3f66a6ded82628e68fe9706adf7676da.ppt
- Количество слайдов: 180



Waterborne Pathogens and Stateof-art Detection Methods Dr Bharat Patel, Associate Professor in Molecular Microbiology & Director, Clinical Microbiology PG Program, School of Biomolecular & Biomedical Sciences, Griffith University, Brisbane Australia

Section I. Indicators of Water Pollution
 2. 1. 1 Concept 3. 1.")
CONTENT 1. The Australian Cooperative Research Centres (CRC) 2. 1. 1 Concept 3. 1. 2 The Five. Water Related CRC 4. 1. 3 CRC Water Quality & Treatment 5. 2. Introduction 6. 2. 1 Microbes on our planet & their role 7. 2. 2 Water as an environment 8. 2. 3 Microbes & their role in water 9. 2. 4 Why monitor water supplies? 10. 2. 5 Ensuring the safety of drinking water. 3. Bacterial Indicators of Pollution 3. 1 What are Bacterial indicators of pollution 3. 2 Total coliforms 3. 3 Changes in coliform definitions 4. Alternatives to Total Coliforms

Section II. Risk Assessment Analysis Framework and Pathogens

CONTENT 1. Epidemiological data on some pathogens. 2. The current list of pathogens 3. How to monitor and assess the risk of pathogens?

SECTION III. Molecular Biology Databases and Tools

CONTENT Molecular Biology Bioinformatics Databases Online tools

SECTION IV. The Biology, Methods for Detection, Identification & Quantitation of Waterborne Pathogens

CONTENT 1. The Biomolecules & Molecular Biology of Cells 2. Biomolecule Based Technics 3. The Biology & Detection Methods of Some Pathogens 4. Modern Techologies a. Polymerase Chain Reaction (PCR) b. Real Time PCR c. Pulse Field Gel Electrophoresis d. New High Throughput Methods

Section I. Indicators of Water Pollution
")
1. The Australian Cooperative Research Centres (CRC)
 in Australia • Participation by")
1. 1 The Concept of Cooperative Research Centres (CRC) in Australia • Participation by industry, universities, CSIRO and State Government bodies • $1 (in cash / in kind contributions) : $1 (cash from Fedral Government • Commercial focus • Skill acquisition and training for the industry. • Usually run by an independent board • 65 CRCs currently on the books ($320 million pa from participants to $140 million pa cash from Fedral Government). • CRCs may have synergies: CRC Water Quality & Treatment CRC Wastewater Treatment CRC Freshwater CRC Microelectronic ecology engineering

1. 2 The Five Water Related CRCs CRC for Coastal Zone, Estuaries and Waterway Management CRC for Waste Management and Pollution Control CRC for Freshwater Ecology CRC for Water Quality And treatment CRC for Catchment Hydrology

1. 3 The Cooperative Research Centre for Water Quality & Treatment 2 nd Round of Funding: 2001 -2008 Participants: 12 Research organisations (8 Universities), 16 Industry & 8 Associate partners Key Objectives: • create a centre of excellence with the capability of pursuing world class research and training. • ensure that participants with their differing disciplines and backgrounds will interact effectively to optimise research outcomes. • increase the human skills base of the water supply industry and to train new post graduate students with specialist water quality skills. • commercialise Project Intellectual Property in such a manner as to ensure that the maximum benefit accrues to the Australian water industry, the Australian environment and the Australian economy generally

2. INTRODUCTION

2. 1 Microbes on our planet & their roles 60% of the organisms are microbial (more microbes than human cells) Surive & thrive in virtually in all environments, often where no other “higher forms” of life exist. 1% have been characterised (24 kingdoms) & 99% remain uncharacterised (the tree of life has been generated using r. RNA as chronometers) Efficient colonisers (rapid growth & doubling) Provide a service to the planet: Ecosystem servicing (biogeochemical cycle, flux) Biotechnology (vitamins, amino acids) Also produce harm: Directly as pathogens Indirectly producing byproducts (toxins) Simple morphology provides very little clues to their identities

NEW Water Microbiology as it Relates to Public Health Human reservoir Animal reservoirs Wastewater Groundwater Aerosols Domestic use Crops Surface water Aerosol Shellfish Land surface Recreation Domestic use Three main routes must be considered to prevent the spread of waterborne (& foodborne) diseases. The particular pathogen, its reservoir and its mode of transmission. The figuree shows the potential route(s) of transmission and the reservoirs. For examples, cows are sources for crypotosporodiosis and poultry are sources for campylosis.

2. 2 Water as a Changable Heterogeneous Environment 1. 2. 3. 4. 5. 6. 7. 8. 9. Climate variability Rainfall Soil erosion Catchment runoff Reservoir Environmental flows Water allocation Irrigation Billabong (wetland) 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. Drinking water Filtration plant Constructed wetland Urban run-offs Wastewater treatment Industrial use Industrial Re-use Bore Water table River sediment Mangrooves Estuary Recreational use

8

2. 3 Microbes & their role in water In nature, microbes live as communities (compete, synergy, complement) They can change the environment for their growth Most natural ecosystems are pristine ie very little nutrients What about reservoirs or dams (man made to maximise storage) A case study of what goes on in a reservoir: Activities affecting a reservoir

Danger Donot enter Farming activities Recreation Forestry activities pump stratification C, N, S, O fluxes & nsformations tra Filtration & treatment ad Le ipe p e INTERACTIVITY & INTERDEPENDENCY pe PVC pi Distribution system ip Copper p Biof ilm nt ? de me elop v Ecology, Environmental & Public Health Microbiology Groups Regulatory Group Transparency Group
: Present in water due to")
2. 4 Why monitor water supplies? Pathogens (produce disease): Present in water due to human / animal fecal contamination Bacteria, virus, protozoa, helminths Diverse types present (eg 100 types of viruses) • Chemical pollutants Carcinogens, toxins, endocrine disruptors & treatment byproducts Present due to industry, microbial activities, geological • Risk to Human Health Dose, host resistance (age, immunity), length of exposure
 Primary assessment: Correct operation of")
2. 5 Ensuring the safety of drinking water (management) Primary assessment: Correct operation of water supply system Verification: Proof that water is safe after supply. This includes monitoring for compliance. Risk assessment: Maximum Acceptable Concentration (MAC). Should be zero but rarely technically & economically feasible. Compliance parameters Compliance & risk assessment may be different for countries, states and applications. Improved awarness: Flexible, transparent, achievable & realistic outcomes

2. 6 Ensuring the safety by monitoring & detection Direct measurement of harmful agents Microbes: Not usually undertaken. Difficult, expensive, time consuming & lack of technology. Risk -> Acute & short-lived Chemicals: Usually undertaken. Technology exists. Risk -> Chronic exposure & delay between sampling, testing & acting on results is okay Monitoring water quality barriers (catchment activities, filtration, disinfection) Complete risk management system for health. Gaining popularity. Currently used indicators of water quality Inadequate, but will be used until “new” & “better” methods tried, tested & ratified. Does not take into account emerging risks (microbes, chemicals). New risks, new ways.

3. Bacterial Indicators of Water Pollution

3. 1 What are bacterial indicators of pollution? Direct pathogen identification / isolation is impractical and / or impossible Alternate indirect “indicator organism” based inference is necessary: • universally present in large nos. in warm blooded animal faeces • readily detectable by simple methods • do not grow in natural waters • persistence in water treatment regimes is similar to that for pathogens
 Coliforms (coli-like, 1880)")
3. 2 Coliforms & E. coli as bacterial indicators (Pre 1948) Coliforms (coli-like, 1880) fulfill these criteria as they indicate fecal pollution and therefore “unsafe water” Total coliforms (Enterobacteriaceae): Escherichia, Klebsiella, Enterobacter & Citrobacter - Ferment lactose, 1% or 109/g human faeces. Used as a standard for testing (assuming that total coliforms = E. coli) PROBLEMS WITH TOTAL COLIFORM RULE • Proportion of E. coli & coliforms as faeces leaves the body. (Coliforms are normal inhabitants of unpolluted soils & water). • Coliforms & waterborne disease outbreaks are not always linked & does not necessarily indicate potential health risk. The current guidelines for drinking water & freshwater recreational waters are shown in the next table as comparisons

Table Bacteriological drinking water & recreational freshwater standards or guidelines Maximum no of indicated organisms permitted / 100 ml of water type Total coliformsa Source of standard WHO Canada Drinking Recreational Thermotolerant coliforms Drinking 1 -10 0 <10 0 Recreational European Economic Community 0 United States 0 200 e <1 -5 200 c <10, 000 d a b C d e Enterococci Turbidtyb (recreational) (NTU) 35 <1 -5 0 -4 <2, 000 d 1 (monthly) < 1 out of the <40 monthly samples analysed or < 5% of the > 40 samples analysed monthly should be positive for coliforms Nephlometric Turbidity Units > 90% are E. coli Compulsory limits, bathing is restricted if >20% samples over 14 day period are positive If 5 samples taken over 30 days are positive
 Rapid methods of")
3. 2 Coliforms & E. coli as bacterial indicators (Post 1948) Rapid methods of identifying were E. coli developed Specific & well known thermotolerant (faecal) coliform test developed. The Total Coliform Rule has been revised, reviewed, reassessed but not dropped (Criteria based on quality & compliance & health risk assessment) • Example 1. US Envrion. Protection agency (USEPA, 1990): The water authority must not find coliforms in > 5% samples. If found, repeat samples within 24 hrs. If repeat samples test positive then it must be analysed for faecal coliforms and E. coli. A positive test signifies Maximum Coliform Limits (MCC) violation & this neccessitates rapid state and public notification. • Example 2 EU Directive, 1998: E. coli, Enterococci & Coliforms 0 / 100 ml. Aesthetic parameters (color, conductivity, chloride, taste & ordour). The parameters should be taken in the context of health risk assessment.

