dda254e0ccddf36af1bde71bb0740279.ppt
- Количество слайдов: 48
 X and Y Chromosomes
X and Y Chromosomes
 Sex Chromosomes • Sex determination is not a highly conserved trait in evolution: there are lots of ways to do it. Sex chromosomes seem to have evolved many times independently. • Mammals (except monotremes) use a chromosomal method of determining sex: XX is female and XY is male. – The gene involved is the SRY gene • Birds use a ZW system: ZZ is male and ZW is female. – the evolutionary origin of mammalian and bird sex chromosomes is different – However, the Y and the W are both small chromosomes with few genes on them. • Some reptiles use developmental temperature to determine sex: depends on the species, but hot is male and cold is female in some. • Drosophila also use an X-Y system (i. e. male is XY and female is XX), but the evolutionary origin and mode of action of Drosophila sex chromosomes is different form mammalian.
Sex Chromosomes • Sex determination is not a highly conserved trait in evolution: there are lots of ways to do it. Sex chromosomes seem to have evolved many times independently. • Mammals (except monotremes) use a chromosomal method of determining sex: XX is female and XY is male. – The gene involved is the SRY gene • Birds use a ZW system: ZZ is male and ZW is female. – the evolutionary origin of mammalian and bird sex chromosomes is different – However, the Y and the W are both small chromosomes with few genes on them. • Some reptiles use developmental temperature to determine sex: depends on the species, but hot is male and cold is female in some. • Drosophila also use an X-Y system (i. e. male is XY and female is XX), but the evolutionary origin and mode of action of Drosophila sex chromosomes is different form mammalian.
 The SRY Gene • The SRY gene, located on the Y chromosome, is the primary determinant of sexual development. Also called Testis Determining Factor. – That is, if a developing embryo has a functional SRY gene in its cells, it will develop as a male. And, if there is no functional SRY, the embryo develops as female. – There is a homolog of SRY on the X: SOX 3. It is highly diverged in sequence from SRY. – SRY makes a transcription factor that activates genes needed to convert the indifferent gonad of the early embryo into testes. • Although the SRY gene is usually on the Y chromosome, it occasionally gets transferred to the X. – People with this condition (46, XX testicular disorder of sexual development) are male, but often with small testes and sometimes genital abnormalities. Also, they are infertile. • Also, sometimes the SRY gene is inactivated by mutation. – Leading to 46, XY females (Swyer syndrome) – it is also possible to have a partially inactive SRY gene, leading to ambiguous genitalia • Amusing bit of history: Aristotle taught that gender was determined by the state of the semen: hot semen produced a male child and cold semen generated a female child.
The SRY Gene • The SRY gene, located on the Y chromosome, is the primary determinant of sexual development. Also called Testis Determining Factor. – That is, if a developing embryo has a functional SRY gene in its cells, it will develop as a male. And, if there is no functional SRY, the embryo develops as female. – There is a homolog of SRY on the X: SOX 3. It is highly diverged in sequence from SRY. – SRY makes a transcription factor that activates genes needed to convert the indifferent gonad of the early embryo into testes. • Although the SRY gene is usually on the Y chromosome, it occasionally gets transferred to the X. – People with this condition (46, XX testicular disorder of sexual development) are male, but often with small testes and sometimes genital abnormalities. Also, they are infertile. • Also, sometimes the SRY gene is inactivated by mutation. – Leading to 46, XY females (Swyer syndrome) – it is also possible to have a partially inactive SRY gene, leading to ambiguous genitalia • Amusing bit of history: Aristotle taught that gender was determined by the state of the semen: hot semen produced a male child and cold semen generated a female child.
 X and Y Homology The X is a large submetacentric chromosome with many genes on it, most of which are unrelated to sex. The Y is acrocentric and much smaller. Only 83 active genes on the Y, most of which are related to sex determination and spermatogenesis. – About 1400 genes on the X However, many homologues exist between the X and the Y, with the Y gene often a pseudogene. This suggests a common evolutionary origin. The tips of the X and Y pair in meiosis, and undergo crossing over. These areas are called the pseudoautosomal regions (PARs). There is another large region of homology between the X and the Y, due to the translocation of a block of Xp 21 onto the Y. – Occurred 3 -4 million years ago, after the human-chimpanzee divergence. – Very few genes here: it is mostly transposable elements and other repeated sequences – No X-Y pairing or recombination here.
X and Y Homology The X is a large submetacentric chromosome with many genes on it, most of which are unrelated to sex. The Y is acrocentric and much smaller. Only 83 active genes on the Y, most of which are related to sex determination and spermatogenesis. – About 1400 genes on the X However, many homologues exist between the X and the Y, with the Y gene often a pseudogene. This suggests a common evolutionary origin. The tips of the X and Y pair in meiosis, and undergo crossing over. These areas are called the pseudoautosomal regions (PARs). There is another large region of homology between the X and the Y, due to the translocation of a block of Xp 21 onto the Y. – Occurred 3 -4 million years ago, after the human-chimpanzee divergence. – Very few genes here: it is mostly transposable elements and other repeated sequences – No X-Y pairing or recombination here.
 Pseudoautosomal Regions • • Unlike the rest of the X and Y chromosomes, the PARs pair with each other in meiosis 1 and undergo crossing over. Because of their behavior in meiosis, genes in these regions are inherited in the same way as autosomal genes, and do not act like sex-linked genes: – Genes in the PARs are not hemizygous (present in only 1 copy) in males: 2 copies, one of the X and one on the Y. – There is no difference in the offspring phenotypes from reciprocal crosses (switching the sexes of the parental phenotypes). • PAR 1 is on the short arms of both X and Y. (Xp and Yp) – About 2. 6 Mbp (the Y is 58 Mbp total length) – Contains 24 genes, all of which are NOT inactivated in X chromosome inactivation – Successful meiosis and spermatogenesis in males requires a crossover between the PAR 1 regions on the X and the Y • PAR 2 is on the long arms of the X and Y. (Xq and Yq) – Much shorter: 320 kb – Only 4 known genes: one of them (SYBL 1) undergoes X chromosome inactivation – Crossing over can occur in meiosis, but is not required. • When different placental mammals are compared, all have pseudoautosomal regions, but which genes are in them, and where the boundaries lie, are quite variable. – Marsupials don’t have PARs or any pairing between X and Y chromosomes
Pseudoautosomal Regions • • Unlike the rest of the X and Y chromosomes, the PARs pair with each other in meiosis 1 and undergo crossing over. Because of their behavior in meiosis, genes in these regions are inherited in the same way as autosomal genes, and do not act like sex-linked genes: – Genes in the PARs are not hemizygous (present in only 1 copy) in males: 2 copies, one of the X and one on the Y. – There is no difference in the offspring phenotypes from reciprocal crosses (switching the sexes of the parental phenotypes). • PAR 1 is on the short arms of both X and Y. (Xp and Yp) – About 2. 6 Mbp (the Y is 58 Mbp total length) – Contains 24 genes, all of which are NOT inactivated in X chromosome inactivation – Successful meiosis and spermatogenesis in males requires a crossover between the PAR 1 regions on the X and the Y • PAR 2 is on the long arms of the X and Y. (Xq and Yq) – Much shorter: 320 kb – Only 4 known genes: one of them (SYBL 1) undergoes X chromosome inactivation – Crossing over can occur in meiosis, but is not required. • When different placental mammals are compared, all have pseudoautosomal regions, but which genes are in them, and where the boundaries lie, are quite variable. – Marsupials don’t have PARs or any pairing between X and Y chromosomes
 Genes in PARs • • During the pre-molecular era in the 20 th century, several Y-linked traits were proposed: hairy ears, porcupine man, etc. All have been disproven. SYBL 1 is in PAR 2, but it is subject to inactivation: only 1 copy is active in both males and females. – Y chromosome copy is inactive in males – A particular allele, a G->C in an intron region that controls splicing, has a strong statistical correlation with bipolar affective disorder • People with bipolar disorder (also called manic depression) have periods of very elevated mood (happy and energetic, often with poorly thought-out decisions) and periods of depression. It is caused by a combination of genetic and environmental factors, but the genetic factors are poorly understood at present. – The protein product (called VAMP 7) is a transmembrane protein involved in vesicle docking of transport vesicles that are moving from endosomes to the lysosomes • • This may affect outgrowth of neurites (projections from the neurons) SHOX (Short stature HOmeobo. X-containing) is involved in bone development. – It is subject to haploinsufficiency: only one copy causes the short stature and minor skeletal deformities on Turner syndrome, and 3 copies seems to cause tallness in people with 3 sex chromosomes. – Mutations in this genes have similar effects. • The Y has a number of genes for sperm development. Mutation sin these genes cause sterility.
