

Лекция роль без вопросов новая.ppt
- Количество слайдов: 63
 Харьковский национальный медицинский университет Кафедра медицинской генетики РОЛЬ НАСЛЕДСТВЕННОСТИ В ПАТОЛОГИИ ЧЕЛОВЕКА
Харьковский национальный медицинский университет Кафедра медицинской генетики РОЛЬ НАСЛЕДСТВЕННОСТИ В ПАТОЛОГИИ ЧЕЛОВЕКА
 n Геномное здоровье –это согласованное взаимодействие генетической информации, взаимодействие генов, эпигенетических механизмов, внешней среды в процессе реализации программы индивидуального развития (онтогенеза).
n Геномное здоровье –это согласованное взаимодействие генетической информации, взаимодействие генов, эпигенетических механизмов, внешней среды в процессе реализации программы индивидуального развития (онтогенеза).
 ВЗАИМОДЕЙСТВИЕ ГЕНОВ Ген 1 ACGTAGCTAG Ядерная ДНК Ген 2 Митохондриальная ДНК ACGTAGCTAG Замена фрагмента гена Ген 2 ACGAGСCTAG ГЕНОМНОЕ ЗДОРОВЬЕ ЭПИГЕНЕТИЧЕСКИЕ ФАКТОРЫ ВНЕШНЕСРЕДОВЫЕ ФАКТОРЫ
ВЗАИМОДЕЙСТВИЕ ГЕНОВ Ген 1 ACGTAGCTAG Ядерная ДНК Ген 2 Митохондриальная ДНК ACGTAGCTAG Замена фрагмента гена Ген 2 ACGAGСCTAG ГЕНОМНОЕ ЗДОРОВЬЕ ЭПИГЕНЕТИЧЕСКИЕ ФАКТОРЫ ВНЕШНЕСРЕДОВЫЕ ФАКТОРЫ
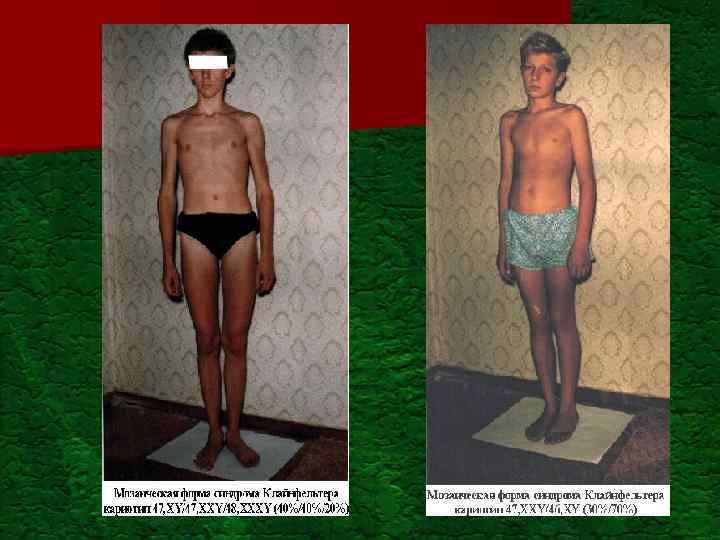
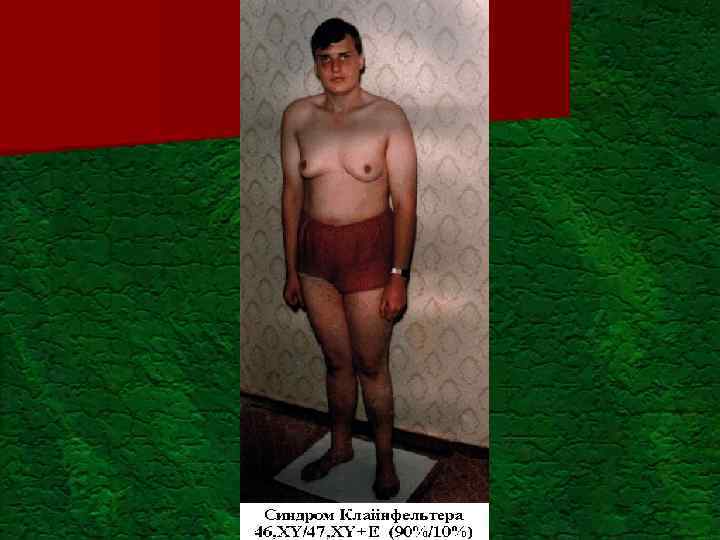
 Постанатальный генетический груз Моногенные болезни всего") Проявление генетического груза популяции (Ю. Е. Вельтищев, 1996) Постанатальный генетический груз Моногенные болезни всего в том числе: аутососмно-доминантные аутососмно-рецессивные Х-сцепленные Мультифакториальные болезни Хромосомные болезни всего Аномалии половых хромосом В том числе: Синдром ломкой Х-хромосомы Синдром Клайнфелтера ХХУ Синдром Шерешевского-Тернера ХО Аномалии аутосом в том числе: Болезнь Дауна (трисомия 21) Синдром Патау (трисомия) Синдром Эдвардса (трисомия 18) 10: 1000 7: 1000 2, 5: 1000 0, 4: 1000 до 25: 100 6: 1000 1: 400 мальчиков 1: 600 девочек 1: 1250 мальчиков 1: 2500 девочек 1: 750 мальчиков 1: 3000 девочек 1: 600 1: 7000 1: 6000
Проявление генетического груза популяции (Ю. Е. Вельтищев, 1996) Постанатальный генетический груз Моногенные болезни всего в том числе: аутососмно-доминантные аутососмно-рецессивные Х-сцепленные Мультифакториальные болезни Хромосомные болезни всего Аномалии половых хромосом В том числе: Синдром ломкой Х-хромосомы Синдром Клайнфелтера ХХУ Синдром Шерешевского-Тернера ХО Аномалии аутосом в том числе: Болезнь Дауна (трисомия 21) Синдром Патау (трисомия) Синдром Эдвардса (трисомия 18) 10: 1000 7: 1000 2, 5: 1000 0, 4: 1000 до 25: 100 6: 1000 1: 400 мальчиков 1: 600 девочек 1: 1250 мальчиков 1: 2500 девочек 1: 750 мальчиков 1: 3000 девочек 1: 600 1: 7000 1: 6000
