся достаточно длинными и появляется гибкость, Тс растет быстрее, чем Тхр и возникает температурный интервал вынужденной эластичности. При дальнейшем росте ММ Тхр даже несколько понижается, что приводит к увеличению интервала вынужденной эластичности. Зависимость температур стеклования и хрупкости от ММ полимера
 Деформационные свойства аморфных полимеров. высокоэластическое состояние, сущность Полимер в ВЭС, т. е. в температурном интервале Тс – Тт характеризуется высокой подвижностью сегментов; способен к большим обратимым деформациям приложении небольших механических нагрузок. Особенно ВЭС выражено у гибкоцепных полимеров с невысоким внутри и межмолекулярным взаимодействием. В случае интенсивного физического взаимодействия (полужесткие полимеры) температурный интервал ВЭС смещается в область повышенных температур. Сущность ВЭД рассматривается с точки зрения термодинамической и молекулярно-кинетической теорий высокоэластического состояния. Сущность ВЭД с точки зрения молекулярной теории состоит в изменении конформации макромолекул под действием внешних сил и возвращении к исходной после снятия нагрузки: статистический клубок – вытянутая макромолекула – статистический клубок. Равновесное состояние гибких макромолекул - молекулярный клубок. При нагружении образца полимера тепловое движение сегментов препятствует растягивающей силе. Однако это противодействие не велико и мол. клубки при небольших нагрузках раскручиваются, что сопровождается большим удлинением образца. После снятия нагрузки макромолекулы за счет теплового движения сегментов возвращаются в наиболее выгодное состояние мол. клубка.
Деформационные свойства аморфных полимеров. высокоэластическое состояние, сущность Полимер в ВЭС, т. е. в температурном интервале Тс – Тт характеризуется высокой подвижностью сегментов; способен к большим обратимым деформациям приложении небольших механических нагрузок. Особенно ВЭС выражено у гибкоцепных полимеров с невысоким внутри и межмолекулярным взаимодействием. В случае интенсивного физического взаимодействия (полужесткие полимеры) температурный интервал ВЭС смещается в область повышенных температур. Сущность ВЭД рассматривается с точки зрения термодинамической и молекулярно-кинетической теорий высокоэластического состояния. Сущность ВЭД с точки зрения молекулярной теории состоит в изменении конформации макромолекул под действием внешних сил и возвращении к исходной после снятия нагрузки: статистический клубок – вытянутая макромолекула – статистический клубок. Равновесное состояние гибких макромолекул - молекулярный клубок. При нагружении образца полимера тепловое движение сегментов препятствует растягивающей силе. Однако это противодействие не велико и мол. клубки при небольших нагрузках раскручиваются, что сопровождается большим удлинением образца. После снятия нагрузки макромолекулы за счет теплового движения сегментов возвращаются в наиболее выгодное состояние мол. клубка.
 Сущность ВЭД с точки зрения термодинамической теории. Допущения: деформируется идеальный каучук без изменения объема при Т= const, при этом ВЭД не сопровождается деформацией течения. Примем, что в ходе растяжения образец идеального каучука длиной l 0 под действием силы f удлинился на величину dl. Поскольку ВЭД является обратимым процессом, происходящим при постоянных Т и V (изохорноизотермический процессом), то согласно второму закону термодинамики изменение свободной энергии d. F (изохорно-изотермического потенциала) в процессе деформирования определяется соотношением: d. F = d. U – Td. S, (1) где d. U и d. S – соответственно изменение внутренней энергии и энтропии в процессе деформации. Изменение свободной энергии d. F, кроме того, может быть выражено через работу растяжения d. A = f dl. Тогда уравнение изменения свободной энергии перепишется в виде: f dl = d. U – Td. S или f = (d. U/dl)T – T(d. S/dl)T (2) Из 2 следует, что в полимере возможно существование двух типов сил, противодействующих внешней силе растяжения: одна связана с изменением внутренней энергии (первый член уравнения), другая – с изменением энтропии (второй член уравнения).
