

Врожденные аномалии, факосы.ppt
- Количество слайдов: 92
 Врожденные пороки мозга и Факоматозы Дисциплина «Невропатология» К. м. н. Королева В. В.
Врожденные пороки мозга и Факоматозы Дисциплина «Невропатология» К. м. н. Королева В. В.
 Врожденные пороки головного мозга • Врожденные пороки головного мозга делятся на несколько категорий: нарушения нормального эмбрионального развития, включающие • нарушения дробления (деления зиготы), • миграции, • сулькации (образования борозд), • пролиферации и • комиссурации (образования спаек). • кистозные поражения, локализующиеся в задней черепной ямки и супратенториально, а также гидранэнцефалия.
Врожденные пороки головного мозга • Врожденные пороки головного мозга делятся на несколько категорий: нарушения нормального эмбрионального развития, включающие • нарушения дробления (деления зиготы), • миграции, • сулькации (образования борозд), • пролиферации и • комиссурации (образования спаек). • кистозные поражения, локализующиеся в задней черепной ямки и супратенториально, а также гидранэнцефалия.
 I. НАРУШЕНИЯ ДРОБЛЕНИЯ II. НАРУШЕНИЯ МИГРАЦИИ И ОБРАЗОВАНИЯ БОРОЗД") Врожденные пороки головного мозга (категории) I. НАРУШЕНИЯ ДРОБЛЕНИЯ II. НАРУШЕНИЯ МИГРАЦИИ И ОБРАЗОВАНИЯ БОРОЗД III. НАРУШЕНИЯ ПРОЛИФЕРАЦИИ IV. НАРУШЕНИЯ КОМИССУРАЦИИ V. ГИДРАНЭНЦЕФАЛИЯ VI. СИНДРОМ ДЭНДИ-УОКЕРА VII. АРАХНОИДАЛЬНЫЕ КИСТЫ
Врожденные пороки головного мозга (категории) I. НАРУШЕНИЯ ДРОБЛЕНИЯ II. НАРУШЕНИЯ МИГРАЦИИ И ОБРАЗОВАНИЯ БОРОЗД III. НАРУШЕНИЯ ПРОЛИФЕРАЦИИ IV. НАРУШЕНИЯ КОМИССУРАЦИИ V. ГИДРАНЭНЦЕФАЛИЯ VI. СИНДРОМ ДЭНДИ-УОКЕРА VII. АРАХНОИДАЛЬНЫЕ КИСТЫ
 I. НАРУШЕНИЯ ДРОБЛЕНИЯ А. Голопрозэнцефалия- нарушение нормального деления переднего мозга эмбриона на две гемисферы. 1. Алобарная голопрозэнцефалия. 2. Семилобарная голопрозэнцефалия. 3. Лобарная голопрозэнцефалия.
I. НАРУШЕНИЯ ДРОБЛЕНИЯ А. Голопрозэнцефалия- нарушение нормального деления переднего мозга эмбриона на две гемисферы. 1. Алобарная голопрозэнцефалия. 2. Семилобарная голопрозэнцефалия. 3. Лобарная голопрозэнцефалия.
 А. Голопрозэнцефалия Этот дефект может быть частичным или полным. С этим видом аномалии связаны самые разнообразные пороки черепа и лицевого скелета: циклопия (единственная орбита, расположенная по средней линии), цебоцефалия (единственный зрительный канал, отсутствие носовой перегородки и резцовой кости) и этмоцефалия (отсутствие носовых костей, носовых раковин, этмоидальной перегородки и резцовой кости).
А. Голопрозэнцефалия Этот дефект может быть частичным или полным. С этим видом аномалии связаны самые разнообразные пороки черепа и лицевого скелета: циклопия (единственная орбита, расположенная по средней линии), цебоцефалия (единственный зрительный канал, отсутствие носовой перегородки и резцовой кости) и этмоцефалия (отсутствие носовых костей, носовых раковин, этмоидальной перегородки и резцовой кости).
 Циклопия
Циклопия
 А. Голопрозэнцефалия В целом, чем сильнее нарушена способность переднего мозга к делению, тем выраженнее черепнолицевые аномалии. Голопрозэнцефалия может сочетаться с прогрессирующей гидроцефалией, требующей лечения.
А. Голопрозэнцефалия В целом, чем сильнее нарушена способность переднего мозга к делению, тем выраженнее черепнолицевые аномалии. Голопрозэнцефалия может сочетаться с прогрессирующей гидроцефалией, требующей лечения.