 Частота наследственных и других врожденных заболеваний у новорожденных, их удельный вес в структуре младенческой смертности и среди госпитализированных детей США в процентах (Ch. J. Epstein 1991) Природа болезней Частота у новорожденных В структуре мл. смертности Среди госпитализированных детей Моногенные 1 8, 5 6, 5 Хромосомные 0, 5 2, 5 0, 5 до 3 20, 0 - Другой природы 1 -2 10, 0 15 -20 Всего 4 -6 40, 0 22 -27 Наследственные: Полигенные
Частота наследственных и других врожденных заболеваний у новорожденных, их удельный вес в структуре младенческой смертности и среди госпитализированных детей США в процентах (Ch. J. Epstein 1991) Природа болезней Частота у новорожденных В структуре мл. смертности Среди госпитализированных детей Моногенные 1 8, 5 6, 5 Хромосомные 0, 5 2, 5 0, 5 до 3 20, 0 - Другой природы 1 -2 10, 0 15 -20 Всего 4 -6 40, 0 22 -27 Наследственные: Полигенные
 ФОРМУЛА ЛОЛЛОНДА
ФОРМУЛА ЛОЛЛОНДА
 Частота після") Типи та частоти генетичних захворювань Частота до 25 років (/1, 000 живонароджених) Частота після 25 років Частота протягом усього життя Хромосомні порушення 1, 8/1, 000 2/1, 000 3, 8/1, 000 1 Порушення одного гену 3, 6/1, 000 16, 4/1, 000 20/1, 000 2 Мультифакторіальні (частково генетичні) порушення 46, 4/1, 000 600/1, 000 646, 4/1, 000 3 Генетичні порушення соматичних клітин 240/1, 000 4 Тип Виключені носії збалансованого структурного перегрупування, що не викликають симптоматики, зокрема числові статеві хромосомні порушення, більшість з яких не діагностовано. 2 Виключені носії премутацій та інших мутацій з декількома симптомами або без них, зокрема мітохондріальні генні мутації. 3 Для природжених вад розвитку, включаючи тільки доведений частково генетичний внесок, зокрема хронічні порушення дозрівання, що є загальною оцінкою частоти. 4 Припускається, що всі ракові пухлини являють собою накопичувальні генетичні мутації і виключено успадковані синдроми ракової пухлини одного гену. 1
Типи та частоти генетичних захворювань Частота до 25 років (/1, 000 живонароджених) Частота після 25 років Частота протягом усього життя Хромосомні порушення 1, 8/1, 000 2/1, 000 3, 8/1, 000 1 Порушення одного гену 3, 6/1, 000 16, 4/1, 000 20/1, 000 2 Мультифакторіальні (частково генетичні) порушення 46, 4/1, 000 600/1, 000 646, 4/1, 000 3 Генетичні порушення соматичних клітин 240/1, 000 4 Тип Виключені носії збалансованого структурного перегрупування, що не викликають симптоматики, зокрема числові статеві хромосомні порушення, більшість з яких не діагностовано. 2 Виключені носії премутацій та інших мутацій з декількома симптомами або без них, зокрема мітохондріальні генні мутації. 3 Для природжених вад розвитку, включаючи тільки доведений частково генетичний внесок, зокрема хронічні порушення дозрівання, що є загальною оцінкою частоти. 4 Припускається, що всі ракові пухлини являють собою накопичувальні генетичні мутації і виключено успадковані синдроми ракової пухлини одного гену. 1