Сущность ВЭД с точки зрения термодинамической теории. Допущения: деформируется идеальный каучук без изменения объема при Т= const, при этом ВЭД не сопровождается деформацией течения. Примем, что в ходе растяжения образец идеального каучука длиной l 0 под действием силы f удлинился на величину dl. Поскольку ВЭД является обратимым процессом, происходящим при постоянных Т и V (изохорноизотермический процессом), то согласно второму закону термодинамики изменение свободной энергии d. F (изохорно-изотермического потенциала) в процессе деформирования определяется соотношением: d. F = d. U – Td. S, (1) где d. U и d. S – соответственно изменение внутренней энергии и энтропии в процессе деформации. Изменение свободной энергии d. F, кроме того, может быть выражено через работу растяжения d. A = f dl. Тогда уравнение изменения свободной энергии перепишется в виде: f dl = d. U – Td. S или f = (d. U/dl)T – T(d. S/dl)T (2) Из 2 следует, что в полимере возможно существование двух типов сил, противодействующих внешней силе растяжения: одна связана с изменением внутренней энергии (первый член уравнения), другая – с изменением энтропии (второй член уравнения).
 ВЭД идеального каучука происходит без изменения объема, валентных углов, длин связей и межмолекулярных расстояний, т. е. внутренняя энергия не изменяется (d. U/dl)T=0. Тогда сила, противодействующая растяжению, связана с изменением энтропии структура каучука становится более ориентированной за счет выпрямления молекулярных клубков. Это означает уменьшение беспорядка в системе, т. е. уменьшение энтропии: f = – T(d. S/dl)T Уменьшение энтропии - не самопроизвольный процесс, поэтому необходимо приложить внешнюю нагрузку для того, чтобы растянуть образец. Т. о. , сущность ВЭД с точки зрения термодинамической теории: напряжение, возникающее в образце при растяжении и противодействующее растяжению связано со стремлением макромолекул перейти в наиболее выгодное состояние молекулярного клубка, которому отвечает максимум энтропии.
ВЭД идеального каучука происходит без изменения объема, валентных углов, длин связей и межмолекулярных расстояний, т. е. внутренняя энергия не изменяется (d. U/dl)T=0. Тогда сила, противодействующая растяжению, связана с изменением энтропии структура каучука становится более ориентированной за счет выпрямления молекулярных клубков. Это означает уменьшение беспорядка в системе, т. е. уменьшение энтропии: f = – T(d. S/dl)T Уменьшение энтропии - не самопроизвольный процесс, поэтому необходимо приложить внешнюю нагрузку для того, чтобы растянуть образец. Т. о. , сущность ВЭД с точки зрения термодинамической теории: напряжение, возникающее в образце при растяжении и противодействующее растяжению связано со стремлением макромолекул перейти в наиболее выгодное состояние молекулярного клубка, которому отвечает максимум энтропии.
 Закономерности ВЭД реальных каучуков: 1. При деформации происходит некоторое уменьшение объема, изменяются расстояния между цепями и энергия межмолекулярного взаимодействия, т. е. деформация сопровождается не только изменением энтропии, но и изменением внутренней энергии, особенно при больших степенях растяжения, когда происходит кристаллизация. 2. Деформация реального сшитого каучука не является чисто высокоэластической. Наряду с ВЭД происходит также деформация течения, приводящая к наличию остаточной деформации. Доля вязкотекучей деформации не велика и зависит от степени сшивания (чем больше степень сшивания, тем меньше течение), а также глубины механохимических процессов (чем ближе к Тс, тем больше число разрывов в основной цепи). 3. В реальных каучуках ВЭД часто носит неравновесный характер. В целом, в системе с подвижными сегментами равновесие по ходу деформации устанавливается относительно быстро и материал в большей мере проявляет свою высокоэластичность. В системе с малоподвижными сегментами равновесие устанавливается очень долго и возможная деформация не успевает развиться, что проявляется в твердости полимерного материала.
Закономерности ВЭД реальных каучуков: 1. При деформации происходит некоторое уменьшение объема, изменяются расстояния между цепями и энергия межмолекулярного взаимодействия, т. е. деформация сопровождается не только изменением энтропии, но и изменением внутренней энергии, особенно при больших степенях растяжения, когда происходит кристаллизация. 2. Деформация реального сшитого каучука не является чисто высокоэластической. Наряду с ВЭД происходит также деформация течения, приводящая к наличию остаточной деформации. Доля вязкотекучей деформации не велика и зависит от степени сшивания (чем больше степень сшивания, тем меньше течение), а также глубины механохимических процессов (чем ближе к Тс, тем больше число разрывов в основной цепи). 3. В реальных каучуках ВЭД часто носит неравновесный характер. В целом, в системе с подвижными сегментами равновесие по ходу деформации устанавливается относительно быстро и материал в большей мере проявляет свою высокоэластичность. В системе с малоподвижными сегментами равновесие устанавливается очень долго и возможная деформация не успевает развиться, что проявляется в твердости полимерного материала.