Genes in PARs • • During the pre-molecular era in the 20 th century, several Y-linked traits were proposed: hairy ears, porcupine man, etc. All have been disproven. SYBL 1 is in PAR 2, but it is subject to inactivation: only 1 copy is active in both males and females. – Y chromosome copy is inactive in males – A particular allele, a G->C in an intron region that controls splicing, has a strong statistical correlation with bipolar affective disorder • People with bipolar disorder (also called manic depression) have periods of very elevated mood (happy and energetic, often with poorly thought-out decisions) and periods of depression. It is caused by a combination of genetic and environmental factors, but the genetic factors are poorly understood at present. – The protein product (called VAMP 7) is a transmembrane protein involved in vesicle docking of transport vesicles that are moving from endosomes to the lysosomes • • This may affect outgrowth of neurites (projections from the neurons) SHOX (Short stature HOmeobo. X-containing) is involved in bone development. – It is subject to haploinsufficiency: only one copy causes the short stature and minor skeletal deformities on Turner syndrome, and 3 copies seems to cause tallness in people with 3 sex chromosomes. – Mutations in this genes have similar effects. • The Y has a number of genes for sperm development. Mutation sin these genes cause sterility.
: long hairs") Traits Once Thought to be Y-linked • Hairy ears (OMIM entry 425500): long hairs growing out of the outer edge of the ear. – A 2004 study by Lee et al. showed that a group of men from southern India with hairy ears had Y chromosomes from several different haplogroups. Since the Y doesn’t recombine (outside of PARs), this observation would require several different independent mutations. This seems unlikely. • Porcupine Man (OMIM 146600: ichthyosis hystrix gravior). The drawing is of Edward Lambert, around 1731. Spiny scales covered most of his body, and also affected 10 other males in 3 generations of descendants. – A 1958 study showed that several females in this family also had the condition, implying that it wasn’t Y-linked.
Traits Once Thought to be Y-linked • Hairy ears (OMIM entry 425500): long hairs growing out of the outer edge of the ear. – A 2004 study by Lee et al. showed that a group of men from southern India with hairy ears had Y chromosomes from several different haplogroups. Since the Y doesn’t recombine (outside of PARs), this observation would require several different independent mutations. This seems unlikely. • Porcupine Man (OMIM 146600: ichthyosis hystrix gravior). The drawing is of Edward Lambert, around 1731. Spiny scales covered most of his body, and also affected 10 other males in 3 generations of descendants. – A 1958 study showed that several females in this family also had the condition, implying that it wasn’t Y-linked.
 Muller’s Ratchet • An important result of sexual reproduction is that mutations can be eliminated by recombination. Two individuals with deleterious mutations in different genes can produce offspring with neither mutation. – Or, to put it more precisely, the heterozygous offspring of those two individuals can produce gametes with neither mutation. • Muller’s ratchet: in the absence of recombination, mutations accumulate: there is no easy way to get rid of them. – Predicts that strictly asexual species won’t survive long unless they share DNA by some other means (like horizontal gene transfer).
Muller’s Ratchet • An important result of sexual reproduction is that mutations can be eliminated by recombination. Two individuals with deleterious mutations in different genes can produce offspring with neither mutation. – Or, to put it more precisely, the heterozygous offspring of those two individuals can produce gametes with neither mutation. • Muller’s ratchet: in the absence of recombination, mutations accumulate: there is no easy way to get rid of them. – Predicts that strictly asexual species won’t survive long unless they share DNA by some other means (like horizontal gene transfer).
 Y Chromosome Evolution • The X and Y started off as the same chromosome, but the Y has become much smaller (both in size and number of genes) and lost the ability to recombine with the X. Several factors are involved: 1. Loss of recombination is due to a series of inversions. Crossing over between an inversion and a normal chromosome results in dead offspring. – No crossing over led to an accumulation of mutations: Muller’s ratchet. The Y contains many pseudogenes, versions of X chromosome genes that have become inactive due to mutation. – The same phenomenon is seen with the Y in Drosophila and the W in birds. – X-degenerate DNA 2. Genes necessary for spermatogenesis are protected from Muller’s ratchet by having 2 copies present in palindromes, kept virtually identical by gene conversions. (ampliconic DNA) 3. Large amounts of transposable elements and repeat sequences have accumulated in certain regions. (heterochromatic DNA) 4. A large block of Xp 21 has been transposed onto the Y (and still exists on the X). (X-transposed DNA)
Y Chromosome Evolution • The X and Y started off as the same chromosome, but the Y has become much smaller (both in size and number of genes) and lost the ability to recombine with the X. Several factors are involved: 1. Loss of recombination is due to a series of inversions. Crossing over between an inversion and a normal chromosome results in dead offspring. – No crossing over led to an accumulation of mutations: Muller’s ratchet. The Y contains many pseudogenes, versions of X chromosome genes that have become inactive due to mutation. – The same phenomenon is seen with the Y in Drosophila and the W in birds. – X-degenerate DNA 2. Genes necessary for spermatogenesis are protected from Muller’s ratchet by having 2 copies present in palindromes, kept virtually identical by gene conversions. (ampliconic DNA) 3. Large amounts of transposable elements and repeat sequences have accumulated in certain regions. (heterochromatic DNA) 4. A large block of Xp 21 has been transposed onto the Y (and still exists on the X). (X-transposed DNA)
 Inversion History • The history of inversions of the Y can be traced by looking at synonymous substitution rates between X and Y homologous genes. – Synonymous substitutions are changes in the DNA of the codons that don’t affect the amino acids (due to the degeneracy of the genetic code). – They should be selectively neutral, since the resulting proteins are the same. • Synonymous substitution rates vary in groups, which suggests blocks of chromosome that became unable to share alleles between the X and Y. – Before the inversions occurred, crossing over kept the X and Y copies identical. – Once crossing over became lethal (due to the inversions), synonymous substitutions accumulated.
Inversion History • The history of inversions of the Y can be traced by looking at synonymous substitution rates between X and Y homologous genes. – Synonymous substitutions are changes in the DNA of the codons that don’t affect the amino acids (due to the degeneracy of the genetic code). – They should be selectively neutral, since the resulting proteins are the same. • Synonymous substitution rates vary in groups, which suggests blocks of chromosome that became unable to share alleles between the X and Y. – Before the inversions occurred, crossing over kept the X and Y copies identical. – Once crossing over became lethal (due to the inversions), synonymous substitutions accumulated.