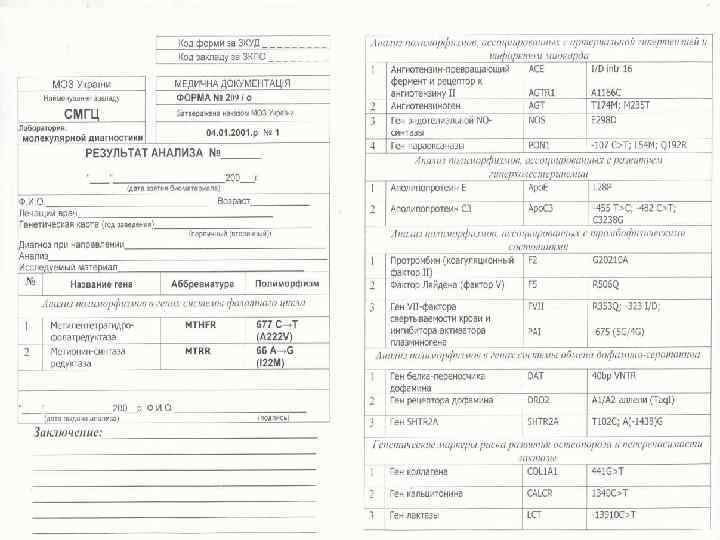
 Частота генетически детерминированных болезней Формы наследственной патологии Частота на 1000 1. Моногенные 4. 5 15. 0 аутосомно доминантные 2. 0 9. 5 аутосомно рецессивные 2. 0 3. 5 Х сцепленные 0. 5 2. 0 2. Хромосомные болезни 5. 0 7, 0 3. Мультифакториально обусловленные* 4. Врожденные пороки развития* 7. 0 10. 0 ВСЕГО 35 54 19. 0 22. 00
Частота генетически детерминированных болезней Формы наследственной патологии Частота на 1000 1. Моногенные 4. 5 15. 0 аутосомно доминантные 2. 0 9. 5 аутосомно рецессивные 2. 0 3. 5 Х сцепленные 0. 5 2. 0 2. Хромосомные болезни 5. 0 7, 0 3. Мультифакториально обусловленные* 4. Врожденные пороки развития* 7. 0 10. 0 ВСЕГО 35 54 19. 0 22. 00
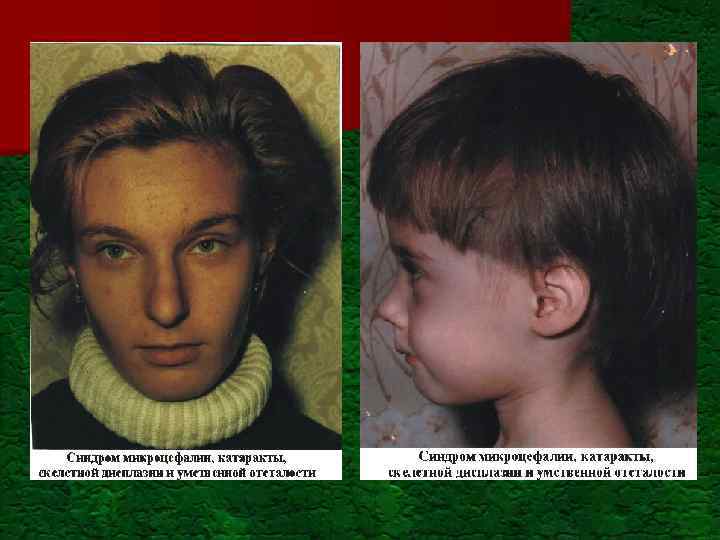
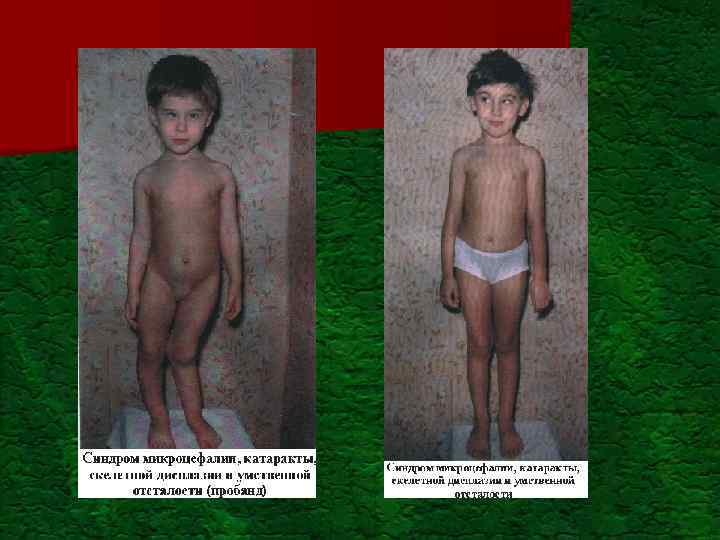
") Вклад генетического компонента в хронические инвалидизирующие врожденные состояния в развитых странах (по материалам ВОЗ) Тип нарушений Частота на 1000 рождений Генетический компонент Умственная недостаточность: - тяжелая - умеренная и слабая Детский церебральный паралич 3, 5 2, 5 Для большинства форм свыше 30% 2, 5 Очень малый Слепота 0, 6 50% Глухота (тяжелая) 1, 0 50% Врожденные пороки развития 50%
Вклад генетического компонента в хронические инвалидизирующие врожденные состояния в развитых странах (по материалам ВОЗ) Тип нарушений Частота на 1000 рождений Генетический компонент Умственная недостаточность: - тяжелая - умеренная и слабая Детский церебральный паралич 3, 5 2, 5 Для большинства форм свыше 30% 2, 5 Очень малый Слепота 0, 6 50% Глухота (тяжелая) 1, 0 50% Врожденные пороки развития 50%
 Эпигенетический контроль – изменение экспрессии генов происходит без изменения первичной последовательности нуклеотидов ДНК. Метилирование ДНК регулирует экспрессию генов через механизм компактизации-декомпактизации хроматина.
Эпигенетический контроль – изменение экспрессии генов происходит без изменения первичной последовательности нуклеотидов ДНК. Метилирование ДНК регулирует экспрессию генов через механизм компактизации-декомпактизации хроматина.
 обобщил особенности эпигенетического программирования генома: n n n") Reick W at all ( 2001) обобщил особенности эпигенетического программирования генома: n n n рисунки метилирования ДНК обладают пространственной, временной и тканевой специфичностью; специфика метилирования ДНК наследуется дочерними клетками; специфические метки метилирования стираются в памяти примордиальных половых клеток; в ходе созревания половых клеток происходит восстановление рисунков метилирования в соответствии с половой принадлежностью организма; после слияния половых клеток происходит деметилирование генома; в процессе эмбриогенеза метилирование соматических клеток происходит де ново.