3. 3 Recent changes in coliform definition Coliforms: Members of the family Enterobactericeae; produce acid & gas from lactose (24 -48 h @ 36± 2 o. C) Thermotolerant (fecal) coliforms: As above but were able to grow & ferment lactose at 44. 5± 0. 2 o. C and include E. coli < Klebsiella, Enterobacter & Citrobacter (E. coli also produce indole from tryptophan). SEE “TESTS FOR DIFFERENTIATING COLIFORMS” SLIDE Report 71, 1994 Bacteriological Examination of Drinking water supplies: biochemical definition changed to “acid-only production from lactose” & therefore increased the numbers of species in the coliform category Enzymes: Lactose fermentation by the presence of -galactosidase is now considered as another modification to the coliform definition. Australiasia, UK, Europe & soon USEPA use commercial enyme kits & these detect coliforms that are not traditionally picked up culture media (Noncultural but viable) hence increasing the numbers of species in the coliform group.

Table showing coliform members by evolving definition Acid & Gas from lactose Escherichia Acid from lactose Escherichia -galactosidase Escherichia Klebsiella, Enterobacter, Citrobacter Yersinia, Serratia, Hafnia, Pantoea, Kluyvera Cedecea Edwingella Moellerella Leclercia Rahnella Yokenella Coliforms that can be present in the environment & in human faeces (bold ) and coliforms that are only environmental (bold & underlined)

Commercial kits based detection methods for microbial indicators Kit Manufacturers: IDEXX: Enterolert, Colisure, Colilert Hach: m-Coli. Blue Bio. Control: Coli. Complete Chromocult: Merck Gelman: Micro. Sure Indicator group Enzyme / (substrate) Total Coliforms -D-galactosidase (o-nitophenyl, 6 -bromo-2 -napthyl, 5 bromo-4 -chloro-3 -indolyl linked to -D-galactoside) SEE NEXT SLIDE E. coli -D-glucoronidase (5 -bromo-4 -chloro-3 -indolyl, 4 methylumbelliferyl linked to -D-glucoronide) SEE NEXT SLIDE Enterococci -D-glucosidase (4 -methylumbelliferyl, indoxyl- linked to -D-glucoroside)

Coliforms E. coli E. aerogenes K. pneumoniae assay for all ferment Lactose Enzyme at If growth at named 35 Elevated temperature o. C uses of -glucoronidase detected with Enzyme 44. 5 o. C designate as named -galactosidase MUG designate as E. coli Fecal coliforms detected with ONPG designate as Total coliforms Tests for differentiating coliforms

4. Alternatives to Coliforms as indicators of water pollution

Faecal coliform absence indicates enetric pathogens most likely absent but does not guarantee absence of viruses & protozoal cysts (survive longer in water & more resistant to disinfection) Enterococci, sulfite-reducing clostridia, Bacteroides fragilis, Bifidobacteria, bacteriophages & non-microbiological indicators (faecal sterols) have been proposed as alternatives to fecal coliforms Entercocci is the most preferred (also as alternative to E. coli) • Common commensals in warm blooded guts • 19 species (faecium, faecalis, durans, hirae dominate) • Survive longer & do not grow in the environment • An order of magnitude less than coliforms • Commercial test available

Section II. Risk Assessment Analysis Framework and Emerging Pathogens

1. Epidemiology of some waterborne pathogens. 2. The current list of pathogens 3. How to monitor and assess the risk of pathogens?

1. Epidemiology of some pathogens.
:")
Information modified 1. 90% water related illness are microbial 2. Canada (1974 – 1987): 32 waterborne outbreaks - Giardia: 10, Norwalk & HAV: 5, 17 unknown origin. 2000: E. coli O 157, 2001 Cryptosporidium. 3. USA (1993 – 1998): Cryptosporidium (Milwaukee, Las Vegas, Nevada) 2001: Microcystin & cylindrospermopsin found in Florida drinking water plant (5 times WHO guideline) 4. Europe (1980 – 1990): Cryptosporidium (UK) 6. Vibrio cholera surveillance in India: 34 k (33 deaths) Flood related since July 2001 5. E. coli 0157 ->feces contaminated soil, to irrigation water, to food (Both E. coli 0157: H 7 and VT 6 gene strains isolated) Swaziland: 1992 (20 k), Missouri: 1989, UK: several outbreaks reported, Wyoming: 1998, NY: 1999 (1 k involved, 2 deaths, Campylobacter also implicated), Canada: 2001 (2 K involved, 7 deathsheavy rainfall & inadequate treatment) 6. Northern Ireland: 2001 Cryptosporidium 7. Portugal: 2001 Cyanobacteria toxins reported

8. Multiagent waterborne disease outbreaks: - Switzerland: 2001, coinfection of small round structured virus (SSRV) + Shigella + Campylobacter - Canada: 2001, E. coli 0157: H 7 & Campylobacter -> 2300 ill, 27 developed haemolytic uraemic syndrome complications (HUS), 7 deaths.

2. Common Waterborne Pathogens

Waterborne Pathogens: are classified as members of domains Bacteria, Eucarya or virus. they differ in: Ømorphologies Øgrowth Øphysiology & metabolism Øfine genetic details • Both classification & Identification is now increasingly based on their molecular events & molecular details (see next figure). • The pathogens listed in the following tables have been detected in water and / or in outbreaks. An attempt has been made to provide their classification on the newly introduced molecular trend. • The biology of a number of the pathogens will be described and the possible targets sites for their identification highlighted.
 ARCHAEA (3) BACTERIA (21) s m er ac t ot eo b")
EUKARYA (7) ARCHAEA (3) BACTERIA (21) s m er ac t ot eo b roc lfu ad s com rru ria Ve cte oba id Ac acter rob Fib s ycete ctom Plan ia ter c Di pl om on ia rob ic Pr occ u s ro di Py s ile ph Ha lo ia rid po os icr M Korarchaeota ia tu ci s cc u co co no et ha M rus Methanopy Thermotoga Thermodesulfobacteria Dictyoglomus Thermales Chrysiogen etes Nitro T spira hermomicrob Cy Defer ia Fir anob ribac m ac ter icu ter tes ia Flagellates Aquifex Brown algae ya Eur Thermococcus Dinoflagellates Ciliates Green algae Plants Red algae Fung i Slime molds Animals Crenarchaeota a eot rcha De su o m o ich Tr d na ob n cti A Evolution of Universal Ancestor (3. 5 billion yrs) The Tree of Life - 16 th November 2000 a ia d my la Ch Fus ifor Sp ms iro Bac ch ter ete oid s es

2. A list of bacterial waterborne pathogens Bacterial pathogen Aermomonas hydrophila Desulfovibrio species Campylobacteria Arcobacter Helicobacteria Leptospira Mycobacteria avium-intracelllare & other species Cyanobacteria (toxins) Enterobacterales P r o t e o b a c t e r i a Feces Urine H Sphingomonas Burkeholdaria E. coli 0157: H 7 (hemorrhagic) E. coli (enteroinvasive) E. coli (enterpathogenic) E. coli (enteroitoxigenic) Salmonellae species Salmonella enterica (serovar typhi) Shigella (S. flexneri, S. sonnei, S. dysenteriae, S. boydii) Plesiomonas shigelloides Vibrio cholera 01 Vibrio cholera non-01 Legionella Pseudomonas Phylum H A Disease A Potential Strain dependent cramps, vomit, diarrhea, fever + + + + - Enterobacterales + - - - Enterobacterales Vibrionales ? + + - ? - ? - Fish & crustaceans Cholera (Asiatic flu, Indian, El Tor) + + - - Water diarrhea Enterobacterales Legionellales Pseudomanadales Aeromoandales Desulovibrionales Watery, bloody diarrhea Typhoid, enteric fever, abdominal pain Shigellosis (bacillary dysentery) Legionellosis Potential Stomach colitis (? ) Spirochaetes Actinobacteria + + + + + - Diarrhea Stomach ulcers Weil’, swineherd’s, hemorrhagic Lung disease Cyanobacteria: taxonomy in flux - - Mycrocystins (60), Cylindrospermopsin Duration of disease is between 1 to 42 days
: Some 50")
Information modified Problems associated with bacterial identification Phylum Cyanobacteria (blue green algae): Some 50 to 60 genera; some produce oligopeptide toxins& are of increasing concern (dermal, cytotoxin, mutantion causing and carcinogens). Lifelong exposure vs short term acute exposure Toxins are produced by (a) nonribosomal peptide synthetase (NRPS) which have iterative catalytic domains. Overproduction of one or several sets up a catalytic reaction leading to production of the toxins. (b) Peptide kinase synthetase (PKS). MALDI-TOF MS shows a large spectrum of oligopeptides & other poorly undertood metabolities from cyanobacteria. Microcystis exist as toxigenic organism in reservoirs & form blooms (summer to late autumn) but reports of non-toxicogenic strains have been reported. Some 60 toxins (collectively called Microcystin) are produced; these are thought to react with chlorine to produce other toxin bye-products They have been traditionally classified on the basis of morphology & physiology which has created confusion. Based on 16 S r. RNA and DNA homology studies, the 23 species have now been identified as belonging to M. aeruginosa Toxin production in strains vary based on growth conditions (in vivo and in situ) causing more confusion.

"Calothrix desertica" PCC 7102. Cylindrospermopsis raciborskii str. AWT 205. "Anabaena variabilis" IAM M-3. Nostoc muscorum PCC 7120. Planktothrix rubescens str. BC-Pla 9303. "Oscillatoria agardhii" str. CYA 18. "Oscillatoria corallinae" str. CJ 1 SAG 8. 92. Nostoc punctiforme PCC 73102. "Anabaena cylindrica" str. NIES 19 PCC 7122. Pseudoanabaena biceps PCC 7367. Lyngbya confervoides PCC 7419. 10% Trichodesmium species Spirulina subsalsa str. M-223. Prochloron didemni. Cyanobacterium stanieri PCC 7202. "Oscillatoria rosea" str. M-220. Merismopedia glauca str. B 1448 -1. Gloeothece membranacea. Microcystis wesenbergii. Microcystis novacekii str. TAC 20. Microcystis viridis. Microcystis ichthyoblabe str. TAC 48. Microcystis aeruginosa. Chamaesiphon subglobosus PCC 7430. Octopus Spring microbial mat DNA Yellow Leptolyngbya boryanum PCC 73110. "Plectonema boryanum" UTEX 485. Leptolyngbya foveolarum str. Komarek 1964/112. Gloeochaete wittrockiana str. SAG B 46. 84 Glaucocystis nostochinearum str. SAG 45 Cyanophora paradoxa (colorless flagellate alga) -- cyanelle. "Oscillatoria limnetica" str. MR 1 Phormidium mucicola str. M-221. Phormidium ambiguum str. M-71. Microcystis holsatica. Microcystis elabens. Prochlorococcus marinus PCC 9511. Synechococcus elongatus. Prochlorothrix hollandica. "Oscillatoria neglecta" str. M-82 Phormidium "ectocarpi" str. N 182. Phormidium minutum str. D 5. The identification of cyanobacteria, the causative agents for a number of toxin-producing illnesses, is in a state of flux. The previous identification by morphology & / or toxin production does not reflect the r. RNA based molecular phylogeny.