 Ks vs. Position on Y Chromosome
Ks vs. Position on Y Chromosome
 Gene Conversion • Gene conversion is an alternative outcome of crossing over in meiosis. – In a crossover, 2 homologues break and rejoin opposite partners, so genes outside the region of the crossover are now connected differently than in the original chromosomes: they go from parental to recombinant. – In gene conversion, a small region (1 -2 kbp) at the site of recombination becomes homozygous, while the homologous chromosomes stay in the parental configuration. – A result of the detailed molecular mechanism of recombination. The lines represent double stranded DNA from the 2 homologues. In the parents (parental configuration), alleles D and F are on one chromosome, while d and f are on the other (coupling phase). Following a crossover, (recombinant configuration) D and f are on one chromosome and d and F are on the other (repulsion phase). In gene conversion, a small region (1 -2 kbp) at the site of recombination becomes homozygous: both homologues end up with the same allele (here, allele e). Note that D and F , and d and f, are still in coupling: the chromosomes are still in the parental configuration.
Gene Conversion • Gene conversion is an alternative outcome of crossing over in meiosis. – In a crossover, 2 homologues break and rejoin opposite partners, so genes outside the region of the crossover are now connected differently than in the original chromosomes: they go from parental to recombinant. – In gene conversion, a small region (1 -2 kbp) at the site of recombination becomes homozygous, while the homologous chromosomes stay in the parental configuration. – A result of the detailed molecular mechanism of recombination. The lines represent double stranded DNA from the 2 homologues. In the parents (parental configuration), alleles D and F are on one chromosome, while d and f are on the other (coupling phase). Following a crossover, (recombinant configuration) D and f are on one chromosome and d and F are on the other (repulsion phase). In gene conversion, a small region (1 -2 kbp) at the site of recombination becomes homozygous: both homologues end up with the same allele (here, allele e). Note that D and F , and d and f, are still in coupling: the chromosomes are still in the parental configuration.
 Gene Conversion in Palindromes • Gene conversion can also occur within a chromosome it there are homologous regions on it. – This is especially true of unpaired chromosomes like the X and Y in humans. • On the Y are 8 large palindromic sequences (inverted repeats), from 9 kbp to 1. 45 Mbp. Each gene in this region is thus duplicated. Gene conversions between these palindromes keeps the two copies almost identical (>99. 9% identical). – This gets around Muller’s ratchet: if mutation inactivates one copy of the gene, there is another good copy, and in some offspring both copies will be converted to the good version. Offspring where both copies are bad will be sterile or die. – Genes in these palindromes are all involved in spermatogenesis.
Gene Conversion in Palindromes • Gene conversion can also occur within a chromosome it there are homologous regions on it. – This is especially true of unpaired chromosomes like the X and Y in humans. • On the Y are 8 large palindromic sequences (inverted repeats), from 9 kbp to 1. 45 Mbp. Each gene in this region is thus duplicated. Gene conversions between these palindromes keeps the two copies almost identical (>99. 9% identical). – This gets around Muller’s ratchet: if mutation inactivates one copy of the gene, there is another good copy, and in some offspring both copies will be converted to the good version. Offspring where both copies are bad will be sterile or die. – Genes in these palindromes are all involved in spermatogenesis.
 Dosage Compensation • • There are over 1000 genes in the X chromosome. How can females have 2 X’s and males only 1 without running into gene dosage problems? Lyon hypothesis (1961): placental mammals randomly inactivate all but 1 X at the 200400 cell embryo stage (blastocyst). The inactivated X's become Barr bodies: late-replicating condensed chromatin sitting on the nuclear membrane (heterochromatin). – Number of Barr bodies is always 1 less than the number of X’s: Seen in XXY, XXX, etc. • why are females rarely colorblind? Many retinal precursor cells present at time of inactivation, so get a fine-grained mosaicism-brain fills in colors
Dosage Compensation • • There are over 1000 genes in the X chromosome. How can females have 2 X’s and males only 1 without running into gene dosage problems? Lyon hypothesis (1961): placental mammals randomly inactivate all but 1 X at the 200400 cell embryo stage (blastocyst). The inactivated X's become Barr bodies: late-replicating condensed chromatin sitting on the nuclear membrane (heterochromatin). – Number of Barr bodies is always 1 less than the number of X’s: Seen in XXY, XXX, etc. • why are females rarely colorblind? Many retinal precursor cells present at time of inactivation, so get a fine-grained mosaicism-brain fills in colors
 Glucose 6 -Phosphate Dehydrogenase • • • Early evidence for X-inactivation: glucose 6 phosphate dehydrogenase (G 6 PD). Gene on the X, with two alleles: normal and G 6 PD deficient. Red blood cells from heterozygous females showed two populations, with and without G 6 PD. Cancerous tumors were shown to originate from a single cell in a similar fashion: in heterozygous females, only one G 6 PD allele was active in the entire tumor. G 6 PD is an enzyme active in red blood cells that shunts glucose off from glycolysis and helps generate NADPH reduces glutathione, which converts hydrogen peroxide into water. – G 6 PD deficiency leads to a buildup of hydrogen peroxide, which can destroy the red blood cells (hemolysis). • • One common allele found in the Mediterranean region is especially susceptible after ingestion of fava beans (favism). The beans contain a compound (vicine) that results in peroxide formation. Mortality rate in the absence of transfusions is about 10%. Malaria parasites also induce peroxide formation, so G 6 PD deficiency helps kill cells that are being infected. – The distribution of G 6 PD deficiency globally matches the distribution of malaria. Very similar to sickle cell anemia.
Glucose 6 -Phosphate Dehydrogenase • • • Early evidence for X-inactivation: glucose 6 phosphate dehydrogenase (G 6 PD). Gene on the X, with two alleles: normal and G 6 PD deficient. Red blood cells from heterozygous females showed two populations, with and without G 6 PD. Cancerous tumors were shown to originate from a single cell in a similar fashion: in heterozygous females, only one G 6 PD allele was active in the entire tumor. G 6 PD is an enzyme active in red blood cells that shunts glucose off from glycolysis and helps generate NADPH reduces glutathione, which converts hydrogen peroxide into water. – G 6 PD deficiency leads to a buildup of hydrogen peroxide, which can destroy the red blood cells (hemolysis). • • One common allele found in the Mediterranean region is especially susceptible after ingestion of fava beans (favism). The beans contain a compound (vicine) that results in peroxide formation. Mortality rate in the absence of transfusions is about 10%. Malaria parasites also induce peroxide formation, so G 6 PD deficiency helps kill cells that are being infected. – The distribution of G 6 PD deficiency globally matches the distribution of malaria. Very similar to sickle cell anemia.
 Tortiseshell and Calico Cats • • • A gene for coat color exists on the X chromosome. It has 2 alleles, orange fur and black fur. Because only X chromosome is active, an individual cell will express either black or orange but not both. Inactivation occurs early in development, and the descendant cells usually stay together to make a patch on the skin Leads to orange and black patches: tortoiseshell. Calico cats also have white patches, due to another (autosomal) gene. Although rare, Klinefelter syndrome cats (XXY) exist: they are male calicos and tortiseshells.
Tortiseshell and Calico Cats • • • A gene for coat color exists on the X chromosome. It has 2 alleles, orange fur and black fur. Because only X chromosome is active, an individual cell will express either black or orange but not both. Inactivation occurs early in development, and the descendant cells usually stay together to make a patch on the skin Leads to orange and black patches: tortoiseshell. Calico cats also have white patches, due to another (autosomal) gene. Although rare, Klinefelter syndrome cats (XXY) exist: they are male calicos and tortiseshells.