Reick W at all ( 2001) обобщил особенности эпигенетического программирования генома: n n n рисунки метилирования ДНК обладают пространственной, временной и тканевой специфичностью; специфика метилирования ДНК наследуется дочерними клетками; специфические метки метилирования стираются в памяти примордиальных половых клеток; в ходе созревания половых клеток происходит восстановление рисунков метилирования в соответствии с половой принадлежностью организма; после слияния половых клеток происходит деметилирование генома; в процессе эмбриогенеза метилирование соматических клеток происходит де ново.
 n Метилирование цитозиновых оснований ДНК предопределяет взаимодействие между ДНК и белками, входящими в состав хроматина. Это взаимодействие через механизм компактизациидекомпактизации хроматина и регулирует экспрессию генов
n Метилирование цитозиновых оснований ДНК предопределяет взаимодействие между ДНК и белками, входящими в состав хроматина. Это взаимодействие через механизм компактизациидекомпактизации хроматина и регулирует экспрессию генов
. n") n Регуляция экспрессии генов обеспечивается химически модифицированными нуклеосомными гистонами (ацетилированными, метилированными или фосфорилированными). n Мобильность хроматина обеспечивают энзимы – амилтрансфераза гистонов и деацетилтрансфераза гистонов.
n Регуляция экспрессии генов обеспечивается химически модифицированными нуклеосомными гистонами (ацетилированными, метилированными или фосфорилированными). n Мобильность хроматина обеспечивают энзимы – амилтрансфераза гистонов и деацетилтрансфераза гистонов.
") Сегмент ДНК (145 оснований)
Сегмент ДНК (145 оснований)
 Структура хроматина с разной степенью конденсации
Структура хроматина с разной степенью конденсации
") Взаимоотношение генетики и эпигенетики (по С. А. Назаренко, 2004)
Взаимоотношение генетики и эпигенетики (по С. А. Назаренко, 2004)
 n Гетерохроматин - пучок фибрилл, диаметр которых варьирует в течение клеточного цикла и зависит от участка хромосомной локализации. n Гетерохроматин – конститутивный и факультативный
n Гетерохроматин - пучок фибрилл, диаметр которых варьирует в течение клеточного цикла и зависит от участка хромосомной локализации. n Гетерохроматин – конститутивный и факультативный
 Конститутивный характеризуется: n стабильностью, n высоким содержанием сателлитных ДНК, выраженным полиморфизмом, n появлением С-бендов после денатурации–ренатурации ДНК. n содержит специфический тип ДНК, который называется сателлитным и состоит из большого количества тандемно расположенных коротких повторов азотистых оснований.
Конститутивный характеризуется: n стабильностью, n высоким содержанием сателлитных ДНК, выраженным полиморфизмом, n появлением С-бендов после денатурации–ренатурации ДНК. n содержит специфический тип ДНК, который называется сателлитным и состоит из большого количества тандемно расположенных коротких повторов азотистых оснований.
 n конденсированным состоянием; n поздней репликацией; n метилированным состоянием; n присутствием гипоацетилированных гистонов.
n конденсированным состоянием; n поздней репликацией; n метилированным состоянием; n присутствием гипоацетилированных гистонов.
 Нарушение эпигенетического статуса отдельных участков") Классификация эпигенетических болезней человека (по С. А. Назаренко, 2004) Нарушение эпигенетического статуса отдельных участков генома (локальный эффект) 1. Болезни, обусловленные унаследованными мутациями, нарушающими моноаллельную экспрессию генов — болезни геномного импринтинга (синдромы Видемана— Беквита, Прадера—Вилли, Энгельмана) Нарушение эпигенетического статуса всего генома (глобальный эффект) 1. Болезни, обусловленные унаследованными мутациями генов, продукты которых во влечены в поддержание уров няметилирования ДНК или модификацию структуры хро матина — Синдромы ICF, Ретта, ATR X, Рубинштей на— Тейби, Коффина—Лаури 2. Болезни, обусловленные нарушенным 2. Болезни, обусловленные глобальным статусом мети лированияотдельных нарушением ме тилированиягенома в генов в результате de novo возникших резу льтатеde novo возникших мутаций в соматических клетках — а) в соматических клетках— Раковые раковые болезни, связанные с потерей болезни, связанные с глобальным импринтинга, приводящей к актива ции гипометилированием генома, неактивного генов или подавлению приводящим к активации он когенов, экспрессии актив ного гена; ретротранспозонов и хромосомной б) раковые болезни, обусловленные нестабильности гиперметилированием промоторов генов опухолевых супрессоров и их инактивацией
Классификация эпигенетических болезней человека (по С. А. Назаренко, 2004) Нарушение эпигенетического статуса отдельных участков генома (локальный эффект) 1. Болезни, обусловленные унаследованными мутациями, нарушающими моноаллельную экспрессию генов — болезни геномного импринтинга (синдромы Видемана— Беквита, Прадера—Вилли, Энгельмана) Нарушение эпигенетического статуса всего генома (глобальный эффект) 1. Болезни, обусловленные унаследованными мутациями генов, продукты которых во влечены в поддержание уров няметилирования ДНК или модификацию структуры хро матина — Синдромы ICF, Ретта, ATR X, Рубинштей на— Тейби, Коффина—Лаури 2. Болезни, обусловленные нарушенным 2. Болезни, обусловленные глобальным статусом мети лированияотдельных нарушением ме тилированиягенома в генов в результате de novo возникших резу льтатеde novo возникших мутаций в соматических клетках — а) в соматических клетках— Раковые раковые болезни, связанные с потерей болезни, связанные с глобальным импринтинга, приводящей к актива ции гипометилированием генома, неактивного генов или подавлению приводящим к активации он когенов, экспрессии актив ного гена; ретротранспозонов и хромосомной б) раковые болезни, обусловленные нестабильности гиперметилированием промоторов генов опухолевых супрессоров и их инактивацией
 Фенотипические особенности, аномалии и пороки развития у пациентов с ХП n кожа и ее производные, n черепно-лицевые дизморфии, n опорно-двигательный аппарат, n нервная система, n сердечно-сосудистая система, n дыхательная система, n пищеварительная система, n мочевыделительная система, n половая система, n эндокринная система.