 каучука представлена") Механическая кривая аморфного полимера в ВЭС Механическая кривая = f ( ) каучука представлена на рис . Рис. Механическая кривая аморфного редко сшитого полимера в ВЭС На начальном линейном участке (I участок) образец деформируется упруго за счет небольшого изменения валентных углов и длин связей. В дальнейшем благодаря высокой подвижности сегментов макромолекул полимер многократно деформируется (растягивается) под действием малых напряжений в результате раскручивания макромолекул в направлении действия силы в пределах узлов вулканизационной сетки (II участок). Разрыву образца предшествует небольшая упругая деформация (III участок).
Механическая кривая аморфного полимера в ВЭС Механическая кривая = f ( ) каучука представлена на рис . Рис. Механическая кривая аморфного редко сшитого полимера в ВЭС На начальном линейном участке (I участок) образец деформируется упруго за счет небольшого изменения валентных углов и длин связей. В дальнейшем благодаря высокой подвижности сегментов макромолекул полимер многократно деформируется (растягивается) под действием малых напряжений в результате раскручивания макромолекул в направлении действия силы в пределах узлов вулканизационной сетки (II участок). Разрыву образца предшествует небольшая упругая деформация (III участок).
 Деформационные свойства аморфных полимеров. вязкотекучее состояние, сущность Вязкотекучее состояние линейных и разветвленных полимеров реализуется в температурном интервале Тт – Тр; в тепловом движении участвуют: атомы и группы атомов, совершающие колебательные движения; сегменты, совершающие поступательные и вращательные движения. В процессе переработки (большинство полимеров перерабатывается в готовые изделия в ВТС) под действием сдвиговых усилий в полимере преимущественно развивается необратимая деформация течения, суть которой – перемещение сегментов в направлении действия силы, что приводит к необратимому смещению центров тяжести макромолекул. Условия реализации деформации течения: – наличие тепловой энергии, достаточной для преодоления сил межмолекулярного взаимодействия и перемещения (“перескоков”) сегментов; - наличие свободного объема ( «дырок» ), куда осуществляется перемещение сегментов. Важная при переработке полимеров характеристика – текучесть. На практике чаще всего пользуются обратной величиной (обратной текучести) – вязкостью, которая отражает способность полимера оказывать сопротивление необратимому изменению формы.
Деформационные свойства аморфных полимеров. вязкотекучее состояние, сущность Вязкотекучее состояние линейных и разветвленных полимеров реализуется в температурном интервале Тт – Тр; в тепловом движении участвуют: атомы и группы атомов, совершающие колебательные движения; сегменты, совершающие поступательные и вращательные движения. В процессе переработки (большинство полимеров перерабатывается в готовые изделия в ВТС) под действием сдвиговых усилий в полимере преимущественно развивается необратимая деформация течения, суть которой – перемещение сегментов в направлении действия силы, что приводит к необратимому смещению центров тяжести макромолекул. Условия реализации деформации течения: – наличие тепловой энергии, достаточной для преодоления сил межмолекулярного взаимодействия и перемещения (“перескоков”) сегментов; - наличие свободного объема ( «дырок» ), куда осуществляется перемещение сегментов. Важная при переработке полимеров характеристика – текучесть. На практике чаще всего пользуются обратной величиной (обратной текучести) – вязкостью, которая отражает способность полимера оказывать сопротивление необратимому изменению формы.
 Механизм течения. Течение осуществляется путем последовательного перемещения сегментов: макромолекула, являющаяся совокупностью сегментов 3, 4, 5, 6, 7, 8 при наличии «дырки» в положении 2 может изгибаться так, что сегмент 3 перейдет в положение 2. Далее при наличии «дырок» по соседству с сегментами 7 или 8 произойдет их перемещение, на освободившееся место перейдут сегменты 5 или 6 и т. д. , это приведет к смещению (вязкому течению) всей макромолекулы. Этот механизм течения называется диффузионным. Наряду с диффузионным у полимеров осуществляется и так называемое "химическое течение", при котором под действием приложенного напряжения и повышенных температур разрываются макромолекулы на отдельные фрагменты, а потом уже осуществляется их перемещение по диффузионному механизму. Особенно химическое течение наблюдается в высоковязких полимерах, при интенсивном внутри- и межмолекулярном взаимодействии. Начинаются разрывы химических связей в основной цепи, возникают свободные радикалы меньшей ММ, способные легко перемещаться под действием внешних сил. По этой причине при переработке высоковязких полимеров могут происходить глубокие химические изменения.