 Mechanism of Inactivation • X inactivation starts at a specific point on the chromosome: Xq 13. 2. Chromosomes lacking this XIC region do not become inactivated. – Pieces of the X translocated to other chromosomes don’t get inactivated: only DNA physically connected to XIC get inactivated. – inactivation is necessary for life: cells (or embryos) with more than one active X (due to mutations in the inactivation mechanism) do not survive. – XIC is also involved in the counting mechanism by which all but 1 X is inactivated. • The XIC region on the inactive X expresses one important gene: XIST (X Inactive Specific Transcript). – – • The XIST RNA is about 18 Kb. It is not translated, but it is spliced and poly-adenylated. It is composed largely of repeated sequences. The inactive X seems to be coated with XIST RNA, which forms the Barr body. If XIST is inactivated during the life of a cell, the X stays inactivated: XIST is needed to initiate inactivation, but not to maintain it.
Mechanism of Inactivation • X inactivation starts at a specific point on the chromosome: Xq 13. 2. Chromosomes lacking this XIC region do not become inactivated. – Pieces of the X translocated to other chromosomes don’t get inactivated: only DNA physically connected to XIC get inactivated. – inactivation is necessary for life: cells (or embryos) with more than one active X (due to mutations in the inactivation mechanism) do not survive. – XIC is also involved in the counting mechanism by which all but 1 X is inactivated. • The XIC region on the inactive X expresses one important gene: XIST (X Inactive Specific Transcript). – – • The XIST RNA is about 18 Kb. It is not translated, but it is spliced and poly-adenylated. It is composed largely of repeated sequences. The inactive X seems to be coated with XIST RNA, which forms the Barr body. If XIST is inactivated during the life of a cell, the X stays inactivated: XIST is needed to initiate inactivation, but not to maintain it.
 XIST RNA in Various Genotypes • The amount of XIST RNA is proportional to the number of X chromosomes minus 1.
XIST RNA in Various Genotypes • The amount of XIST RNA is proportional to the number of X chromosomes minus 1.
 More Inactivation Mechanism • Other events happen after XIST is active: – XIST RNA recruits “silencing protein complexes” to complete the inactivation. – Histones on the inactive X are under-acetylated; histone acetylation is found near active genes. – the 5’ end of the XIST gene on the active X is heavily methylated, but the 5’ end of XIST on the inactive X is not methylated. • The other strand of the XIST gene is also transcribed, called TSIX is thus antisense to the XIST RNA, and TSIX RNA represses XIST expression. TSIX is not expressed by the inactive X, but is expressed by the active X. • It has recently been shown that the XIC regions of different X’s pair up transiently at the time of inactivation, suggesting that the choice of which X stays active depends on a cis-acting mechanism.
More Inactivation Mechanism • Other events happen after XIST is active: – XIST RNA recruits “silencing protein complexes” to complete the inactivation. – Histones on the inactive X are under-acetylated; histone acetylation is found near active genes. – the 5’ end of the XIST gene on the active X is heavily methylated, but the 5’ end of XIST on the inactive X is not methylated. • The other strand of the XIST gene is also transcribed, called TSIX is thus antisense to the XIST RNA, and TSIX RNA represses XIST expression. TSIX is not expressed by the inactive X, but is expressed by the active X. • It has recently been shown that the XIC regions of different X’s pair up transiently at the time of inactivation, suggesting that the choice of which X stays active depends on a cis-acting mechanism.
 More X Chromosome Inactivation • the X from the sperm is inactivated in the zygote and very early embryo (a case of imprinting). – It remains inactive in the extra-embryonic tissues: only the maternal X is active here. – The imprinting is removed in the morula stage, so both X’s have an equal chance of being inactivated in the developing embryo. – also reactivation in the oogonia: cells that will undergo meiosis in the female. • • • In marsupial mammals, the paternal X is always inactivated. Only the maternal X is used during development. There is also some reactivation of the X during aging. Some genes on the X escape inactivation, especially genes that have functional homologues on the Y. Thus, 2 copies of these genes are active in all cells. – Most, but not all, of these genes are in the pseudoautosomal regions. – They account for the Turner and Klinefelter syndrome phenotypes.
More X Chromosome Inactivation • the X from the sperm is inactivated in the zygote and very early embryo (a case of imprinting). – It remains inactive in the extra-embryonic tissues: only the maternal X is active here. – The imprinting is removed in the morula stage, so both X’s have an equal chance of being inactivated in the developing embryo. – also reactivation in the oogonia: cells that will undergo meiosis in the female. • • • In marsupial mammals, the paternal X is always inactivated. Only the maternal X is used during development. There is also some reactivation of the X during aging. Some genes on the X escape inactivation, especially genes that have functional homologues on the Y. Thus, 2 copies of these genes are active in all cells. – Most, but not all, of these genes are in the pseudoautosomal regions. – They account for the Turner and Klinefelter syndrome phenotypes.
 Human Sexual Development
Human Sexual Development
 Early Gonad Development • Before 6 -7 weeks of development, the gonad is indifferent: neither male nor female. • It develops from the same tissue as the kidneys and adrenal glands. • Also developing by this time: 2 sets of ducts that will eventually lead to the outside world. – Wolffian ducts = male – Mullerian ducts = female
Early Gonad Development • Before 6 -7 weeks of development, the gonad is indifferent: neither male nor female. • It develops from the same tissue as the kidneys and adrenal glands. • Also developing by this time: 2 sets of ducts that will eventually lead to the outside world. – Wolffian ducts = male – Mullerian ducts = female
 Gonad Differentiation • If SRY is present in the indifferent gonad at 6 weeks, it gets activated. This in turn activates other genes, and the indifferent gonad is converted to a testes. • In the absence of SRY, a different set of genes is activated, and the indifferent gonad becomes an ovary. • The germ cells, which actually become sperm or eggs, migrate into the gonad about this time.
Gonad Differentiation • If SRY is present in the indifferent gonad at 6 weeks, it gets activated. This in turn activates other genes, and the indifferent gonad is converted to a testes. • In the absence of SRY, a different set of genes is activated, and the indifferent gonad becomes an ovary. • The germ cells, which actually become sperm or eggs, migrate into the gonad about this time.
 Development of Phenotypic Sex • The cells of the newly formed testes start secreting the steroid hormone testosterone. – Testosterone secretion peaks about week 16, with levels similar to those found in adult males. After this, the testosterone level drops to about the same level as female fetuses. – The testes also secrete another hormone: Mullerian inhibiting substance (MIS) (aka anti. Mullerian hormone, AMH). (a glycoprotein hormone) • Another important process in the developing male: during the last trimester of pre-natal life, the testes migrate (“descend”) from the kidney region into the scrotum. • The developing ovary secretes estrogen, which is important after birth, but estrogen from the mother completely swamps it out before birth.
Development of Phenotypic Sex • The cells of the newly formed testes start secreting the steroid hormone testosterone. – Testosterone secretion peaks about week 16, with levels similar to those found in adult males. After this, the testosterone level drops to about the same level as female fetuses. – The testes also secrete another hormone: Mullerian inhibiting substance (MIS) (aka anti. Mullerian hormone, AMH). (a glycoprotein hormone) • Another important process in the developing male: during the last trimester of pre-natal life, the testes migrate (“descend”) from the kidney region into the scrotum. • The developing ovary secretes estrogen, which is important after birth, but estrogen from the mother completely swamps it out before birth.
 • • Internal Ducts In the early embryo, two duct systems form. After the gonad differentiates into a testis or ovary, one set of ducts develops further while the other set degenerates. Testosterone causes the Wolffian ducts to develop into male structures: epididymus, vas deferens, seminal vesicles. – In the absence of testosterone, the Wolffian ducts disappear (except a bit becomes the adrenal glands in both sexes) • Mullerian inhibiting substance causes the Mullerian ducts to disappear. – In the absence of MIS, the Mullerian ducts develop into the Fallopian tubes, uterus, and upper vagina.