Фенотипические особенности, аномалии и пороки развития у пациентов с ХП n кожа и ее производные, n черепно-лицевые дизморфии, n опорно-двигательный аппарат, n нервная система, n сердечно-сосудистая система, n дыхательная система, n пищеварительная система, n мочевыделительная система, n половая система, n эндокринная система.
") Хромосомный полиморфизм (15 pstk)
Хромосомный полиморфизм (15 pstk)
 XY, 9 gh+, 15 pstk, 2% хр. нест. XX, 15 pstk, 1% нест. хр.
XY, 9 gh+, 15 pstk, 2% хр. нест. XX, 15 pstk, 1% нест. хр.
") Хромосомный полиморфизм (9 qh+)
Хромосомный полиморфизм (9 qh+)
 XY, 9 gh+, G окр. , С окр. , 1% хр. нест. XY, 22 pstk, 9 gh+ XY, 9 gh+, 1% хр. нест.
XY, 9 gh+, G окр. , С окр. , 1% хр. нест. XY, 22 pstk, 9 gh+ XY, 9 gh+, 1% хр. нест.
 Удельный вес различных хромосомных вариантов
Удельный вес различных хромосомных вариантов
 Частота проявления фенотипических признаков у пациентов с хромосомным полиморфизмом Группа признаков 46, ХХ 46, XY Контр 46, ХХ, 1 qh+ 46, XY, 1 qh+ n P 46, ХХ, 9 qh+ 46, XY, 9 qh+ n 46, ХХ, 16 qh+ 46, XY qh+ P n P n P <0. 01 0 5 <0. 01 26 <0. 01 Спутнич ный поли морфизм Кожа и её производные 74 23 <0. 01 28 Черепно лицевые дизморфии 225 89 <0. 01 133 <0. 01 12 <0. 01 50 <0. 01 164 <0. 01 Опорно двигатель ный аппарат 112 61 <0. 01 67 <0. 01 14 <0. 01 117 <0. 01 Нервная система 10 15 <0. 01 19 <0. 01 4 <0. 01 18 <0. 01 0
Частота проявления фенотипических признаков у пациентов с хромосомным полиморфизмом Группа признаков 46, ХХ 46, XY Контр 46, ХХ, 1 qh+ 46, XY, 1 qh+ n P 46, ХХ, 9 qh+ 46, XY, 9 qh+ n 46, ХХ, 16 qh+ 46, XY qh+ P n P n P <0. 01 0 5 <0. 01 26 <0. 01 Спутнич ный поли морфизм Кожа и её производные 74 23 <0. 01 28 Черепно лицевые дизморфии 225 89 <0. 01 133 <0. 01 12 <0. 01 50 <0. 01 164 <0. 01 Опорно двигатель ный аппарат 112 61 <0. 01 67 <0. 01 14 <0. 01 117 <0. 01 Нервная система 10 15 <0. 01 19 <0. 01 4 <0. 01 18 <0. 01 0
 Группа признаков 46, ХХ 46, XY Контр. 46, ХХ, 1 qh+ 46, XY, 1 qh+ 46, ХХ, 9 qh+ 46, XY, 9 qh+ 46, ХХ, 16 qh+ 46, XY qh+ Спутни чный поли мор физм n P n P n P Сердечно сосудистая система 12 2 <0. 01 6 >0. 99 1 0. 95 3 0. 75 5 >0. 99 Дыхательная система 1 1 <0. 01 7 0. 25 0 0 1 >0. 99 Пищеварительная система 16 5 >0. 99 10 >0. 99 0 4 0. 50 15 0. 025 Мочевыделитель ная система 13 4 >0. 99 8 >0. 99 0 1 0. 90 10 0. 75 Половая система 8 4 0. 05 7 0. 025 1 0. 05 0 13 <0. 01 Эндокринная система 10 2 >0. 99 0 2 0. 75 7 >0. 99