Механизм течения. Течение осуществляется путем последовательного перемещения сегментов: макромолекула, являющаяся совокупностью сегментов 3, 4, 5, 6, 7, 8 при наличии «дырки» в положении 2 может изгибаться так, что сегмент 3 перейдет в положение 2. Далее при наличии «дырок» по соседству с сегментами 7 или 8 произойдет их перемещение, на освободившееся место перейдут сегменты 5 или 6 и т. д. , это приведет к смещению (вязкому течению) всей макромолекулы. Этот механизм течения называется диффузионным. Наряду с диффузионным у полимеров осуществляется и так называемое "химическое течение", при котором под действием приложенного напряжения и повышенных температур разрываются макромолекулы на отдельные фрагменты, а потом уже осуществляется их перемещение по диффузионному механизму. Особенно химическое течение наблюдается в высоковязких полимерах, при интенсивном внутри- и межмолекулярном взаимодействии. Начинаются разрывы химических связей в основной цепи, возникают свободные радикалы меньшей ММ, способные легко перемещаться под действием внешних сил. По этой причине при переработке высоковязких полимеров могут происходить глубокие химические изменения.
 Особенности текучего состояния полимеров: 1. Полимеры по сравнению с НМВ имеют очень высокую вязкость – от нескольких тысяч до 1010 Па*с. Вследствие этого полимеры называют вязкотекучими. 2. В вязкотекучем состоянии у линейных и разветвленных полимеров наряду с необратимой деформацией течения проявляются упругая и ВЭД, поэтому текучие полимеры называют вязкоупругими. Общая деформация является их суммой общ = у + ВЭД + т. По мере течения упругая и ВЭД достигают постоянного значения, а необратимая равномерно увеличивается во времени. 3. Процесс течения сопровождается механохимическими явлениями, так как течение осуществляется при повышенных температурах и под действием механической нагрузки. Механохимический крекинг вызывает уменьшение ММ и увеличение скорости течения. Наиболее склонны к химическому течению полимеры, которые в основной цепи содержат менее прочные связи, чем С-С, например, полисульфидные каучуки.
Особенности текучего состояния полимеров: 1. Полимеры по сравнению с НМВ имеют очень высокую вязкость – от нескольких тысяч до 1010 Па*с. Вследствие этого полимеры называют вязкотекучими. 2. В вязкотекучем состоянии у линейных и разветвленных полимеров наряду с необратимой деформацией течения проявляются упругая и ВЭД, поэтому текучие полимеры называют вязкоупругими. Общая деформация является их суммой общ = у + ВЭД + т. По мере течения упругая и ВЭД достигают постоянного значения, а необратимая равномерно увеличивается во времени. 3. Процесс течения сопровождается механохимическими явлениями, так как течение осуществляется при повышенных температурах и под действием механической нагрузки. Механохимический крекинг вызывает уменьшение ММ и увеличение скорости течения. Наиболее склонны к химическому течению полимеры, которые в основной цепи содержат менее прочные связи, чем С-С, например, полисульфидные каучуки.
 Деформационные свойства полимера в ВТС Деформационные свойства полимеров в ВТС характеризуются кривыми течения (реологическими кривыми), т. е. зависимостями т = f ( *); = f ( т) или = f ( *). При воздействии на расплав полимера нагрузки, вызывающей течение, образец одновременно испытывает простой сдвиг, одноосное растяжение и всестороннее сжатие, при этом превалирующим является простой сдвиг. Если плоскость А в массе полимера смещается относительно неподвижного элемента Б, например, стенки канала, в котором происходит течение, то деформация сдвига определяется отношением = dl/l, -величина безразмерная. Скорость деформации сдвига * = d /dt определяет изменение величины деформации во времени и имеет размерность с-1. В простейшем случае напряжение сдвига т пропорционально скорости сдвига (прямолинейная зависимость): т = * (1) или в логарифмической форме lg т = lg + lg * где т напряжение сдвига, Н/м 2, * - скорость сдвига, с-1, – коэффициент пропорциональности или вязкость Н*с/м 2, характеризует сопротивление полимера сдвигу. Уравнение (1) – уравнение Ньютона, которое количественно характеризует течение НМВ, а также некоторых полимеров с узким ММР.