• • Internal Ducts In the early embryo, two duct systems form. After the gonad differentiates into a testis or ovary, one set of ducts develops further while the other set degenerates. Testosterone causes the Wolffian ducts to develop into male structures: epididymus, vas deferens, seminal vesicles. – In the absence of testosterone, the Wolffian ducts disappear (except a bit becomes the adrenal glands in both sexes) • Mullerian inhibiting substance causes the Mullerian ducts to disappear. – In the absence of MIS, the Mullerian ducts develop into the Fallopian tubes, uterus, and upper vagina.
 Another Duct Picture
Another Duct Picture
 Development of the External Genitalia • This process is controlled by the presence or • • absence of dihydrotestosterone (DHT). Testosterone gets converted into DHT by the enzyme 5 -alpha reductase, which is found in the testes and the skin. Both sexes start out with the same structures, which develop along different lines under the influence of testosterone and DHT. The default condition in female: in the absence of DHT, the external genital structures develop along female lines. DHT also causes hair loss: male pattern baldness. Testosterone is converted to DHT locally. Rogaine (the trademark name for minoxidil, a hair-restoring drug) works by blocking 5 -alpha reductase
Development of the External Genitalia • This process is controlled by the presence or • • absence of dihydrotestosterone (DHT). Testosterone gets converted into DHT by the enzyme 5 -alpha reductase, which is found in the testes and the skin. Both sexes start out with the same structures, which develop along different lines under the influence of testosterone and DHT. The default condition in female: in the absence of DHT, the external genital structures develop along female lines. DHT also causes hair loss: male pattern baldness. Testosterone is converted to DHT locally. Rogaine (the trademark name for minoxidil, a hair-restoring drug) works by blocking 5 -alpha reductase
 External Development • • In the absence of DHT, the genital swellings form the labia majora; the genital folds remain unfused and form the labia minora; the genital tubercle forms the clitoris and the urogenital sinus forms the lower part of the vagina. With DHT present, the genital swellings migrate and become the scrotum; the urogenital folds enlarge and enclose the penile urethra and become the shaft of the penis; the genital tubercle becomes the glans penis; and the urogenital sinus forms the prostate gland
External Development • • In the absence of DHT, the genital swellings form the labia majora; the genital folds remain unfused and form the labia minora; the genital tubercle forms the clitoris and the urogenital sinus forms the lower part of the vagina. With DHT present, the genital swellings migrate and become the scrotum; the urogenital folds enlarge and enclose the penile urethra and become the shaft of the penis; the genital tubercle becomes the glans penis; and the urogenital sinus forms the prostate gland
 Variant Conditions • • The large majority of people develop as either completely male or completely female. However, 1% or more of the population has some variant condition. A few important terms: – Gynecomastia: development of breasts in a male – Hypospadia: the urethra exits the male body at the base of the penis instead of at the tip, due to failure of the urethra to become enclosed by the urogenital folds. – Intersex: a person whose genitalia are ambiguous or a mixture of male and female • Used to be called hermaphrodites, a term still used in describing animals and plants with both male and female sex organs – a true hermaphrodite has both testicular and ovarian tissue. Very rare, associated with chimeras and mosaics. – Ovotestes are gonads with both male and female tissue in the same organ. – Ambiguous genitalia can be quantified on the Prader scale.
Variant Conditions • • The large majority of people develop as either completely male or completely female. However, 1% or more of the population has some variant condition. A few important terms: – Gynecomastia: development of breasts in a male – Hypospadia: the urethra exits the male body at the base of the penis instead of at the tip, due to failure of the urethra to become enclosed by the urogenital folds. – Intersex: a person whose genitalia are ambiguous or a mixture of male and female • Used to be called hermaphrodites, a term still used in describing animals and plants with both male and female sex organs – a true hermaphrodite has both testicular and ovarian tissue. Very rare, associated with chimeras and mosaics. – Ovotestes are gonads with both male and female tissue in the same organ. – Ambiguous genitalia can be quantified on the Prader scale.
 Chromosomal Variants • • Meiosis, the form of cell division that generates the sperm and eggs, carefully puts exactly 1 copy of each chromosome pair into each cell. Sometimes meiosis goes wrong and puts 0 or 2 copies of some chromosome into a sperm or egg cell. – the best example of this: Down syndrome, which starts with a sperm or egg with 2 copies of chromosome 21. – Maternal age effect: more frequent in older mothers • • The sex chromosomes are quite tolerant of variants. Most common types involve 45 or 47 chromosomes There are many other, rarer types, with 48 or even 49 chromosomes, such as 49, XXXXY. Such conditions almost always lead to serious mental deficiencies. The general rule: if the Y is present, the person is internally and externally male.
Chromosomal Variants • • Meiosis, the form of cell division that generates the sperm and eggs, carefully puts exactly 1 copy of each chromosome pair into each cell. Sometimes meiosis goes wrong and puts 0 or 2 copies of some chromosome into a sperm or egg cell. – the best example of this: Down syndrome, which starts with a sperm or egg with 2 copies of chromosome 21. – Maternal age effect: more frequent in older mothers • • The sex chromosomes are quite tolerant of variants. Most common types involve 45 or 47 chromosomes There are many other, rarer types, with 48 or even 49 chromosomes, such as 49, XXXXY. Such conditions almost always lead to serious mental deficiencies. The general rule: if the Y is present, the person is internally and externally male.
 Klinefelter Syndrome: 47, XXY • • • Occurs about 1 per 500 male births. It is the most common type of sex chromosome variant. The presence of the Y chromosome causes a 47, XXY person to be male, both externally and internally, because the testes are formed. Root symptom: small testes, leading to low testosterone levels. Most, but not all, are sterile. • At puberty, reduced facial and body hair, broader hips, breast development. • 47, XXY children tend to be taller, less physically strong and coordinated, and more quiet and shyer than their peers. Some language and learning problems are common: often slow to learn to speak and read. • Testosterone replacement therapy helps with some of the physical symptoms. Speech therapy and educational services also help. 46, XX males, with the SRY gene on the X, have the Klinefelter appearance.
Klinefelter Syndrome: 47, XXY • • • Occurs about 1 per 500 male births. It is the most common type of sex chromosome variant. The presence of the Y chromosome causes a 47, XXY person to be male, both externally and internally, because the testes are formed. Root symptom: small testes, leading to low testosterone levels. Most, but not all, are sterile. • At puberty, reduced facial and body hair, broader hips, breast development. • 47, XXY children tend to be taller, less physically strong and coordinated, and more quiet and shyer than their peers. Some language and learning problems are common: often slow to learn to speak and read. • Testosterone replacement therapy helps with some of the physical symptoms. Speech therapy and educational services also help. 46, XX males, with the SRY gene on the X, have the Klinefelter appearance.