2. A list of protozoal waterborne pathogens Protozoa Source Disease Animal feces Nonfecal Entamoeba histolytica Rare No Amebiasis (dysentry, enetritis, colitis) Giardia lamblia Yes No Giardiasis (hikers disease) Cryptosporidium parvum Yes No Cryptosporodiosis (cramp, vomit, fever, diarrhea) Microsporidia: Enterocytozoon Septata Yes ? Cyclospora cayatenensis ? ? Toxoplasma gondii Yes No Acanthamoeba No Yes Blantidium coli Yes Cramp, vomit, fever, diarrhea Abdominal pain, bloody diarrhea
, polio, coxsackies")
2. A List of viral waterborne pathogens Virus Cytopathogenic human orphan (ECHO), polio, coxsackies Hepatitis A Virus (HAV) Hepatitis E Virus (HEV) Rotavirus A Rotavirus B Nowalk virus Snow mountain Astrovirus 100’s of others (Developing new method to work with them? ) Small small round structured virus (SSRV) Group Entero Hepatitis Faecal Source Human Animal Yes No Calicivirus Yes Yes Yes No Pigs ? Yes ? Astrovirus Yes No Picorna, Corona, parvo, picobirna, picotrirna ? ? Rotavirus Disease Aseptic meningitis, infantile diarrhea, polio Infectious Hepatitis Acute gastroenteritis Uncertain
 as")
Viruses: Role of some human enteric & respiratory viruses (& some animal viruses) as waterborne pathogens has been well established Most are nonenveloped (except corona & picobirnaviruses) – more ressistant to physical & chemical agents then the lipid containing enveloped viruses Potential transmission route directly or indirectly from animal human & this is of concern

3. How to prioritise the list of pathogens for further studies? By using risk assessment analysis frame work
, Epidemiological")
Table 2 Ecology / occurrence framework for waterborne pathogens Occurrence determinants: Incidence, Lifecycle(s), Epidemiological data – worldwide, reservoirs of agents (animal / human), geological distribution Detection: General, viable? , temperature (water pollution) Water-based vs water borne: Secondary hosts Biofilm Treatment barriers: Source water quality Watershed management Treatment process configuration (driven by source water quality) Distribution concerns Microbial adaptation: Treatment chain Distribution system Pathways: Ingestion Dermal Inhalation

Table 3 Treatment framework for waterborne pathogens Organism properties & origins: Physical: Size, surface properties, (charge, hydrophobicity, affinity for adsobtion), surface structure, settling rate, aggregration, spore-formation Oxidant: Mechanism of action Origin: human, animal, naturally occurring Disinfection kinetics: Disinfection sensitivity (chlorine/chloroamine, chlorine dioxide, ozone, advanced oxidation processes (AOP), UV, pottasium permanganate Synergistic / sequential Contact time Organism survivability: Survival/transport Inactivation/injury/culturability Survival in sludges Organism growth / regrowth: Regrowth Growth in filters Microbial protection / antagonism: Engineering Plant operation: Source basin (size, settling rate, residence time, turnover) intake characteristics (level, position, hydrology), filter operations, line breaks / replacements Maintenance practices (flushing) Water Quality Characteristics: Particulates Chemical & physical (p. H, temp, NOM, hardness, alkalinity) Watershed management: Human activity (sewage inputs) Animal & environmental sources

Conclusions from discussion on pathogens Many pathogens cause water-borne diseases Complex habitats for their growth Pathogenic bacteria, virus & protozoa may co-exist Symptoms similar but causative agents may be different. Therefore assisted diagnosis is not always possible Identification essential as patient treatment regimes depend on the type of causative agent (bacteria vs virus vs protozoa) Alternative methods to assess the risk of the pathogens present in water are necessary which can be achieved by using various frameworks

The Need for Molecular methods for the identification & detection of pathogens • Current US$380 million market & a 20% annual increase is expected • Emerging sophisticated gene technologies (indicators & pathogens) • Skilled (bioinformatics, genomics, phenomics) staff required. • Multicomprehension (ecology, environmental etc) required • Method rapid flexible, reproducible & can be ariticulated to particular needs of different countries • Initial research & development outlay is expensive (research costs)

Next? Finding molecular biology “information libraries” Understanding the principles of molecular biology Finding & using tools for molecular methods

SECTION III. Molecular Biology Databases and Tools

CONTENT Molecular Biology Bioinformatics Databases Online tools Microbial Genomes

Molecular Biology Data. Bases A. Biologists have been very successful in finding DNA & protein sequences: B. - high-speed automated DNA sequencing equipme C. - the Microbial Genomes (and eucaryotic genomes D. - bulk sequences of c. DNAs (ESTs) especially for eucaryotic genomes. Why? - Bioinformatics scientists collect, organize and make sequence data that is generated, available to all biologists - Today data is shared and integrated between the three major data depositories, namely, Gen. Bank, which forms part of the NCBI, European Molecular Biology Laboratory (EMBL) and the DNA Database of Japan (DDBJ). - During Oct. 1996, Gen. Bank contained 1, 021, 211 sequence records = 652, 000 bases of DNA sequence = 3. 1 gigabytes of computer storage space. In June 1997 this escalated to 1, 491, 000 records and 967, 000 bases. Check the sequence record out for 2000 - The contents of Gen. Bank are now doubling in less than a year, and the doubling rate is accelerating ie the data generated and collected is growing exponentially. - Whole genome data has been generated with 32 microbial genomes sequenced. A list of completely sequenced genomes and ongoing genome

- Even simple computation or searching these enormous database requires a huge amount of computer power. What will be needed in 5 to 10 years time is hard to image. B. The Resources at NCBI Established in 1988 as a national resource for molecular biology information. It creates public databases, conducts research in computational biology, develops software tools for analyzing genome data, and disseminates biomedical information - all for the better understanding of molecular processes affecting human health and disease. The NCBI can be summarised as having 3 arms: • Gen. Bank Data Base: The Gen. Bank Database is a sequence database and has a collecti on of publically available sequence data. It is part of National Institute of He alth (NIH), USA. Gen. Bank, Data. Bank of Japan (DDBJ) and European Molecular Biolog y Laboratory (EMBL) have formed the International Nucleotide Sequence Database C ollaboration project under which the 3 organisations exchange data on a daily basis. • In this database, new protein and nucleic acids sequences are deposited by researchers. These sequences are annotated and placed in the sequence database for access and public viewing. The database can also be searched.

• Literature Data Base: This is refered to as Pub. Med. The database holds the abstracte of published articles. The various Sequence Data Bases and Pub. Med literature Data Base are linked via ENTREZ is at the core of the search and retrieval system that integrates and links th e various databases. In order to maximise the benfits of the various databases it is imperative that you read and learn from the ENTREZHELP FILE • Bioinformatics Tools: The most commonly used tool is known as BLAST and enables the user to input a sequence and search for the most similar sequences in the Data Base. C The Ribosomal Data. Base Project (RDP): Contains downloadable Gen. Bank formatted aligned and unaligned small subunit ribosomal r. RNA sequences. Mainly extracted from the Gen. Bank Data Base - is a Gen. Bank subset specialist Data Base. It also conatins a set of integrated online analysis bioinformatics tools useful for aligning user input sequences based on r. RNA secondary structural constraints and for constructing phylogeny. D. KEGG Data Base:

Some Useful Online Molecular Biology Tools 1. Search launchers at http: //searchlauncher. bcm. tmc. edu/ 2. Computational Biology at EMBL: http: //www. emblheidelberg. de/Services/index. html 3. National Centre for Genome Research: http: //www. ncgr. org/ 4. UC Sac Diego Motif Search & alignment tools: http: //meme. sdsc. edu/meme/website/ 5. The tools at Info. Biogen, France: http: //www. infobiogen. fr/services/deambulum/english/index. html 6. The tools at the University of Pennsylvania: http: //www. cbil. upenn. edu/ 7. Compilation of tools & references at the University of California, Santa Cruz: http: //www. cse. ucsc. edu/~karplus/compbio_pages. html

Microbial Genomes Why study microbial genomes? n n n until whole genome analysis became viable, life sciences have been based on a reductionist principle – dissecting cell and systems into fundamental components for further study studies on whole genomes and whole genome sequences in particular give us a complete genomic blueprint for an organism we can now begin to examine how all of these parts operate cooperatively to influence the activities and behavior of an entire organism – a complete understanding of the biology of an organism microbes provide an excellent starting point for studies of this type as they have a relatively simple genomic structure compared to higher, multicellular organisms studies on microbial genomes may provide crucial starting points for the understanding of the genomics of higher organisms

n n analysis of whole microbial genomes also provides insight into microbial evolution and diversity beyond single protein or gene phylogenies in practical terms analysis of whole microbial genomes is also a powerful tool in identifying new applications in for biotechnology and new approaches to the treatment and control of pathogenic organisms

History of microbial genome sequencing n n n 1977 - first complete genome to be sequenced was bacteriophage X 174 - 5386 bp first genome to be sequenced using random DNA fragments - Bacteriophage - 48502 bp 1986 - mitochondrial (187 kb) and chloroplast (121 kb) genomes of Marchantia polymorpha sequenced early 90’s - cytomegalovirus (229 kb) and Vaccinia (192 kb) genomes sequenced 1995 - first complete genome sequence from a free living organism - Haemophilus influenzae (1. 83 Mb) late 1990’s - many additional microbial genomes sequenced including Archaea (Methanococcus jannaschii 1996) and Eukaryotes (Saccharomyces cerevisiae - 1996)

Microbial genomes sequenced to date n n currently there are 32 complete, published microbial genomes – 25 domain Bacteria, 5 Domain Archaea, 1 domain Eukarya (www. tigr. org) around 130 additional microbial genome and chromosome sequencing projects underway

Laboratory tools for studying whole genomes n n n conventional techniques for analysing DNA are designed for the analysis of small regions of whole genomes such as individual genes or operons many of the techniques used to study whole genomes are conventional molecular biology techniques adapted to operate effectively with DNA in a much larger size range. An example is that of pulsed field gel electrophoresis (PFGE), the principle of which will be discussed in detail under Molecular Methods section. PFGE is utilised routinely for epidemiological studies and for fingerprinting of E. coli and Neisseria meningitidis genomes. A potential useful tool for studying species, strain and serovariants

Characteristics of sequenced genomes n n n the 32 complete genome sequences currently available cover a diverse range in terms of phylogeny and environments (eg. human pathogens, plant pathogens, extremophiles etc. ) what conclusions can be made by comparing the genomes of these organisms regarding specific adaptations to proliferation in remarkably different environments? What conclusions can be made about evolutionary relationships between these organisms?