Деформационные свойства полимера в ВТС Деформационные свойства полимеров в ВТС характеризуются кривыми течения (реологическими кривыми), т. е. зависимостями т = f ( *); = f ( т) или = f ( *). При воздействии на расплав полимера нагрузки, вызывающей течение, образец одновременно испытывает простой сдвиг, одноосное растяжение и всестороннее сжатие, при этом превалирующим является простой сдвиг. Если плоскость А в массе полимера смещается относительно неподвижного элемента Б, например, стенки канала, в котором происходит течение, то деформация сдвига определяется отношением = dl/l, -величина безразмерная. Скорость деформации сдвига * = d /dt определяет изменение величины деформации во времени и имеет размерность с-1. В простейшем случае напряжение сдвига т пропорционально скорости сдвига (прямолинейная зависимость): т = * (1) или в логарифмической форме lg т = lg + lg * где т напряжение сдвига, Н/м 2, * - скорость сдвига, с-1, – коэффициент пропорциональности или вязкость Н*с/м 2, характеризует сопротивление полимера сдвигу. Уравнение (1) – уравнение Ньютона, которое количественно характеризует течение НМВ, а также некоторых полимеров с узким ММР.
 Деформационные свойства полимера в ВТС Течение большинства полимеров не подчиняется закону Ньютона, т. е. зависимость напряжения сдвига от скорости сдвига оказывается криволинейной. Течение в этом случае описывается уравнением Оствальда де Вила: т = *n, (2) или в логарифмичесой форме lg т = lg + n lg * Полимеры, течение которых подчиняется уравнению 2, называются псевдопластичными жидкостями; уравнение 2 применимо для ограниченного интервала значений скорости сдвига. Уравнения 1 и 2 в логарифмических координатах – прямые линии, но тангенс угла первой равен единице, а второй - n. Показатель степени n носит название индекса течения и характеризует степень отклонения течения от ньютоновского. Кривые течения в логарифмических координатах для ньютоновской (1) и псевдопластичной жидкости (2)
Деформационные свойства полимера в ВТС Течение большинства полимеров не подчиняется закону Ньютона, т. е. зависимость напряжения сдвига от скорости сдвига оказывается криволинейной. Течение в этом случае описывается уравнением Оствальда де Вила: т = *n, (2) или в логарифмичесой форме lg т = lg + n lg * Полимеры, течение которых подчиняется уравнению 2, называются псевдопластичными жидкостями; уравнение 2 применимо для ограниченного интервала значений скорости сдвига. Уравнения 1 и 2 в логарифмических координатах – прямые линии, но тангенс угла первой равен единице, а второй - n. Показатель степени n носит название индекса течения и характеризует степень отклонения течения от ньютоновского. Кривые течения в логарифмических координатах для ньютоновской (1) и псевдопластичной жидкости (2)
; = f (") Кривые течения полимеров, т. е. зависимости т = f ( *); = f ( т) или = f ( *) Течение Ньютоновских жидкостей: зависимость т = f ( *) прямолинейная; вязкость (при Т= const) не зависит от напряжения сдвига. Течение псевдопластичных жидкостей: зависимость т = f ( *) имеет Sобразный вид; на II участке течение не подчиняется закону Ньютона: т растет быстрее сростом скорости сдвига, чем это следует из закона Ньютона; вязкость при достижении критического Реологические кривые: т = f ( *); = f ( т) значения не является величиной т ньютоновской (а) и псевдопластичной (б) постоянной (эффективная вязкость). жидкостей I участок - в области низких скоростей сдвига выполняется закон Ньютона; вязкость наибольшая. II участок – зависимость т от * нелинейна; область аномалии вязкости. III участок – при больших скоростях сдвига – зависимость т от * - линейна; область наименьшей ньютоновской вязкости.
Кривые течения полимеров, т. е. зависимости т = f ( *); = f ( т) или = f ( *) Течение Ньютоновских жидкостей: зависимость т = f ( *) прямолинейная; вязкость (при Т= const) не зависит от напряжения сдвига. Течение псевдопластичных жидкостей: зависимость т = f ( *) имеет Sобразный вид; на II участке течение не подчиняется закону Ньютона: т растет быстрее сростом скорости сдвига, чем это следует из закона Ньютона; вязкость при достижении критического Реологические кривые: т = f ( *); = f ( т) значения не является величиной т ньютоновской (а) и псевдопластичной (б) постоянной (эффективная вязкость). жидкостей I участок - в области низких скоростей сдвига выполняется закон Ньютона; вязкость наибольшая. II участок – зависимость т от * нелинейна; область аномалии вязкости. III участок – при больших скоростях сдвига – зависимость т от * - линейна; область наименьшей ньютоновской вязкости.