 Turner Syndrome: 45, X • • Only one X chromosome, sometimes called XO. Since there is no Y chromosome, the primary gonad is the ovary, and 45, X people are female. About 1 in 2500 live female births. – 10% of all spontaneous abortions (miscarriages) are due to Turner syndrome; about 98% of all Turner’s embryos die before birth • • Ovaries completely non-functional, so 45, X women are sterile, with no production of sex hormones and development of secondary sexual characteristics at puberty. Some characteristic physical abnormalities: short stature, low hairline, webbed skin at neck. Kidney and circulatory system problems Often have problems with spatial reasoning and mathematics. Also social difficulties: inability to understand others’ emotions. Can be treated with growth hormone and estrogen. You need 2 X chromosomes fo proper ovarian development. 46, XY females (non-functional SRYgene) resemble Turner’s
Turner Syndrome: 45, X • • Only one X chromosome, sometimes called XO. Since there is no Y chromosome, the primary gonad is the ovary, and 45, X people are female. About 1 in 2500 live female births. – 10% of all spontaneous abortions (miscarriages) are due to Turner syndrome; about 98% of all Turner’s embryos die before birth • • Ovaries completely non-functional, so 45, X women are sterile, with no production of sex hormones and development of secondary sexual characteristics at puberty. Some characteristic physical abnormalities: short stature, low hairline, webbed skin at neck. Kidney and circulatory system problems Often have problems with spatial reasoning and mathematics. Also social difficulties: inability to understand others’ emotions. Can be treated with growth hormone and estrogen. You need 2 X chromosomes fo proper ovarian development. 46, XY females (non-functional SRYgene) resemble Turner’s
 47, XYY • • • About 1 in 1000 live male births. Most XYY’s are never detected: a very mild condition. – since 1960, newly discovered chromosome variants aren’t given the discoverer’s name It was once thought to create hyper-aggressive males with a tendency towards criminal behavior. – Richard Speck, the killer of eight student nurses in 1966, pretended (falsely) to be an XYY to obtain leniency. – A 1968 letter to the Lancet claimed that XYY men were in prison at a rate "25 -60 times as high as the prevalence in the general population”, based on finding 2 XYY’s. – the plot of Aliens 3 involves a prison planet for XYY’s are generally normal in appearance, but with average height about 7 cm above expected and normal build. Perhaps acne is more common than average, but this is disputed. They are often more physically active, somewhat delayed in emotional maturity, and have a slight increase in learning and speech problems. Fertile, normal sex drive, normal testosterone elvels, very rarely pass 2 Y’s to sons. 1970’s British TV series: He had an extra Y, which made him a macho , yet patriotic, criminal!
47, XYY • • • About 1 in 1000 live male births. Most XYY’s are never detected: a very mild condition. – since 1960, newly discovered chromosome variants aren’t given the discoverer’s name It was once thought to create hyper-aggressive males with a tendency towards criminal behavior. – Richard Speck, the killer of eight student nurses in 1966, pretended (falsely) to be an XYY to obtain leniency. – A 1968 letter to the Lancet claimed that XYY men were in prison at a rate "25 -60 times as high as the prevalence in the general population”, based on finding 2 XYY’s. – the plot of Aliens 3 involves a prison planet for XYY’s are generally normal in appearance, but with average height about 7 cm above expected and normal build. Perhaps acne is more common than average, but this is disputed. They are often more physically active, somewhat delayed in emotional maturity, and have a slight increase in learning and speech problems. Fertile, normal sex drive, normal testosterone elvels, very rarely pass 2 Y’s to sons. 1970’s British TV series: He had an extra Y, which made him a macho , yet patriotic, criminal!
 47, XXX • About 1 in 1000 live female births. So mild as to be only rarely detected. Also called triplo-X. • Originally called “superfemale” (early 1960’s).
47, XXX • About 1 in 1000 live female births. So mild as to be only rarely detected. Also called triplo-X. • Originally called “superfemale” (early 1960’s).
 • • Both terms refer to people who have 2 different chromosome sets in different cells. For example, a 46, XX/47, XXY person has some cells with 46 chromosomes and other cells with 47. A mosaic starts out with a single fertilized egg. During an early cell division in the embryo, one cell gained or lost a chromosome. (This is non-disjunction, the same event that happens in meiosis to generate Klinefelter’s, etc. ) A chimera starts out with two separate fertilized eggs, fraternal twins. The two embryos fuse together to form a single individual. – It is not uncommon to have fraternal twins sharing some blood cells, a “blood chimera” – fused embryo chimeras are very rare: there about 30 -40 known XX/XY chimeras (and undoubtedly an equal number same sex chimeras). “tetragametic chimera” – Chimerism is probably the way most true hermaphrodites, who have both ovarian and testicular tissue, are formed. However, actual XX/XY chimeras have been everything from normal male, through various degrees of ambiguous genitalia, to normal female. Sexual development can be quite variable in such people, because the characteristics depend on which cells have which chromosome complement. Mosaics and Chimeras
• • Both terms refer to people who have 2 different chromosome sets in different cells. For example, a 46, XX/47, XXY person has some cells with 46 chromosomes and other cells with 47. A mosaic starts out with a single fertilized egg. During an early cell division in the embryo, one cell gained or lost a chromosome. (This is non-disjunction, the same event that happens in meiosis to generate Klinefelter’s, etc. ) A chimera starts out with two separate fertilized eggs, fraternal twins. The two embryos fuse together to form a single individual. – It is not uncommon to have fraternal twins sharing some blood cells, a “blood chimera” – fused embryo chimeras are very rare: there about 30 -40 known XX/XY chimeras (and undoubtedly an equal number same sex chimeras). “tetragametic chimera” – Chimerism is probably the way most true hermaphrodites, who have both ovarian and testicular tissue, are formed. However, actual XX/XY chimeras have been everything from normal male, through various degrees of ambiguous genitalia, to normal female. Sexual development can be quite variable in such people, because the characteristics depend on which cells have which chromosome complement. Mosaics and Chimeras
 Gene Mutations • • The variants up to now all involve whole chromosomes, which have lots of genes on them. The effects of changing the dosage of many genes tend to be widespread but mild. (or completely lethal, as with most nonsex chromosomes). Now we are going to look at several gene mutations. In these cases, only one gene is affected, but it is completely knocked out. This can lead to large effects, but limited to a few subsystems in the body. Rates are different: for chromosome changes, about 1 in 1000 births is a typical frequency. For gene mutations, each parent needs to contribute a mutated copy of the gene, so rates are usually 1 in 10, 000 births or less. Inheritance is also a factor here: most chromosomal variants are spontaneous events and don’t run in families. Gene mutations are usually inherited variants: there is often a family/community history of the variant type. – New mutations do occur spontaneously, but it’s rare. Most gene variants are inherited from the parents.
Gene Mutations • • The variants up to now all involve whole chromosomes, which have lots of genes on them. The effects of changing the dosage of many genes tend to be widespread but mild. (or completely lethal, as with most nonsex chromosomes). Now we are going to look at several gene mutations. In these cases, only one gene is affected, but it is completely knocked out. This can lead to large effects, but limited to a few subsystems in the body. Rates are different: for chromosome changes, about 1 in 1000 births is a typical frequency. For gene mutations, each parent needs to contribute a mutated copy of the gene, so rates are usually 1 in 10, 000 births or less. Inheritance is also a factor here: most chromosomal variants are spontaneous events and don’t run in families. Gene mutations are usually inherited variants: there is often a family/community history of the variant type. – New mutations do occur spontaneously, but it’s rare. Most gene variants are inherited from the parents.