Horizontal gene transfer n n n before microbial genome sequences became available most of the focus of microbial evolution was on ‘vertical’ transmission of genetic information – mutation recombination and rearrangement within the clonal lineage of a single microbial population genome sequences have demonstrated that horizontal transfer of genes (between different types of organisms) are widespread and may occur between phylogentically diverse organisms generally speaking, essential genes (such as 16 S r. RNA) are unlikely to be transferred because the potential host most likely already contains genes of this type that have co-evolved with the rest of its cellular machinery and cannot be displaced genes encoding non-essential cellular processes of potential benefit to other organisms are far more likely to be transferred (eg. those involved in catabolic processes) clearly, lateral transfer of genomic information has enormous potential in improving an microorganisms ability to compete effectively - this may explain why horizontally transferred genes appear so frequently and ubiquitously in microbial genomes an example of this is horizontally transferred genes has been found in pathogenic microbes

Whole genome phylogenetic analysis n n n most of the evolutionary relationships between microorganisms are inferred by comparison of single genes – usually 16 s r. RNA genes although extremely effective, single gene phylogenetic trees only provide limited information which can make determining broad relationships between major groups difficult phylogenetic relationships can be determined by whole genome comparisons of the observed absence or presence of protein encoding gene families in effect this is similar to using the distribution of morphological characteristics to determine phylogeny – without the problem of convergent evolution trees produced using this method are similar to 16 s r. RNA trees, however, as more genome sequences become available more detailed conclusions can be drawn using this method

Species and strain specific genetic diversity n n n although genome sequencing and analysis is very useful when comparing phylogenetically distant taxa, it is also of interest to examine the genomes of very closely related microorganisms this allows a more quantitative approach for examining the relationships between genotype and phenotype complete genome sequences have been determined for two species of the genus Chlamydia (pneumoniae and trachomatis) although the overall genome structure was quite similar, C. pneumoniae contained an additional 214 genes most of which have an unknown function two strains of the bacterium Helicobacter pylori have been completely sequenced (26695 and J 99) overall the two strains were very similar genetically with only 6% of genes being specific to each strain

Case study - Neisseria meningitits n n n n N. meningititis causes bacterial meningitis and is therefore an important pathogen genome is 2. 2 megabases in size 2121 ORF’s were identified with many having extremely variable G+C% (recently acquired genes) many of these recently acquired genes are identified as cell surface proteins there is a remarkable abundance and diversity of repetitive DNA sequences nearly 700 neisserial intergenic mosaic elements (NIME’s) 50 to 150 bp repeat elements these repeat elements may be involved in enhancing recombinase specific horizontal gene transfer

Case study - Borellia burgdorferi n n n B. burgdorferi is a spirochaete which causes Lyme disease it has a 0. 91 megabase linear genome and at least 17 linear and circular plasmids which total 0. 53 megabases 853 predicted ORF’s identified - these encode a basic set of proteins for DNA replication, transcription, translation and energy metabolism no genes encoding proteins involved in cellular biosynthetic reactions were identified - appears to have evolved via gene loss from a more metabolically competent precursor there is significant amount of genetic redundancy in the plasmid sequences although a biological role has not been determined it is possible these plasmids undergo frequent homologous recombination in order to generate antigenic variation in surface proteins
")
What can we learn from microbial genomes? Comparative Genomics: Multiple Pathogenecity Associated Islands (PAI) of 4 uropathogenic E. coli strains against the backdrop of E. coli strain K-12. The PAIs of 25 to 190 k, are inserted within or adjacent to t. RNA genes & contain a different % GC content to the genomic DNA. Transfer mechanism(s)? leu. X 190 kb 97 min phe. R 94 min ~25 kb sel. C 82 min 70 kb phe. V >170 kb 64 min thrw 5. 6 min ~25 kb Strain # 535 536 J 96 E. coli K-12 Chromosome met. V 27 min 60 kb 44 min asn. T 45 kb CFT 073

What can we learn from microbial genomes? Another case study of a microbial genome

Summary n n Microbial genome sequencing and analysis is a rapidly expanding and increasingly important strand of microbiology important information about the specific adaptations and evolution of an organism can be determined from genome sequencing however, genome sequencing merely a strong starting point on road to completely understanding the biology of microorganisms further characterisation of ORF’s of unknown function, in combination with gene expression analysis and proteomics is required

SECTION IV. The Biology, Methods for Detection, Identification & Quantitation of Waterborne Pathogens

CONTENT 1. The Biomolecules & Molecular Biology of Cells 2. Biomolecule Based Technics 3. The Biology & Detection Methods of Some Pathogens 4. Modern Techologies a. Polymerase Chain Reaction (PCR) b. Real Time PCR c. Pulse Field Gel Electrophoresis d. New High Throughput Methods

1. The biomolecules & molecular biology of cells
 • Genome size")
DNA RNA TOTAL DNA: • Mol%G+C • Restriction Patterns (RFLP, PFGE) • Genome size • DNA homology DNA SEGMENTS: • PCR based fingerprinting (ribotyping, ARDDRA, RAPD, AFLP, AP-PCR, rep-PCR) • DNA probes • DNA sequencing • r. RNA sequencing • LMW RNA profiles 23 S S 16 5 S t. RNA Plasmid DNA • Electrophoretic patterns of total cellular or cell envelope proteins (1 D or 2 D) PROTEINS m. RNA • Multienzyme patterns (multilocus enzyme electrophoresis) CHEMOTAXONOMIC MARKERS • Cellular fatty acids (FAME) • Mycolic acids • Polar lipids • Quinones • Polyamines • Cell wall compounds • Exopolysaccharides EXPRESSED FEATURES • Morphology • Physiology (Biolog, API, …) • Enzymololgy (APIzyme) • Serology (monoclonal, polyclonal) DIFFERENT TARGETS FOR MICROBIAL IDENTIFICATION
: 200,")
New Selection of Different Targets 1. Cell surface: a. proteins (receptors, porins, siderophores): 200, 000 / cell b. Polysaccharides (LPS): 2 million in Gram –ve cells 2. Cytoplasmic: a. Ribosomes (rproteins & r. RNA): 20, 000 in dividing cells. b. Non-ribosomal RNA: 100 – 1, 000 / cell (depending on rate of transcription or rate of degradation) c. Non-iobosomal proteins (RNA polymerase): 3, 000 / cell The target concentrations in a 1 ml sample will be 0. 03 attomolar(3, 000 molecules / cell) to 20 attomolar (2 million / cell)

2. Biomolecule based Technology

F G es Sp i ec n s u en St ra i Technique ily am Restriction Fragment Length Polymorphism (RFLP) Low frequency restriction fragment analysis (PFGE) Phage and bacteriocin typing Serological techniques Ribotyping DNA amplification (AFLP, AP-PCR, RAPD) Zymograms (multilocus enzymes) Total cellular protein electrophoretic patterns DNA homology Mol% G+C DNA amplification (ARDRA) t. DNA-PCR Chemotaxonomic markers Cellular fatty acid fingerprinting (FAME) r. DNA / r. RNA sequencing DNA probes DNA sequencing Highthrougput assays (Microarrays, Cantilever arrays) The limits of resolution of various techniques in microbial identification

3. The biology & detection methods of some pathogens
 of Water-borne Pathogens Virulence Factors: • VF encoded by genes •")
Virulence Factors (VF) of Water-borne Pathogens Virulence Factors: • VF encoded by genes • their presence makes the microbe pathogenic • Most E. coli in human/animals not pathogenic as VF genes are absent • Aquatic environment may be reservoir where “virulence breed” by Plasmids/phage transmissision of VF (E. coli, Y. eneterocolitica & A. hydrophila) Viruses: • Virus multiplication • Most non-enveloped. Antigenic shift & drift in capsid proteins Bacteria: • Salmonella – O (in LPS, endotoxin) & Vi (capsule) antigens • E. coli may contain > 1 VFs: -EIEC enteroinvasive: Shiga-like toxin (SLT), -ETEC enterotoxigenic: Vibrio like heat labile/stable toxin (ST, LT), ID > 1 million cells. Interfere with Na & Cl across CM, travelers diarrhea. -EPEC, enteropathogenic: Adhesive VF for GI epithelia. , infantile diarrhea in developing countries -EHEC, enterohemorrhagic: Shiga-like toxin (SLT), ID < 1000 cells, Since 1982, strain O 157: H 7 has affected 20, 000 in US (>100 deaths), Found in ground beef & now in cider & fruit juices. • Vibrio cholera: Cholera txin resides on plasmids which are transferred by phage

Protozoal Parasites: Detection in water supplies is a challenge Biology remains unstudied, biomarkers unavailable Methods have limitation & cannot differentiate: • human species form animal species • infectious forms from noninfectious forms Techniques such as Microscopy, PCR & RFLP of limited use for diagnostics Characteristics: • Entamoeba histolytica: a long history as a waterborne pathogen (no US major outbreaks reported for decades, no major nonhuman reservoir) Cryptosporidium parvum: Major problem. • • Microsporidia: unknown. Ubiquitous parasite of insects, human & animals. Significance

Diagnostic Methods 1. Recovery and Concentration: To increase pathogen concentration by physical, chemical or enrichments. 2. Purification & Separation: Methods use knowledge of pathogen size, shape, density etc surface properties (hydrophilicity, reactivity, receptors), growth stages (spores, capsules, ooocytes) for this. 3. Assay & Characterisation: Differentiate pathogens from all others: Qualitative / quantitative, viable / nonviable. Cultural, immunological and NA based [ NA amplification (PCR), NA identification & characterisation methods (hybridisation by gene probes, RFLP & nucleotide sequencing)]. NA based methods are specific & sensitive but incapable of differentiating live but inactivated cells from dead / noninfectious ones.