Группа признаков 46, ХХ 46, XY Контр. 46, ХХ, 1 qh+ 46, XY, 1 qh+ 46, ХХ, 9 qh+ 46, XY, 9 qh+ 46, ХХ, 16 qh+ 46, XY qh+ Спутни чный поли мор физм n P n P n P Сердечно сосудистая система 12 2 <0. 01 6 >0. 99 1 0. 95 3 0. 75 5 >0. 99 Дыхательная система 1 1 <0. 01 7 0. 25 0 0 1 >0. 99 Пищеварительная система 16 5 >0. 99 10 >0. 99 0 4 0. 50 15 0. 025 Мочевыделитель ная система 13 4 >0. 99 8 >0. 99 0 1 0. 90 10 0. 75 Половая система 8 4 0. 05 7 0. 025 1 0. 05 0 13 <0. 01 Эндокринная система 10 2 >0. 99 0 2 0. 75 7 >0. 99
 Цитогенетическая, фенотипическая и метаболическая характеристика пациентов с хромосомным полиморфизмом Метаболические Кариотип Возраст Фенотип изменения 13 ps+ 1 год МД, МАР, атаксия, ГГААУ, аланинурия, 46, XY, 13 ps+ 8 макроцефалия, ренальная глюкозурия, фруктозурия, 3% ХН месяцев дисплазия мальтозурия Низкая масса, пупочная грыжа, деформированная грудная клетка, гиперэластичность Пролинурия, глицинурия, 46, XY, 13 ps+ кожи, гемангиоматоз, 13 лет аспарагинурия, 1% ХН преаурикулярные фистулы, орнитинурия микроцефалия, эписиндром, МАР Микроцефалия, Пролинурия, глицинурия, долихоцефалия, лордоз, 46, XХ, 13 ps+ 21 год аспарагинурия, 4% ХН генитальная гипоплазия, орнитинурия вторичная аменорея глицинемия СТД, МАР, брахицефалия, Пролинемия, , серинемия, , 46, XХ, 13 ps+ 10 мес диспластическая кардиопатия валинемия 4% ХН глюкозурия 46, XХ, 13 ps+ 25 лет МАР, брахицефалия, дисплазия 3% ХН почек ГГААУ
Цитогенетическая, фенотипическая и метаболическая характеристика пациентов с хромосомным полиморфизмом Метаболические Кариотип Возраст Фенотип изменения 13 ps+ 1 год МД, МАР, атаксия, ГГААУ, аланинурия, 46, XY, 13 ps+ 8 макроцефалия, ренальная глюкозурия, фруктозурия, 3% ХН месяцев дисплазия мальтозурия Низкая масса, пупочная грыжа, деформированная грудная клетка, гиперэластичность Пролинурия, глицинурия, 46, XY, 13 ps+ кожи, гемангиоматоз, 13 лет аспарагинурия, 1% ХН преаурикулярные фистулы, орнитинурия микроцефалия, эписиндром, МАР Микроцефалия, Пролинурия, глицинурия, долихоцефалия, лордоз, 46, XХ, 13 ps+ 21 год аспарагинурия, 4% ХН генитальная гипоплазия, орнитинурия вторичная аменорея глицинемия СТД, МАР, брахицефалия, Пролинемия, , серинемия, , 46, XХ, 13 ps+ 10 мес диспластическая кардиопатия валинемия 4% ХН глюкозурия 46, XХ, 13 ps+ 25 лет МАР, брахицефалия, дисплазия 3% ХН почек ГГААУ
 Цитогенетическая, фенотипическая и метаболическая характеристика пациентов с хромосомным полиморфизмом Кариотип Возраст Фенотип 9 qh+ 46, XY, 9 qh+ 30 лет 46, XY, 9 qh+ 19 лет 46, XY, 9 qh+ 34 года 46, XY, 9 qh+ 2 года 2 6% ХН месяца 46, XY, 9 qh+ 27 лет 46, XY, 9 qh+ 11 лет 4% ХН Метаболические изменения ГГАА, тирозин, МД триптофан, аланин, аспарагиновая кислота Валинемия, аланинемия, пролинемия, глицинемия, МД, СТД, МАР, флюктуация Х аспарагиновая кислота, ребра, оксипролин галактозурия, мальтозурия (синдром Штиллера) Брахицефалия, множественные очаги депигментации, Пролинурия, карцинома яичка, нефроптоз, глицеринурия, аланинемия диспластические изменения в почках Акроцефалия, киста сосудистого Валинемия, аланинемия, сплетения, дермоидная киста аспарагиновая кислота Аланинемия, У жены замершая беременность пролинемия, глицинемия, в 9 недель лизинемия Кифоз, СТД, проксимальная девиация V пальцев верхних Аланинурия, пролинурия, конечностей, вальгусная деформация коленных суставов, глицинурия, орнитинурия вирусная деформация голени
Цитогенетическая, фенотипическая и метаболическая характеристика пациентов с хромосомным полиморфизмом Кариотип Возраст Фенотип 9 qh+ 46, XY, 9 qh+ 30 лет 46, XY, 9 qh+ 19 лет 46, XY, 9 qh+ 34 года 46, XY, 9 qh+ 2 года 2 6% ХН месяца 46, XY, 9 qh+ 27 лет 46, XY, 9 qh+ 11 лет 4% ХН Метаболические изменения ГГАА, тирозин, МД триптофан, аланин, аспарагиновая кислота Валинемия, аланинемия, пролинемия, глицинемия, МД, СТД, МАР, флюктуация Х аспарагиновая кислота, ребра, оксипролин галактозурия, мальтозурия (синдром Штиллера) Брахицефалия, множественные очаги депигментации, Пролинурия, карцинома яичка, нефроптоз, глицеринурия, аланинемия диспластические изменения в почках Акроцефалия, киста сосудистого Валинемия, аланинемия, сплетения, дермоидная киста аспарагиновая кислота Аланинемия, У жены замершая беременность пролинемия, глицинемия, в 9 недель лизинемия Кифоз, СТД, проксимальная девиация V пальцев верхних Аланинурия, пролинурия, конечностей, вальгусная деформация коленных суставов, глицинурия, орнитинурия вирусная деформация голени
 Хирургические болезни Всего больных Количество синдромов абс. % Ортопедическая") Синдромология хирургических болезней (собственные данные) Хирургические болезни Всего больных Количество синдромов абс. % Ортопедическая патология (косолапость, деформация грудной клетки, сколиоз, лучевая косорукость, псевдоартрозы, врожденный вывих бедра) 76 44 58 Аноректальные дефекты (атрезия ануса и прямой кишки, стенозы и эктопия ануса) 19 12 62 Патология пищевода (атрезии и стенозы пищеводы, желудочно-пищеводный рефлюкс) 26 9 34
Синдромология хирургических болезней (собственные данные) Хирургические болезни Всего больных Количество синдромов абс. % Ортопедическая патология (косолапость, деформация грудной клетки, сколиоз, лучевая косорукость, псевдоартрозы, врожденный вывих бедра) 76 44 58 Аноректальные дефекты (атрезия ануса и прямой кишки, стенозы и эктопия ануса) 19 12 62 Патология пищевода (атрезии и стенозы пищеводы, желудочно-пищеводный рефлюкс) 26 9 34
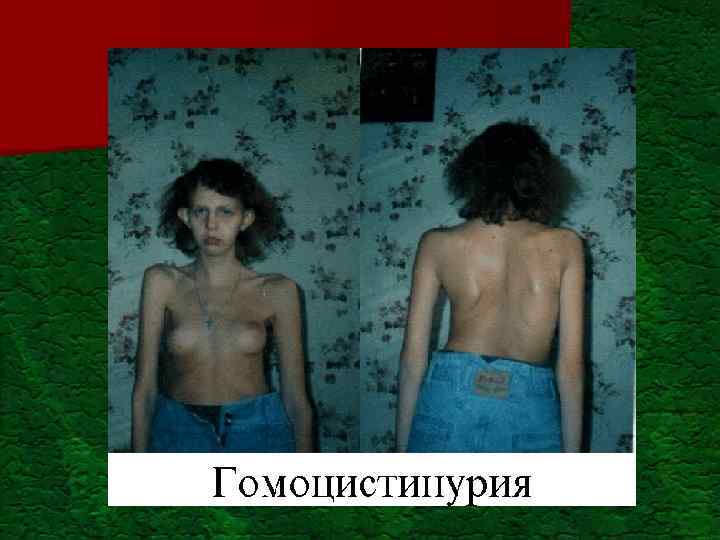