 • • 5 -alpha reductase is the enzyme") 5 -alpha Reductase Deficiency (5 -ARD) • • 5 -alpha reductase is the enzyme that converts testosterone into DHT. If both copies of the gene that makes this enzyme are defective, the person has 5 -ARD. – Recall that DHT is responsible for the development of male external genitalia At birth, people with 5 -ARD have undescended testes and male ducts (with no female ducts), but genitalia that appear somewhere between female and ambiguous, including a very small penis with hypospadias (which appears to be an enlarged clitoris), and a short vagina. Often raised as girls At puberty, the increase in testosterone is large enough that some DHT gets made, and they develop a male appearance: the testes descend, the penis enlarges, facial hair appears, the voice deepens, muscles develop. Large group in the Dominican Republic: maybe 1 in 90 men. Called Guevedoces, a corruption of “huevos a los doce” (eggs--testicles- at age 12). Raised as girls, they easily switch to the male role. – Other groups found in Malta, Jordan, Pakistan, New Guinea
5 -alpha Reductase Deficiency (5 -ARD) • • 5 -alpha reductase is the enzyme that converts testosterone into DHT. If both copies of the gene that makes this enzyme are defective, the person has 5 -ARD. – Recall that DHT is responsible for the development of male external genitalia At birth, people with 5 -ARD have undescended testes and male ducts (with no female ducts), but genitalia that appear somewhere between female and ambiguous, including a very small penis with hypospadias (which appears to be an enlarged clitoris), and a short vagina. Often raised as girls At puberty, the increase in testosterone is large enough that some DHT gets made, and they develop a male appearance: the testes descend, the penis enlarges, facial hair appears, the voice deepens, muscles develop. Large group in the Dominican Republic: maybe 1 in 90 men. Called Guevedoces, a corruption of “huevos a los doce” (eggs--testicles- at age 12). Raised as girls, they easily switch to the male role. – Other groups found in Malta, Jordan, Pakistan, New Guinea
 Guevodoces Case
Guevodoces Case
 Androgen Insensitivity • The testes secrete testosterone, but the cells lack a receptor for it. No receptor = no response to the hormone. Complete androgen insensitivity, CAIS. – 46, XY with normal (undescended) testes – Used to be called “testicular feminization”. – Incidence about 1 in 20, 000 births • As a result, the male ducts (vas deferens, epididymus, seminal vesicles) are not present. However, the testes secrete MIS, which causes the female ducts (uterus, fallopian tubes, upper vagina) to degenerate. • CAIS people develop female external genitalia, including the lower 2/3 of the vagina. – External genitalia develop as male if DHT is present, but testosterone and DHT use the same receptor. • At puberty, the testes again secrete testosterone. The enzyme aromatase converts some of it into estradiol. Thus, female secondary sexual characteristics develop. Often “voluptuously feminine”. No menstruation of course: no ovaries and no uterus. Pubic and armpit hair is usually scant or absent. – Occasionally, the undescended testes can become cancerous, so they are often surgically removed after puberty is complete (so as to get normal female development).
Androgen Insensitivity • The testes secrete testosterone, but the cells lack a receptor for it. No receptor = no response to the hormone. Complete androgen insensitivity, CAIS. – 46, XY with normal (undescended) testes – Used to be called “testicular feminization”. – Incidence about 1 in 20, 000 births • As a result, the male ducts (vas deferens, epididymus, seminal vesicles) are not present. However, the testes secrete MIS, which causes the female ducts (uterus, fallopian tubes, upper vagina) to degenerate. • CAIS people develop female external genitalia, including the lower 2/3 of the vagina. – External genitalia develop as male if DHT is present, but testosterone and DHT use the same receptor. • At puberty, the testes again secrete testosterone. The enzyme aromatase converts some of it into estradiol. Thus, female secondary sexual characteristics develop. Often “voluptuously feminine”. No menstruation of course: no ovaries and no uterus. Pubic and armpit hair is usually scant or absent. – Occasionally, the undescended testes can become cancerous, so they are often surgically removed after puberty is complete (so as to get normal female development).
 Partial Androgen Insensitivity • Sometimes, the testosterone receptors work inefficiently, due to less drastic mutations than in CAIS. In these cases, the body cells respond in a variable manner to testosterone, leading a a wide variety of ambiguous genitalia. PAIS = partial androgen insensitivity. Also called Reifenstein syndrome. – there is also mild androgen insensitivity (MAIS), which leads to completely male appearance internally and externally, but with some impairment of masculinization at puberty. • • • Variable symptoms: can be predominantly male (with hypospadia, abnormal scrotum, small penis), predominantly female (with enlarged clitoris, fused labia, separate vaginal and urethral openings), or ambiguous genitalia (microphallus--less than 1 cm long), labia-like scrotum, hypospadia, gynecomastia. Similar variability in male internal ducts; females ducts are usually absent due to MIS secretion. Sometimes people with PAIS change gender identity after puberty, in either direction.
Partial Androgen Insensitivity • Sometimes, the testosterone receptors work inefficiently, due to less drastic mutations than in CAIS. In these cases, the body cells respond in a variable manner to testosterone, leading a a wide variety of ambiguous genitalia. PAIS = partial androgen insensitivity. Also called Reifenstein syndrome. – there is also mild androgen insensitivity (MAIS), which leads to completely male appearance internally and externally, but with some impairment of masculinization at puberty. • • • Variable symptoms: can be predominantly male (with hypospadia, abnormal scrotum, small penis), predominantly female (with enlarged clitoris, fused labia, separate vaginal and urethral openings), or ambiguous genitalia (microphallus--less than 1 cm long), labia-like scrotum, hypospadia, gynecomastia. Similar variability in male internal ducts; females ducts are usually absent due to MIS secretion. Sometimes people with PAIS change gender identity after puberty, in either direction.
 Congenital Adrenal Hyperplasia • • • Also called 21 -hydroxylase deficiency. The adrenal glands sit on top of the kidneys and secrete a variety of steroid hormones, including cortisone (stress response), aldosterone (salt balance) androgens (male sex hormones). Steroid hormones are made from cholesterol through a series of biochemical steps. Any one of these steps can be inactivated by mutation. However, about 95% of CAH cases involve defects in the enzyme 21 -hydroxylase is needed to make cortisol and aldosterone (but not androgens). Cortisol is secreted in response to the pituitary hormone ACTH, in a feedback loop. So, if there isn’t enough cortisol being made, more ACTH is made, and this causes the adrenal gland to grow larger (hyperplasia). And, all of those steroid molecules that were destined to become cortisol and aldosterone get diverted into male sex hormones (androstendione and testosterone), which don’t need the 21 -hydroxylase. Very little effect on male fetus, which is already making testosterone, except that after birth the lack of salt regulation can lead to death from excess salt secretion (salt-wasting).
Congenital Adrenal Hyperplasia • • • Also called 21 -hydroxylase deficiency. The adrenal glands sit on top of the kidneys and secrete a variety of steroid hormones, including cortisone (stress response), aldosterone (salt balance) androgens (male sex hormones). Steroid hormones are made from cholesterol through a series of biochemical steps. Any one of these steps can be inactivated by mutation. However, about 95% of CAH cases involve defects in the enzyme 21 -hydroxylase is needed to make cortisol and aldosterone (but not androgens). Cortisol is secreted in response to the pituitary hormone ACTH, in a feedback loop. So, if there isn’t enough cortisol being made, more ACTH is made, and this causes the adrenal gland to grow larger (hyperplasia). And, all of those steroid molecules that were destined to become cortisol and aldosterone get diverted into male sex hormones (androstendione and testosterone), which don’t need the 21 -hydroxylase. Very little effect on male fetus, which is already making testosterone, except that after birth the lack of salt regulation can lead to death from excess salt secretion (salt-wasting).
 21 -Hydroxylase Deficiency
21 -Hydroxylase Deficiency
 fetuses with 21 -hydroxlase deficiency have some problems due to") • Female (XX) fetuses with 21 -hydroxlase deficiency have some problems due to the flood of androgens released by the adrenal gland. The ovaries are normal, and the female (Mullerian) ducts are also normal (since no MIS is made). • Main effects are on the external genitalia: enlarged clitoris, sometimes with an enclosed urethra (i. e. like the penis), labia can fuse and become scrotum-like, vaginal opening can be partly or completely closed. • Appearance at birth varies a lot. Some appear to be normal male with undescended (because nonexistent) testes. However, the chromosomes are XX, the gonads are ovaries, and the uterus and fallopian tubes are usually intact. • Normally, very little androgen is made in childhood. But, CAH causes excess androgens throughout life, leading to rapid growth, but an early closure of the bone growth plates: a very short adult. Also: early puberty, with menstrual problems (and poor sperm production in males). • The other hormones, aldosterone and cortisol, need to be replaced. The cortisol replacement calms the ACTH activity, leading to less androgen production. CAH in females CAH is the most frequent cause of non-standard genitals in genetically female (XX) children.