4. Modern Techologies
")
a. Polymerase Chain Reaction (PCR)

Figure 1 B Figure 1 A Figure 1 C DNA Fundamentals

A short video clip to show the principle of PCR
. Xeroxing (copying) of")
2. What is PCR? DNA replication in a tube (in vitro). Xeroxing (copying) of DNA. 3. The Components of PCR The basic components of a PCR reaction are - one or more molecules of target DNA - two oligonucleotide primers - thermostable DNA polymerase - d. NTPs 4. The Process of PCR Each PCR cycle requires three temperature steps to complete a round of DNA synthesis:

Cycle 1: The original DNA template will continue to be copied by the DNA polymerase until it stops or the process is interrupted by the start of the next cycle. Cycle 2: Amplicons of intermediate lengths produced Cycle 3: Amplicons of defined lengths will be produced. Cycle 4 onwards: Target sequence will be amplified exponentially The final number of copies of the target sequences is expressed as: (2 n-2 n)x where n = no of cycles, 2 n = 1 st product obtained after cycle 1 & 2 nd products obtained after cycle 2 with undefined length and x= no. of copies of the original template

PCR target molecules accumulate as a function of cycle number with the exponential phase lasting for about 30 cycles under standard reactions conditions. The plateau phase results from limiting amounts of enzyme and reduced enzyme activity. The production of 1 billion copies of the specific targeted DNA from 1 template during the 30 cycles is theoretically possible but never practically achieved because of lack of 100% PCR efficiency. The products formed during the process could be mixtures of specific and non-specific products and these factors reduce PCR efficiency from theoretical 100%.

5. The Factors Affecting PCR I. Generalities: • pipette water first, followed by the other ingredients. • work “on ice” in order to minimise primers binding to the DNA template and to prevent functioning of the polymerase (even theoretically) prior to the first denaturing step. • avoid aerosols while pipetting (or use aerosol-ressistant pipette tips) & work under laminar flow hoods. • Be very accurate when dealing with small volumes. (Multiplex PCR of two different genomic DNA samples can be very susceptible to errors in pipetting). II Thermocyclers and PCR vials: • The same PCR program will work slightly different on different thermocyclers (temperature and time profiles may differ) and therefore the PCR results using the same primer pair may vary. • New PCR machine designs accommodate thin-walled 0. 2 ml PCR vials (and/or 96 wells microtiter dishes). Contact between the metal and plastic is very good and aided by the downward pressure from the heated lid. • Older machines accommodated 0. 5 or 1. 5 ml vials and the contact between the vials and the metal block is not always perfect because of slight differences in shape and wall thickness amongst manufacturers, often resulting in reduced or no amplification

II. Denaturing temperature and time: • 150 m. M Na. Cl decreases the melting (denaturing) temperatures of 91 -97 o. C (not 100 o. C) • Taq polymerase has a half-life of 30 min at 95 o. C. Denaturation temp as short as possible • 2 -5 minutes initial denaturing step prior to the start of cycling is not necessary • 1 min at 94 o. C (usually between 15 sec to 30 secs) avoids loss of Taq enzyme activity III. Choosing and Primer Design: • 17 -28 bases. Longer primers 30 -35 bp for multiplexing • both primers have a close melting temperature or Tm of within 5 o. C. • G+C content of 40 -60% (Tms between 55 -80 o. C are preferred). • primer sequence with 1 -2 GC pairs at the start and end improves priming efficiency • three or more Cs or Gs at the 3'-ends of primers may promote mispriming • Check for primer-primer interactions: 3'-ends of primers not complimentary • Check for primer self-complementarity (ability to form 2 o structures such as hairpins) • primer sequences checked against DNA database (using BLAST programs) for “uniqueness” • Useful ON-LINE programs for primer design can be found at the following URLs Primer 0. 5: http: //trishul. sci. gu. edu. au/Ja. MBW/5/2/index. html Web. Primer: http: //genome-www 2. stanford. edu/cgi-bin/SGD/web-primer Primer 3: http: //www-genome. wi. mit. edu/cgi-bin/primer 3_www. cgi IV. Primer Annealing Temperature: • Calculate Tm of primer using “rule of thumb” 4(G + C) + 2(A + T)o. C. • Temperature of annealing (Ta) should be about < 5 o. C below the lowest Tm of the pair of primers V. Extension or Polymerisation: • Taq polymerase incorporates about 2000 nucleotides/minute at optimal temperature 72 -78 o C • Rule of thumb “ 1 min for a 1 kb product, 2 min for a two kb”
 Vent (Thermococcus")
Some Commercial thermostable DNA polymerases and their sources: Deep Vent (Pyrococcus GB-D) Vent (Thermococcus litoralis) Ul. Tma (Thermotoga maritime) Tth (Thermus thermophilus) Amplitaq (Thermus aquaticus) Amplitaq Stoffel (Thermus aquaticus) Hot Tub – (Thermus flavus) Pyrostase (Thermus flavus) Tbr (Thermus brockianus) Tfl (Thermus flavus) Pfu – (Pyrococcus furiosus) Pwo – (Pyrococcus wosei) Recombinant Recombinant Natural Natural Properties of some thermostable DNA polymerases:

VI. Reaction Volumes: • thin walled, 0. 2 ml plastic vials for 96 well thermocyclers have heated lids (no oil). Reaction volumes (5, 25 or 100 ul) okay • PCR product yield is higher in 5 µL compared to 100 µL volumes (product can be visualised) VII. Primer Amount in PCR: • 100 -500 n. M each primer • Purchased as 10 -25 m. M; 0. 5 -1 ml primer is sufficient for 25 -100 ml PCR reactions VIII. Template concentrations: • Within limits, increasing primer and template concentration may improve the outcome of the PCR reaction, and should be considered as a way to optimize PCR reactions IX. Nucleotides (d. NTP): • Stock of 25 m. M each stored as small aliquots (2 -5 µl) at -20 o C. • Centrifuge long term stored solutions as water condenses on the walls changing conc • Dilute stocks in buffered water (10 m. M Tris p. H 7. 7 -8. 0) as acid p. H hydrolysis d. NTP to d. NDP and d. NMP X. Relationship between Mg. Cl 2 and d. NTP concentration: • 200µM d. NTP each and 1. 5 m. M Mg. Cl 2 is recommended with Taq polymerase, (Perkin Elmer Cetus). Theoretically 6 -6. 5 µg of DNA is synthesised from 25 µl reaction. Besides magnesium bound by the d. NTP and the DNA, Taq polymerase requires free magnesium. This is probably the reason why small increases in the d. NTP concentrations can rapidly inhibit the PCR reaction (Mg gets "trapped") whereas increases in magnesium concentration often have positive effects.

XI. Adjuvants in PCR Reactions: • Between 5 -10% DMSO or glycerol promotes increase in PCR yield • 0. 8µg/µl BSA promotes better yield than DMSO or glycerol Agarose Gels: Gel electrophoresis for detecting PCR products • Nu. Sieve agarose separates short products better than the regular agarose. More expensive but use less for the same gel strength as regular agarose. • All regular agarose, irrespective of the brand, behave the same • 600 bp separation: Run very fast (3 -4 h for a 15 -20 cm long 2 -3% agarose gel). Bands are sharper Non-denaturing PAA gels: • 6 -10% PAA gels used for PCR products differing in only a few bp in length Denaturing PAA gels: • 6% PAA/7 M urea sequencing gel is used to separate radiolabeled multiplex PCR products Real Time detection of PCR products • No gels required. Recent method. Relies on the ability of a dye, SYBR Green, to intercalate with double stranded amplicons produced during PCR, to produce fluorescence which is detected in a flurometer. (Dealt with in a subsequent section)

New Distinguishing between PCR & Real Time PCR Real. Time-PCR Primer design & annealing Primer Design & annealing Probe Design internal to PCR amplicons & Annealing PCR cycling parameters PCR Cycling Parameters 30 cycles Detection of fluorescence every cycle (annealing and / or extension) Detection by Gel electrophoresis Subsequent Gel Electrophoresis (if necessary)

b. Real. Time-PCR

Real Time PCR 1. Introduction 2. General Principles & Concepts A. What are Fluorescent dyes? B. What is Fluorescence Resonance Energy Transfer (FRET)? C. Some commonly used flurophores for labeling probes D. Quantitating Fluorescence E. Improving Fluorescence Signal Detection (new) 3. Instruments A. Light. Cycler (Idaho Technologies Roche) B. Rotor-Gene (Corbett Research) C. i. Cycler (Bio. Rad) D. Mx 4000™ Multiplex Quantitative PCR System (Stratagene) E. ABI Prism 7700 (Perkin-Elmer-Applied-Biosystem) F. Smart. Cycler (Cephid)

4. Types of probes & design A. DNA binding dyes B. Oligonucleotide Hybridisation Probes I. Hydrolysis Probes (Taq. Man) II. Strand Displacement Probes A. Roche Dual probe B. Hair Pin Probes Molecular Beacon Sunrise Uni. Primer Scorpion • • • Stem & Loop Duplex FRET Duplex C. Peptide Nucleic Acid Probes (PNA) 5. Applications

1. Introduction

What is Real Time PCR? Real Time PCR is a technique in which fluoroprobes bind to specific target regions of amplicons to produce fluorescence during PCR. The fluorescence, measured in Real Time, is detected in a PCR cycler with an inbuilt filter flurometer.