 Дифференционная диагностика признаков и симптомов НБО (по Nyhan W. L. , Ozand P. T. (1998), G. Hoffmann et al. (2002)) Признак Алопеция Ангиокератомы Артрит Возможные НБО Врожденная ангидротическая эктодермальная дисплазия Недостаточность биотин Синдром Конради-Гюнерманна Множественная недостаточность карбоксилазы (недостаточность голокарбоксилазы и недостаточность биотинидазы) Узлуватый трихорексисаргиноянтарная ацидурия Нарушение витамин D-зависимого рахита-рецептора Болезнь Фабри Фукозидоз Галактосиалидоз Ганглиозидоз GM 1 Сиалидоз Алкаптонурия Болезнь Фабри Болезнь Гоше I типа Подагра-недостаточность гипоксанингуанинфосфорибозилтрансферазы (HPRT) (болезнь Леша-Нимена); повышенная активность фосфорибозилпрофосфатсинтетазы (PRPPS) Гомоцистинурия I-клеточная болезнь (муколипидоз II) Болезнь Леша-Нимена Муколипидоз III Мукополисахаридоз (МШС) тип IS (синдром Хурлера) или тип IIS (синдром Шайе)
Дифференционная диагностика признаков и симптомов НБО (по Nyhan W. L. , Ozand P. T. (1998), G. Hoffmann et al. (2002)) Признак Алопеция Ангиокератомы Артрит Возможные НБО Врожденная ангидротическая эктодермальная дисплазия Недостаточность биотин Синдром Конради-Гюнерманна Множественная недостаточность карбоксилазы (недостаточность голокарбоксилазы и недостаточность биотинидазы) Узлуватый трихорексисаргиноянтарная ацидурия Нарушение витамин D-зависимого рахита-рецептора Болезнь Фабри Фукозидоз Галактосиалидоз Ганглиозидоз GM 1 Сиалидоз Алкаптонурия Болезнь Фабри Болезнь Гоше I типа Подагра-недостаточность гипоксанингуанинфосфорибозилтрансферазы (HPRT) (болезнь Леша-Нимена); повышенная активность фосфорибозилпрофосфатсинтетазы (PRPPS) Гомоцистинурия I-клеточная болезнь (муколипидоз II) Болезнь Леша-Нимена Муколипидоз III Мукополисахаридоз (МШС) тип IS (синдром Хурлера) или тип IIS (синдром Шайе)
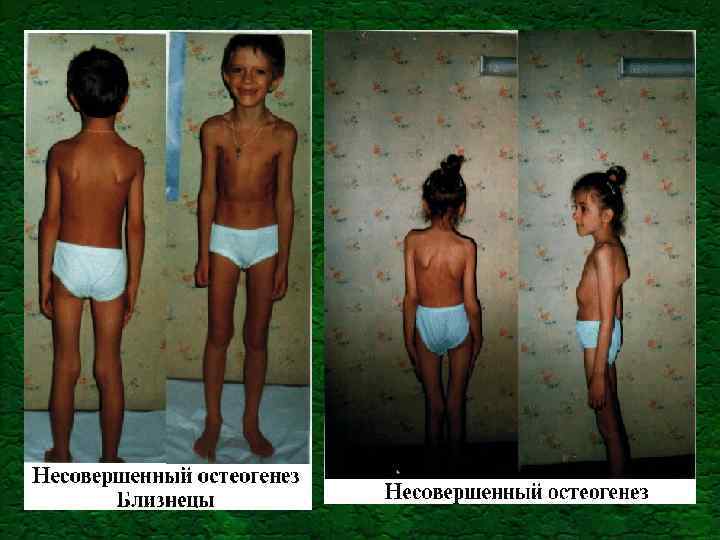
 Синдром Марфана
Синдром Марфана


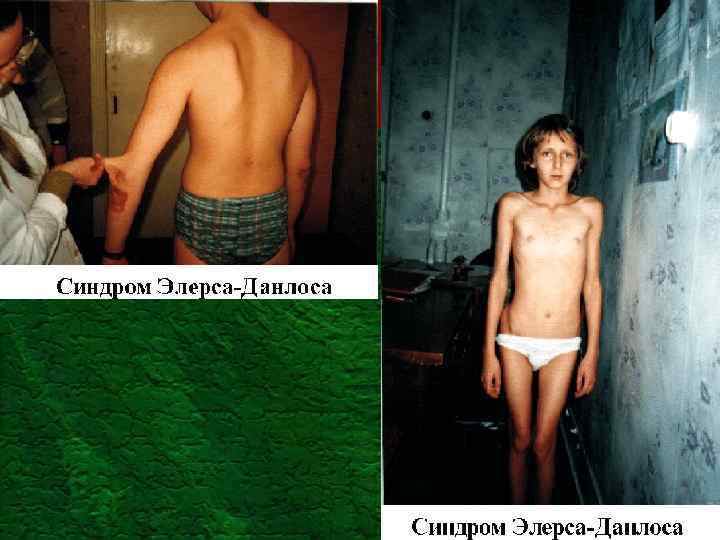
 Синдром Элерса - Данлоса
Синдром Элерса - Данлоса

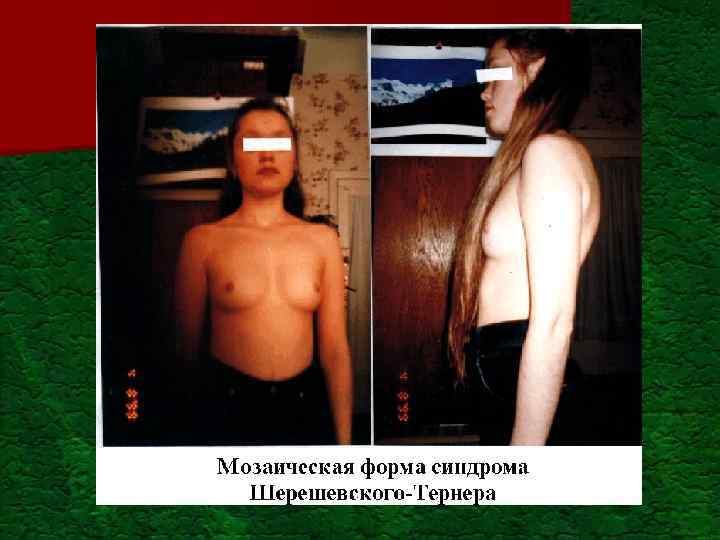
 Синдром Шерешевского-Тернера
Синдром Шерешевского-Тернера





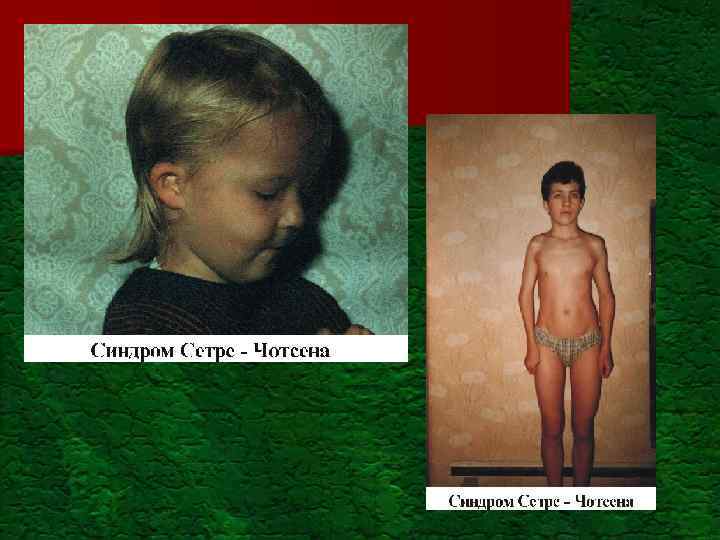
") MNGIE-синдром (митохондриальная нейро-желудочнокишечная энцефалопатия)
MNGIE-синдром (митохондриальная нейро-желудочнокишечная энцефалопатия)
 Родинний випадок синдрому Кернса-Сейра
Родинний випадок синдрому Кернса-Сейра
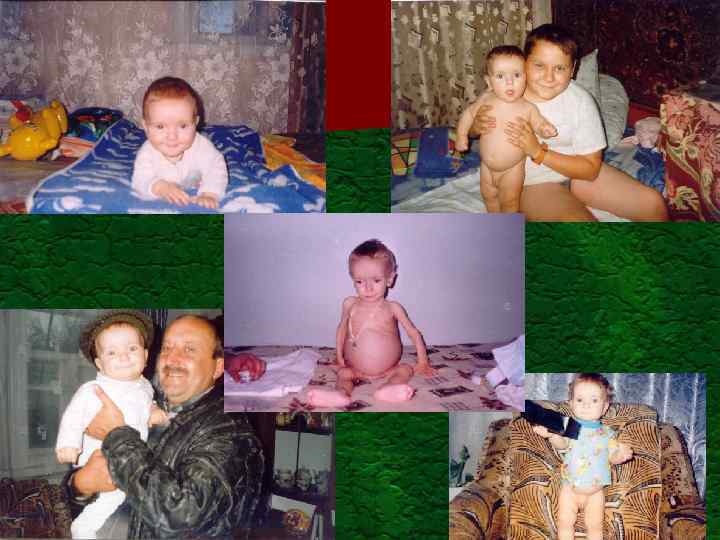


, 8472") Мітохондріальна хвороба Просеквенований ген т. РНК – лізин. Знайдені мутації 8836 A/G (met/val), 8472 C/T(pro/leu 2), 8614 T/C
Мітохондріальна хвороба Просеквенований ген т. РНК – лізин. Знайдені мутації 8836 A/G (met/val), 8472 C/T(pro/leu 2), 8614 T/C