 Причина аномалии вязкости - последовательное исключение предельно напряженных макромолекул из процесса течения, так как в ходе течения макромолекулы дополнительно подвергаются ВЭД и раскручиваются в направлении сдвига. При малых скоростях сдвига (I участок) полимеры текут подобно ньютоновским жидкостям, при этом развивается ВЭД (100 – 200 %), вязкость в системе наибольшая. Начиная с некоторого критического значения скорости сдвига, макромолекулы с наибольшей ММ оказываются предельно напряженными вследствие ВЭД в направлении сдвига (500 – 600 %). В первую очередь раскручиваются самые большие макромолекулы, т. к. скорости перемещения их концов сильно различаются. Развернутые напряженные молекулы перестают участвовать в течении, что приводит к снижению вязкости (начало II участка). Дальнейшее увеличение скорости сдвига приводит к тому, что макромолекулы с меньшей ММ выключаются из сегментального движения; вязкость снижается. На третьем участке в течении (перемещении сегментов) участвуют низкомолекулярные фракции, причем в соответствии с законом Ньютона, при этом вязкость – наименьшая (III участок). При дальнейшем увеличении скорости сдвига поток становится упругим, нетекучим, т. е. расплав ведет себя как квазисшитая система. В результате теряется контакт со стенками канала, струя расплава отрывается от стенок и быстро проходит канал. Отмечается резкий рост количества полимера, прошедшего через канал при данной скорости сдвига, т. е. скачок расхода.
Причина аномалии вязкости - последовательное исключение предельно напряженных макромолекул из процесса течения, так как в ходе течения макромолекулы дополнительно подвергаются ВЭД и раскручиваются в направлении сдвига. При малых скоростях сдвига (I участок) полимеры текут подобно ньютоновским жидкостям, при этом развивается ВЭД (100 – 200 %), вязкость в системе наибольшая. Начиная с некоторого критического значения скорости сдвига, макромолекулы с наибольшей ММ оказываются предельно напряженными вследствие ВЭД в направлении сдвига (500 – 600 %). В первую очередь раскручиваются самые большие макромолекулы, т. к. скорости перемещения их концов сильно различаются. Развернутые напряженные молекулы перестают участвовать в течении, что приводит к снижению вязкости (начало II участка). Дальнейшее увеличение скорости сдвига приводит к тому, что макромолекулы с меньшей ММ выключаются из сегментального движения; вязкость снижается. На третьем участке в течении (перемещении сегментов) участвуют низкомолекулярные фракции, причем в соответствии с законом Ньютона, при этом вязкость – наименьшая (III участок). При дальнейшем увеличении скорости сдвига поток становится упругим, нетекучим, т. е. расплав ведет себя как квазисшитая система. В результате теряется контакт со стенками канала, струя расплава отрывается от стенок и быстро проходит канал. Отмечается резкий рост количества полимера, прошедшего через канал при данной скорости сдвига, т. е. скачок расхода.
 Факторы, оказывающие влияние на Тт полимера Температура текучести Тт полимера зависит от: 1. ММ полимера. С ростом ММ Тт увеличивается. Влияние молекулярной массы полимера на величины Тхр, Тс, Тт 2. гибкости макромолекул. Чем выше гибкость, тем ниже Тт. Полимеры с гибкими цепями (эластомеры) начинают течь при 370 – 390 К; жесткоцепные – 420 – 500 К, но для таких полимеров повышается и Тс , область ВЭС сужается или “вырождается”.
Факторы, оказывающие влияние на Тт полимера Температура текучести Тт полимера зависит от: 1. ММ полимера. С ростом ММ Тт увеличивается. Влияние молекулярной массы полимера на величины Тхр, Тс, Тт 2. гибкости макромолекул. Чем выше гибкость, тем ниже Тт. Полимеры с гибкими цепями (эластомеры) начинают течь при 370 – 390 К; жесткоцепные – 420 – 500 К, но для таких полимеров повышается и Тс , область ВЭС сужается или “вырождается”.