WHAT IS THE DIFFERENCE BETWEEN PCR AND REALTIME PCR ? Fluorescence is measured every cycle The signal is proportional to the amount of product Observed in Real Time during PCR

WHAT CAN BE DONE WITH Real Time PCR? Specific quantification of: DNA RNA Protein

2. General Principles & Concepts

2 A. What are Fluorescent dyes? New When a population of fluorochrome molecules is excited by light of an appropriate wavelength, fluorescent light is emitted. The light intensity can be measured using a flurometer or by measuring a pixel-by-pixel digital image of the sample. In the later case, image analysis software, makes it possible to view, measure, render, and quantitate the resulting image. Excitation and Emission: Fluorodyes absorb light at one ê level (wavelength) & thereby boosts an electron to a higher energy shell (an unstable, excited state). • The excited electron falls back to the ground state and the flurophore re-emits light but at a second lower ê, longer wavelength. • This shift makes it possible to separate excitation light from emission light with the use of optical filters. • The wavelength (nm) where photon energy is most efficiently captured is defined as the Absorbancemax & the wavelength (nm) where light is most efficiently released is defined as the Emissionmax. • The difference in absorbed & emitted wavelength = Stoke’s shift ( ). can be a large or small number depending on the loss of energy during fluorescence process.
 New • The wavelegth range for which")
2 A. What are Fluorescent dyes? (cont’d) New • The wavelegth range for which flurodyes absorb light is small (~ < 50 nm) and light outside this range will not cause the molecule to fluoresce. 2. Linearity: Theintensity of the emitted fluorescent light is a linear function of the amount of fluorochrome present when the illuminating light has a constant wavelength and intensity (for example, using a controlled laser light source). The signal becomes nonlinear at very high fluorochrome concentrations. 3. Brightness: Fluorochromes differ in how much intensity they are capable of producing. This is important because a dull fluorochrome is a less sensitive probe than a bright fluorochrome. The brightness depends on two properties of the fluorochrome • Its ability to absorb light (extinction coefficient). • The efficiency with which it converts absorbed light into emitted fluorescent light (quantum efficiency). 4. Environmental factors: Environmental conditions can affect the brightness or the wavelength of the absorption or emission peaks. Such fluorochromes are useful for analyzing changes in H+, Mg 2+, or Ca 2+ concentration & detecting lipids or double-stranded DNA. Photodestruction (photobleaching) of photosensitive dyes (eg fluorescein) is caused by intense light. Use antifade agents or lower the laser power

Fluorescent dyes have become the preferred method of detection for nucleic acids in Molecular Biology. They are used as single conjugated dyes to oligonucleotides for: • Automated fluorescent DNA sequencing, • Fluorescent genotyping & Terminal Fragment Restriction Length Polymorphism (TFRLF) AND As double or multiple conjugated dyes to oligonucleotides for simultaneous detection, identification and quantitative techniques in Real Time PCR (Molecular Beacons) based on the principle of Fluorescence Resonance Energy Transfer (FRET) or quenching.
? FRET is a distance")
Modified 2 B. What is Fluorescence Resonance Energy Transfer (FRET)? FRET is a distance dependent interaction between the excited states of 2 dye molecules in which excitation is transferred from a donor molecule to an acceptor molecule without emission of a photon
: The Donor and Acceptor in close physical proximity (10 100")
2 B. FRET (cont’d): The Donor and Acceptor in close physical proximity (10 100 Angstrom) can lead to FRET or Quenching hv hv A A D D A D (a) Physical proximity + hv (b) No physical proximity + hv (c) No hv Hybridization probes (FRET +ve) hv hv R R Q Q (d) Physical proximity + hv (Quenching) (e) No Physical proximity + hv (Quenching released) Taq. Man & Beacon Probes

2 C. Some Commonly used flurophores for labeling probes FLUORESCEIN TAMARA 494 / 518 nm TET CY 5 650 / 690 nm CY 3 TEXAS RED HEX LC-RED 595 / 615 nm • Consider the cost, ease of synthesis, proprietary, delivery time etc

2 D. Quantitating Fluorescence A flurometer exploits the principles of fluorescence to quantitate fluorescent (dye) molecules in the following way: A strong light source which produces light within a specific light range ( eg xenon arc lamp) is focused down to a tight beam. The tight beam of light is sent through a filter which removes most of the light outside of the target wavelength range for a particular fluorescent molecule. The filtered light beam passes through the liquid target sample striking some of the fluorescent molecules in the sample. Light emitted from the fluorescent molecules that is traveling orthogonal to the excitation light beam pass through a secondary filter that removes most of the light outside of the target wavelength range.

The filtered light then strikes a photodetector or photomultiplier which allows the instrument to give a relative measurement of the intensity of the emitted light. Fluorescent molecules can be detected at concentrations below a level visible to the subjective human eye & as fluorescence intensity vs. concentration is a linear relationship, dye concentrations can be determined with a good degree of accuracy

2 E. Improving Fluorescence Signal Detection A number of ways are available to improve detection and measurement of the emitted fluorescent signal. a. Elimination of the excited light from the collection pathway by several methods: • Orienting the excitation light path so that the light does not shine into the collection pathway. • Inserting optical filters into the collection pathway to reject the excitation wavelength. • Delaying collection until after a pulse of excitation light has disappeared. b. The fluorescent signal can also be enhanced by increasing the dwell time or by scanning the sample multiple times and mathematically processing the signals to reduce random noise. Such methods are useful and practical for increasing the sensitivity at the low end. c. A band-pass optical filter can be used to reject broad-spectrum background emissions. This type of filter rejects wavelengths shorter and longer than the selected band, while allowing wavelengths in the selected wavelength range (centered around the fluorescent emissions of the sample) to pass through to the collection pathway.
 Wide variety: Fluorochromes with a wide variety of")
2 F. Advantages of Fluorescence (New) Wide variety: Fluorochromes with a wide variety of characteristics are available, including fluorochromes that • Respond to p. H or ion concentrations. • Localize based on hydrophobic and hydrophilic interactions. • Can be cross-linked to proteins, NA, lipids, or polysaccharides. Commercial available: Fluorochromes are available crosslinked to many other molecules (eg fluorescently labeled monoclonal and polyclonal antibodies with a choice of fluorochrome, fluorescently labeled enzyme substrates, such as fluorescent chloramphenicol for chloramphenicol acetyl transferase (CAT) assays and fluorescein digalactoside for b-galactosidase assays (lac. Z gene). Multiple-label possibility: A significant advantage of fluorescent labeling over other methods is the possibility of recording the fluorescence of two or more fluorochromes separately using optical filters and a fluorochrome separation algorithm. Thus, components can be labeled specifically and identified separately in the sample or lane (EG Real Time PCR applications)
 (New) Stability: The long shelf life compared to")
2 F. Advantages of Fluorescence (cont’d) (New) Stability: The long shelf life compared to radiolabeled molecules. Fluoromonoclonal antibodies, oligonucleotide hybridization probes, and PCR primers can be stored for six months or more but antibodies labeled with 125 I and 32 P-labeled nucleotides and oligonucleotides become unusable in a month and a week respectively. Reagent batches can be standardized and used for extended periods in antigen localization, ELISAs, enzyme assays (such as CAT and kinase), PCR-based genetic typing assays (such as STR analyses), DNA sizing and quantitation, DNA sequencing, protein sizing and quantitation. Low hazard: Most fluorochromes are easy to handle, however, proper care should be observed (eg gloves) with DNA and RNA stains (mutagenic as they bind to these molecules). In contrast, lead or acrylic shields are required for handling radioactive materials and require special disposal protocols (eg shielded storage, long-term decay, or regulated land-fill disposal) Lower cost: The long shelf life and cheaper transportation and disposal costs for fluorochromes make fluorescent labeling, in many cases, less expensive than radiolabeling.

3. Real Time PCR Instruments

General Description of Instruments 1. PCR cycler: 1. 96 well format, 8 tube format, capillary (glass) 2. Air or block heater 3. Temeperature ramp, temperature gradient 2. Fluorescence emission & detection : 1. Fluorometer 2. CCD camera 3. Excitation source: xenon, halogen, laser 3. Fluorescent Dye Labeling of: 1. Oligonucleotides 2. Peptide Nucleic acids (PNA) 4. Near Infra Red Dyes: 1. Available but no commercial labeling service available
 Glass capillaries + Air (not metal block) =")
Xenon Arc lamp (250 -1000 continuous) Glass capillaries + Air (not metal block) = rapid Idaho Light. Cycler

The Lightcycler performs PCR in small-volume glass capillary tubes, contained within a rotor-like carousel, that are heated and cooled in an airstream. The carousel is rotated past a blue lightemitting diode, and fluorescence is read by three photodetection diodes with different wavelength filters that allow the use of spectrally distinct fluorescent probes. Assays based on DNAbinding dyes, hydrolysis probes, molecular beacons and dual hybridisation probes are possible. Up to 32 reactions are typically carried out in 5– 20 µl volumes and PCR is completed in less than 20 min. The fluorescence readings taken at every cycle of the PCR reaction are displayed immediately after each measurement, allowing amplification runs to be terminated or extended, as appropriate, during individual runs.

Dual Light source Excitation: 470 & 530 32 x 0. 2 ml plastic tubes Air heated, centrifugal mixing Rotor-Gene from Corbett Research

Real Time Detection 1 a. Excitation filters 1 b. Emission filters Tungsten halogen light source (350 - 1000 nm continuous) Microplate format Cycler i. Cycler from Bio. Rad

Biorad Instruments have recently launched an optical module that fits their standard thermal cycler and transforms it into a realtime RT-PCR system. This instrument is capable of generating and detecting a wider range of excitation frequencies than either the ABI 7700 or the Lightcycler. At present, it can monitor up to four different fluorescent reporters at any one time and can be used for any one of of the alternative fluorescent RT-PCR strategies. Furthermore, unlike the ABI 7700 which scans its 96 samples sequentially, this instrument can scan up to 96 samples simultaneously, with a sampling frequency that can be defined by the user.
 96 well plates")
Quartz tungsten halogen lamp (excitation range of 350 to 750 nm) 96 well plates Fast cycling (90 min) CCD camera for capturing fluorescence Mx 4000™ Multiplex Quantitative PCR System from Stratagene
 contains a builtin thermal cycler with 96 -well")
The ABI Prism 7700 (Perkin-Elmer–Applied Biosystems) contains a builtin thermal cycler with 96 -well positions, and is able to detect fluorescence between 500 nm and 660 nm. Fluorescence is induced during the PCR by distributing laser light to all 96 samples contained in thinwalled reaction tubes via a multiplexed array of optical fibres. The resulting fluorescent emission returns via the fibres and is directed to a spectrograph with a charge-coupled device (CCD) camera. Because each well is irradiated sequentially, the dimensions of the CCD array can be used for spectral resolution of the fluorescent light. This instrument can be used for assays based on DNA-binding dyes, molecular beacons and hydrolysis probes.