• Female (XX) fetuses with 21 -hydroxlase deficiency have some problems due to the flood of androgens released by the adrenal gland. The ovaries are normal, and the female (Mullerian) ducts are also normal (since no MIS is made). • Main effects are on the external genitalia: enlarged clitoris, sometimes with an enclosed urethra (i. e. like the penis), labia can fuse and become scrotum-like, vaginal opening can be partly or completely closed. • Appearance at birth varies a lot. Some appear to be normal male with undescended (because nonexistent) testes. However, the chromosomes are XX, the gonads are ovaries, and the uterus and fallopian tubes are usually intact. • Normally, very little androgen is made in childhood. But, CAH causes excess androgens throughout life, leading to rapid growth, but an early closure of the bone growth plates: a very short adult. Also: early puberty, with menstrual problems (and poor sperm production in males). • The other hormones, aldosterone and cortisol, need to be replaced. The cortisol replacement calms the ACTH activity, leading to less androgen production. CAH in females CAH is the most frequent cause of non-standard genitals in genetically female (XX) children.
 Some Environmental Causes • Progestin-induced virilization. Progestin was used to prevent miscarriages in the 1950’s and 60’s. Related to this is the use of androgens to treat endometriosis during that time period, and occasional accidental use of androgens. 160 known cases. – XX fetuses develop as normal females with functioning ovaries, but they may develop some male secondary characteristics and often have enlarged clitorises. Effects are very similar to CAH. • Freemartin: usually seen in cattle: female and male twins, with testosterone from male leaking over to the female due to a shared placenta. Normal female appearance, but undeveloped ovaries and masculinized behavior. Rare or unknown in humans. – Aldous Huxley’s book Brave New World has human freemartins created by hormone treatment of fetuses.
Some Environmental Causes • Progestin-induced virilization. Progestin was used to prevent miscarriages in the 1950’s and 60’s. Related to this is the use of androgens to treat endometriosis during that time period, and occasional accidental use of androgens. 160 known cases. – XX fetuses develop as normal females with functioning ovaries, but they may develop some male secondary characteristics and often have enlarged clitorises. Effects are very similar to CAH. • Freemartin: usually seen in cattle: female and male twins, with testosterone from male leaking over to the female due to a shared placenta. Normal female appearance, but undeveloped ovaries and masculinized behavior. Rare or unknown in humans. – Aldous Huxley’s book Brave New World has human freemartins created by hormone treatment of fetuses.
 Maybe We Will Do The Following
Maybe We Will Do The Following
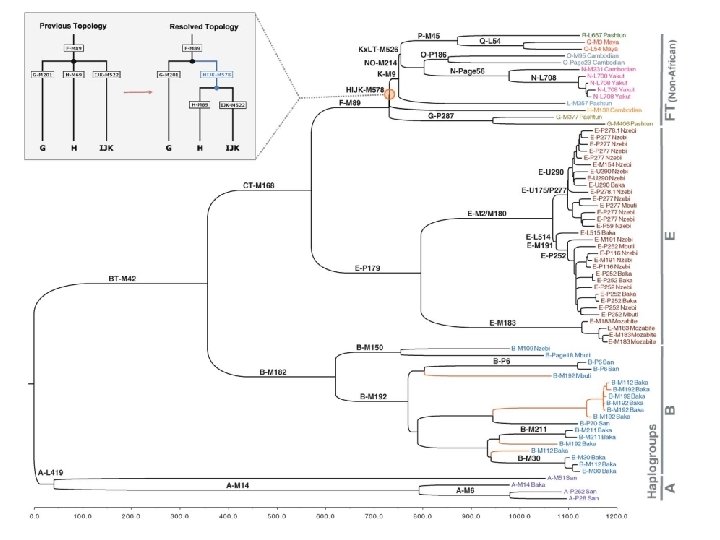
 Y Chromosome Adam • Most of our Y chromosome lineages can be traced to a man who lived about 84, 000 years ago. – This man is our Y-chromosomal most recent common ancestor (Y-MRCA). – Or, relating to the Biblical story of the first man, Y-Adam. This is a strictly metaphorical relationship, implying no scientific basis for any part of the Biblical story! • However, as more mitochondrial DNAs are sequenced, especially from Africa (or descended from there), more unusual lineages have been found. – Haplogroup A, found mostly among the San people in central Africa, contains lineages that don’t converge with everyone else’s until about 200, 000 years ago. (2013 data) – Haplogroup A 00, found in an African-American, and then among a few of his relatives in the Mbo tribe in Cameroon, has an estimated divergence about 338, 000 years ago. This is well before the appearance of the first anatomically modern human fossils.
Y Chromosome Adam • Most of our Y chromosome lineages can be traced to a man who lived about 84, 000 years ago. – This man is our Y-chromosomal most recent common ancestor (Y-MRCA). – Or, relating to the Biblical story of the first man, Y-Adam. This is a strictly metaphorical relationship, implying no scientific basis for any part of the Biblical story! • However, as more mitochondrial DNAs are sequenced, especially from Africa (or descended from there), more unusual lineages have been found. – Haplogroup A, found mostly among the San people in central Africa, contains lineages that don’t converge with everyone else’s until about 200, 000 years ago. (2013 data) – Haplogroup A 00, found in an African-American, and then among a few of his relatives in the Mbo tribe in Cameroon, has an estimated divergence about 338, 000 years ago. This is well before the appearance of the first anatomically modern human fossils.
.") Methods and Rationale • Method: The Y chromosome doesn’t recombine (outside of the PARs). It is much bigger than mitochondrial DNA (60 Mbp vs. 16, 000 bp), and the Y mutation rate is lower. • A mutation occurs, and is then shared with all male-line (patrilineal) descendants. Compare humans and chimpanzees, and find groups that share mutations. • Molecular clock: average rate of mutation stays constant • Calibrate with know archeological events: peopling of Americas (about 12, 000 YBP) and Australia (50, 000 YBP). • Haplogroup A is the most diverged lineage (most basal in the phylogenetic tree). Found primarily among the San, but also scattered throughout Africa. – who are hunter-gatherers in southern Africa, also known as Bushmen, featured in The Gods Must Be Crazy movie, one sub-tribe is the !Kung. – They also have the oldest mitochondrial DNA lineages • Haplogroup BT is found in most Africans and nearly everyone else outside of Africa. Many subgroups.
Methods and Rationale • Method: The Y chromosome doesn’t recombine (outside of the PARs). It is much bigger than mitochondrial DNA (60 Mbp vs. 16, 000 bp), and the Y mutation rate is lower. • A mutation occurs, and is then shared with all male-line (patrilineal) descendants. Compare humans and chimpanzees, and find groups that share mutations. • Molecular clock: average rate of mutation stays constant • Calibrate with know archeological events: peopling of Americas (about 12, 000 YBP) and Australia (50, 000 YBP). • Haplogroup A is the most diverged lineage (most basal in the phylogenetic tree). Found primarily among the San, but also scattered throughout Africa. – who are hunter-gatherers in southern Africa, also known as Bushmen, featured in The Gods Must Be Crazy movie, one sub-tribe is the !Kung. – They also have the oldest mitochondrial DNA lineages • Haplogroup BT is found in most Africans and nearly everyone else outside of Africa. Many subgroups.