4. Probe types & Design

A. Fluorescent DNA Binding Dyes
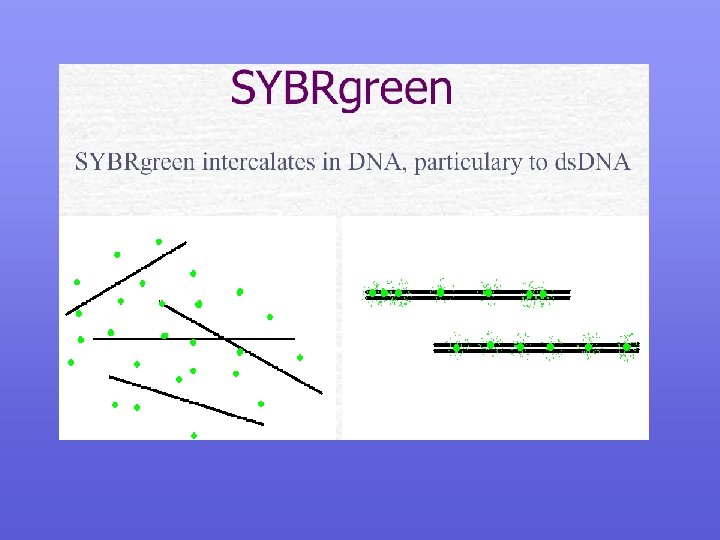
")
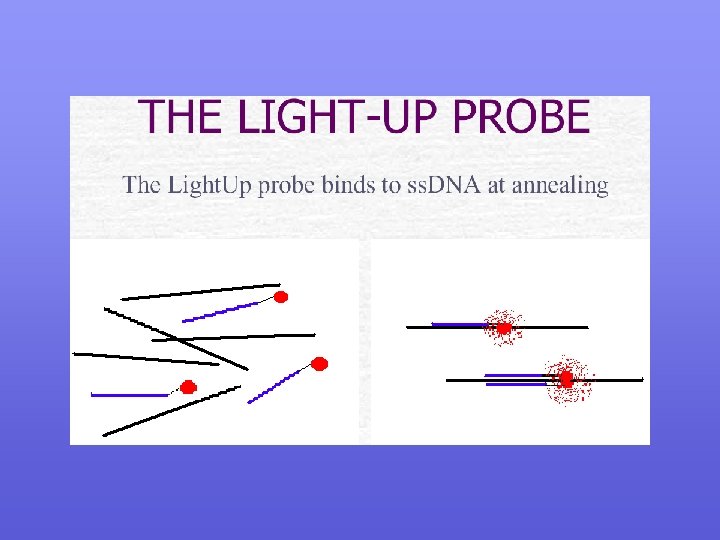
B. Oligonucleotide Hybridisation Probes I. Hydrolysis Probes (Taq. Man)

Taq. Man Probe Forward Primer R Probe Q This strand is not shown below R Q Forward Primer Reverse Primer R Q Probe The Taq. Man probe binds to ss. DNA at a combined annealing and elongation step. It is degraded by the polymerase which releases the reporter dye (R) from the quencher (Q).

New Designing Taq. Man Probes Taq. Man® Probe Design: • Keep the G-C content in the 30— 80% range. • Avoid runs of an identical nucleotide especially Guanine • Do not put Gs on the 5' end. • Select the strand that gives the probe more Cs than Gs. • For single-probe assays, Tm should be 68— 70 °C Primer Design: • Choose the primers after designing the probe. • Design the primers as close as possible to the probe without overlapping the probe. • Keep the G-C content in the 30— 80% range. • Avoid runs of an identical nucleotide especially Guanine • The Tm should be 58— 60 °C. • The five nucleotides at the 3' end should have no more than two G and/or C bases. NOTE: Applied Biosystems provide Primer Express® for design of primers and probes in real time with Real Time Quantitative PCR systems (7700, 5700)

B. Oligonucleotide Hybridisation Probes II. Strand Displacement Probes A. Roche Dual probe

Microbe using identification 16 S r. RNA genes Adjacent probes
 Identify useful Primer / probe regions (use")
New Designing Dual Adjacent Hybridisation Probes (i) Identify useful Primer / probe regions (use any of the Primer software) (ii) Check primer / probe specificity using Fasta against Genbank database (iii) Check Tm using http: //alces. med. umn. edu/rawtm. html (iv) Check propenisity of probe to self anneal using Oligo Selection Program (v) Probe Tm’s should be near equal and 5 -10 C greater than primer Tm’s (vi) The 3’ end of the upstream probe should be labeled by fluorescein, which serves as the donor in the FRET and blocks extension from the probe (vii) the 5’ end of the downstream probe should be labeled by Cy 5, which serves as the acceptor in the FRET, and the 3’ end of the probe should be phosphorylated to block extension the probes should be separated by one base (viii) the probes should be placed on one strand near a primer on the opposite strand.


Designing r. RNA gene directed fluorroprobes for detection & identification of Campylobacter & Arcobacter by Real Time PCR Forward PCR primer FITC probe Reverse PCR Cy 5 probe

2 1 Figure 1: Continuous monitoring of fluorescence during PCR in which DNA templates from A. butzleri ATCC (- -), C. jejuni ATCC (-+-) and A. skirrowii ATCC (-х-) show a significant increase in fluorescence emission whereas templates from E. coli (- -), C. upsaliensis (- -) and C. hyointestinalis (-o-) show only a marginal increase when compared to no DNA template control (-◊-) which shows no increase. Template DNA was prepared using the rapid boiling method Figure 2. Derivative melting curves (-d. F/d. T) determined by the dissociation of fluorogenic adjacent hybridisation probes from the target amplicons enables discrimination of A. butzleri ATCC (Tm 68 o. C), A. skirrowii (Tm 64 o. C), C. jejuni ATCC and C. coli (Tm 66 o. C) from each other. E. coli, C. upsaliensis, and C. hyointestinalis and template DNA produce no Tm.

Other Virulence Factor genes

Designing virulence gene directed fluorroprobes for detection, identification & differentiation of Campylobacter coli & Campylobacter jejuni from other species by Real Time PCR Forward PCR primer Cy 5 probe Reverse PCR primer
,")
2 1 Figure-1 - Real time detection of hippuricase gene for Campylobacter jejuni (-■-), Campylobacter coli (-▲-), Campyloacter hyointestinalis (-×-), Campylobacter upsaliansis(- -), E. coli (-●-) and Negative control (-+-) (No template Figure 2: Melting temperature of hippuricase gene for Campylobacter species

Quantitation of gene copy numbers

B. Oligonucleotide Hybridisation Probes II. Strand Displacement Probes B. Hair Pin Probes Molecular Beacon Sunrise Uni. Primer Scorpion Stem & Loop Duplex FRET Duplex

MOLECULAR BEACONS Molecular Beacons are hairpin structures composed of a nucleotide base paired stem and a target specific nucleotide loop. The loop consists of target specific nucleotide (probe) sequences The stem is formed by annealing of complementary nucleotide bases of the probe sequence. A fluorescent moiety (reporter)is attached to one end of the arm and a non-fluorescent quenching moiety is attached to the other arm. The stem keeps both the moieties in close proximity so that fluorescence is quenched (Loop) Stem

5’ 3’ 3’ 5’ 5’ 3’ 5’ R 5’ Q Denaturation 5’ 3’ 5’ 5’ Primer molecular beacon annealing 3’ 5’ Extension 5’ 3’ Q 5’ Operation of Molecular Beacon (MB): MB is non-fluorescent due to close proximity of the non-fluorescent quencer (Q) and the fluorescent Reporter. However when the probe denatures and the loop anneals to the target sequence of the amplicon, a conformational reorganization occurs separating the quencher from the fluorophore and thereby producing fluorescence which is proportional to the amplicons produced during PCR

SUNRISE UNIPRIMER PROBE Similar to Molecular Beacon except that the stem contains a poly A (15 mer) tai. This tail is complimenatry to the poly. T tail of one f the primers. AAAAAAAA Q Poly. A Tail

Primer with poly. T tail TT TT AA AA AA TTTT TT TT Sunrise Probe with poly. A tail binds to the primer poly. T tail at annealing. R Q hv Q R AAAAAAA The Sunrise probe changes conformation during denaturation & quenching by DABCYL is removed allowing FITC to fluoresce Sunrise Uni. Primer Probe is a modification of Molecular Beacon

The template & probe denature The primer is part of the Scorpion probe Scorpion stem-loop format Primer, stopper to prevent read PCR through, probe sequence, fluorophore & quencher (detection system). The primer is extended The primer binds to the target The probe binds to the complimentary sequence of the DNA

Duplex Scorpion Format Similar to the Ste-loop Scorpion except the probe sequence is part of the stem. There is no loop in this case.

FRET Duplex Scorpions with 3 different versions of the quencher oligonucleotides
 Probes New In general, design complexty: Dual adjacent")
Designing Stem Loop Molecular Beacon (MB) Probes New In general, design complexty: Dual adjacent > Taqman > Stem Loop Ideally, MBs should hybridise at their annealing temps (fluorescent) & free MBs should be closed (nonfluorescent) Use Oligo 4. 0 or “percent GC rule” to calculate that the loop sequence length (usually 15 -30 nucleotides) is such that it dissociates from its target at temperatures 7 -10 o. C higher then the annealing temp of the PCR. Add two complimentary arms on either side of the loop probe sequence. (usually 5 -7 nucleotides; 5 GC rich stems melt between 55 & 60, 6 between 60 & 65 and 7 between 65 & 70). In order that it remains closed in the absence of the target, the length & GC content should be 7 -10 o. C higher then the annealing temp of the PCR. The melting temperature of the stem cannot be predicted by the “GC rule” as the stems forms by an intramolecular hybridisation event There should be no in between conformational changes, ie should always be the intended hairpin structure an d nothing in between. Commercial program available for making MB probes. . . My DocumentsMy PicturesBD 100 Tour. exe

Probe Detection of the different fluorescent probes during Real Time PCR , Dual probe

SOME REALTIME-PCR APPLICATIONS Simultaneous identification, detection and quantitation of pathogenes in human/food/water/animal samples Gene expression analysis (splicing variants for example) Single Nucleotide Polymorphism (SNP) analysis Protein expression analysis Chromosome aberrations

Application: Bacterial Pathogen detection • Listeria monocytogenes • Campylobacter jejuni group • Arcobacter group • Leptospira group Application: Bacterial Non-Pathogen detection • Thermoanaerobacter species • Caloramator species • Fervidobacterium species
: 1. Rapid")
Advantages of Adjacent Probe Technique with Real Time PCR (Idaho -> Roche): 1. Rapid requiring < 30 mins in a Light Cycler 2. r. RNA and / or r. RNA genes can be used = flexible 3. Simultaneous detection, identification & quantitation 4. PCR primer design + probe design = extremely specific assays possible 5. Different flurodyes available. Multiplexing possible 6. Population dynamics in an ecosystem can be followed 7. Forms a powerful tool when used in conjunction with r. RNA sequencing & FISH
")
c. Pulse Field Gel Electrophoresis (PFGE)
 n n agarose gel electrophoresis is a fundamental technique")
Pulsed Field Gel Electrophoresis (PFGE) n n agarose gel electrophoresis is a fundamental technique in molecular biology but is generally unable to resolve fragments greater than 20 kilobases in size (whole microbial genomes are usually greater than 1000 kilobases in size) PFGE (pulsed field gel electrophoresis) is a adaptation of conventional agarose gel electrophoresis that allows extremely large DNA fragments to be resolved (up to megabase size fragments) essential technique for estimating the sizes of whole genomes/chromosomes prior to sequencing and is necessary for preparing large DNA fragments for large insert DNA cloning and analysis of subsequent clones also a commonly used and extremely powerful tool for genotyping and epidemiology studies for pathogenic microorganisms

Principle of PFGE n n n n two factors influence DNA migration rates through conventional gels - charge differences between DNA fragments - ‘molecular sieve’ effect of DNA pores DNA fragments normally travel through agarose pores as spherical coils, fragments greater than 20 kb in size form extended coils and therefore are not subjected to the molecular sieve effect the charge effect is countered by the proportionally increased friction applied to the molecules and therefore fragments greater than 20 kb do not resolve PFGE works by periodically altering the electric field orientation the large extended coil DNA fragments are forced to change orientation and size dependent separation is reestablished because the time taken for the DNA to reorient is size dependent

Principle of PFGE

Principle of PFGE n n the most important factor in PFGE resolution is switching time, longer switching times generally lead to increased size of DNA fragments which can be resolved switching times are optimised for the expected size of the DNA being run on the PFGE gel switch time ramping increases the region of the gel in which DNA separation is linear with respect to size a number of different apparatus have been developed in order to generate this switching in electric fields however most commonly used in modern laboratories are FIGE (Field Inversion Gel Electrophoresis) and CHEF (Contour. Clamped Homogenous Electrophoresis)

CHEF Switch Time Electric Field 1 - Electric Field 2 - - - + + + +

Preparation of DNA for PFGE n n ideally a genomic DNA preparation that contains a high proportion of completely or almost completely intact genome copies would be suitable for PFGE conventional means of DNA preparation are unsuitable for PFGE as mechanical shearing and low-level nuclease activity will result in fragmented DNA with an average size much smaller than an entire microbial genome (usually less than 200 kb in size) the solution to this is to prepare genomic DNA from whole cells in a semisolid matrix (ie. agarose) that eliminates mechanical shearing a very high concentration of EDTA is also used at all times in order to eliminate all nuclease activity
 intact cells are mixed with")
Preparation of DNA for PFGE n n n 1) intact cells are mixed with molten LMT agarose and set in a mold forming agarose ‘plugs’ 2) enzymes and detergents diffuse into the plugs and lyse cells 3) proteinase K diffuses into plugs and digests proteins 4) if necessary restriction digests are performed in plugs (extensive washing or PMSF treatment is required to remove proteinase K activity) 5) plugs are loaded directly onto PFGE and run

Preparation of DNA for PFGE n n n for restriction digests, conventional enzymes are unsuitable as they cut frequently on an entire genome sequence producing DNA fragments that are far too small ‘rare cutter’ restriction endonucleases cut genomic DNA with far less frequency than conventional restriction enzymes such as Hind. III, Bam. HI etc. many rare cutter RE’s have 6 -bp (or longer) recognition sites eg. Not. I GC GGCCGC in many cases the frequency of cutting is highly species dependent eg. Bam. HI will cut far less frequently on a low GC% genome when compared to a intermediate or high GC content genome suitable rare cutter enzymes therefore have to be determined experimentally for each new species being studied

d. New High Throughput Methods

a. DNA Micro. Arrays

DNA Microarray n n n a completely annotated microbial genome sequence, whilst a powerful scientific tool, still doesn’t provide all of the information needed to understand the complete biology of an organism as it essentially a static picture of the genome for truly complete characterisation, the dynamic nature of gene expression within a microbial cell needs to be determined microarray technology allows whole organism gene expression to be investigated PCR products of every gene from a complete genome sequence are bound in a high density array on a glass slide these arrays are probed with fluorescently labelled c. DNA prepared from whole RNA under specific environmental conditions the level of c. DNA for each ORF is then quantified using high resolution image scanners

DNA MICROARRAYS TECHNIQUES: The development based on bioinformatics knowledge genes & genomes; High throughput & can analysise complex gene expression profiles There are different formats for DNA high density microarrays: • c. DNA arrays (Stanford University development 1999): 0. 5 – 5 kb • Oligonucleotide synthesised (Genechip®) arrays (Affymetrix, 1998): 20 -25 base • Oligomers / PNA, in situ or spotted An example of how a microarray is made by in situ synthesis approach is shown as a movie. . . My DocumentsGene. Chip. mov

STEPS IN THE DNA ARRAY TECHNIQUE: 1. Probe Selection - c. DNA / oligo with known identify: Small oligos, c. DNA, chromosome 2. Chip Fabrication – Putting probes on the chip: photolithography, pipette, drop-touch, piezoelectric (inkjet), electric 3. Target – fluroscently labeled sample: RNA (m. RNA) to c. DNA 4. Assay: Hybridisation, ligase, base addition, electric, electrophoresis, fluocytometry, PCR-DIRECT, Taq. Man 5. Readout: Flurorescence 6. Informatics: Robotic controls, image processing, DBMA, WWW, bioinformatics EXAMPLES: Bio. Merieux is developing for a water company a 4 h fecal indicator test using Affymetrix technology 1 cm 2 has 400 k oligonucleotide probes, small volumes of sample required limits the usefulness Taq. Man type: Leptospira for WHO regional reference laboratory, Campylobacter & Arcobacter for QHSS, Brisbane.

An example of Microarray hybridisation n a microarray containing 97% of the predicted ORF’s from Mycobacterium tuberculosis was used to investigate the response to the antituberculosis drug isoniazid (INH) INH was found to induce several genes related to outer lipid envelope biosynthesis – consistent with the drugs physiological mode of action a number of additional genes were also induced which may provide potential drug targets in the future

INH untreated - green INH treated - red Overlay Yellow = Red + Green (no change in expression) Green (untreated controls) ie expressed without INH treatment Red = expressed as a result of INH treatment The effect of Isoniazid (INH) on the gene expression of Mycobacterium tuberculosis using DNA microarray technique

The Future of DNA Microarrays New 1. Studies on the mechanism of toxicity of drugs to humans: INH is very safe, but like any medicine it can sometimes cause side effects. (yellowish skin, dark urine, vomiting, loss of appetite, nausea, changes in eyesight, unexplained fever, unexplained fatigue & stomach cramps). Human DNA arrays can be used to investigate the mechanism of toxicity on human which may not be possible using animal models. 2. Studies on cyanobacterial toxins extracted from nature blooms: A human DNA array can be used for testing water which has been contaminated by cyanobacterial blooms before and after treatment. This will provide useful information on exposure levels (time & concentration). 3. The role of “uncultured” viruses on different human cell lines: This will provide a rapid method for identifying the most susceptible cell lines that can be used to isolate the “offending” pathogen.

Cantilever arrays

What are cantilever arrays? Cantilever arrays are produced by microfabrication in silicon using dry- and wet-ething techniques. The cantilevers are 500 µm long, 100 µm wide and about 1 µm thick. The spring constant is 20 milli. Newton per meter, resulting in a resonance frequency of about 4 k. Hz. The reproducibility of the resonance frequency from cantilever to cantilever within the array is better than 2%. Owing to their high flexibility, such cantilevers are appropriate for measuring tiny changes in surface stress. For such applications as mass determination, we have designed special cantilevers with a thickness of about 8 µm, producing a resonance frequency of around 50 k. Hz.

Cantilevers are used for imaging in scanning force microscopy but is now being tested as a nanotech sensor. A thin flexible beam made of silicon coated with a sensor layer serves as a chemical sensor. Eight cantilevers aligned in a row form a nanomechanical cantilever sensor array, which can detect small amounts of analytes via very specific reactions. The analyte can also be characterized via its diffusion properties through the coating, e. g. a polymer layer. Nanomechanical cantilever array sensor If analyte molecules dock on the surface of the cantilevers the surface stress at the interface changes, causing the cantilever to bend. The amount of bending is quantitated.

DNA hybridisation cantilever array The binding of two complementary single stranded oligonucleotides can be observed in a setup of two cantilevers, each functionalized with a different synthetic oligonucleotide (red and blue). If the complementary oligonucleotide (green) is injected, it binds preferably to the red oligomer, but not to the blue one. This hybridization process involves bending of the cantilever due to steric and charging effects. If the complementary sequence to the blue strand is injected, the second cantilever bends.

Thank you for your attention
3f66a6ded82628e68fe9706adf7676da.ppt