 г Внешний вид ребенка с голопрозэнцефалией
г Внешний вид ребенка с голопрозэнцефалией
 1. Алобарная голопрозэнцефалия • моновентрикулярный мозг без разделения на доли или полушария. • В задних отделах может иметься диспластичная мембрана мозгового плаща. • Имеются обонятельные луковицы и тракты. • Головной мозг обычно маленького размера.
1. Алобарная голопрозэнцефалия • моновентрикулярный мозг без разделения на доли или полушария. • В задних отделах может иметься диспластичная мембрана мозгового плаща. • Имеются обонятельные луковицы и тракты. • Головной мозг обычно маленького размера.
 Гидроцефалия
Гидроцефалия
 2. Семилобарная голопрозэнцефалия • возникает в результате частичного разделения переднего мозга при неполном образовании долей. • Имеется межполушарная щель. • Полосатые тела сращены по средней линии.
2. Семилобарная голопрозэнцефалия • возникает в результате частичного разделения переднего мозга при неполном образовании долей. • Имеется межполушарная щель. • Полосатые тела сращены по средней линии.
 3. Лобарная голопрозэнцефалия - имеются хорошо сформированные доли и межполушарная щель. Часто сочетается с пороками развития глаз, варьирующими от циклопии до колобомы (дефектов ткани глаза).
3. Лобарная голопрозэнцефалия - имеются хорошо сформированные доли и межполушарная щель. Часто сочетается с пороками развития глаз, варьирующими от циклопии до колобомы (дефектов ткани глаза).
 • Б. Пахигирия •") II. НАРУШЕНИЯ МИГРАЦИИ И ОБРАЗОВАНИЯ БОРОЗД • А. Лиссэнцефалия (агирия) • Б. Пахигирия • В. Полимикрогирия (микрополигирия) • Г. Нейрональная гетеротопия
II. НАРУШЕНИЯ МИГРАЦИИ И ОБРАЗОВАНИЯ БОРОЗД • А. Лиссэнцефалия (агирия) • Б. Пахигирия • В. Полимикрогирия (микрополигирия) • Г. Нейрональная гетеротопия
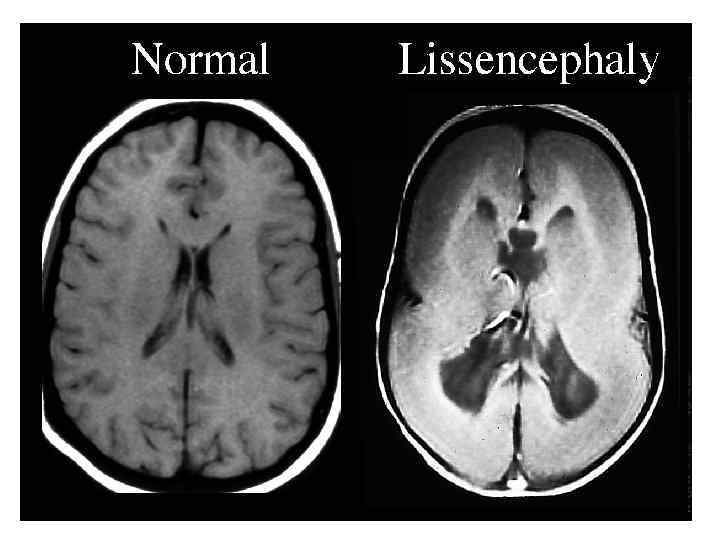
 • характеризуется отсутствием мозговых извилин, что придает мозгу гладкий вид. •") А. Лиссэнцефалия (агирия) • характеризуется отсутствием мозговых извилин, что придает мозгу гладкий вид. • Сильвиева борозда обычно имеется. • Эта аномалия может быть односторонней или двусторонней. • Головной мозг в типичных случаях имеет маленькие размеры при вентрикуломегалии разной степени выраженности.
А. Лиссэнцефалия (агирия) • характеризуется отсутствием мозговых извилин, что придает мозгу гладкий вид. • Сильвиева борозда обычно имеется. • Эта аномалия может быть односторонней или двусторонней. • Головной мозг в типичных случаях имеет маленькие размеры при вентрикуломегалии разной степени выраженности.
 Б. Пахигирия менее тяжелая форма лиссэнцефалии. • Эти две формы часто смешивают, что неправильно. • При пахигирии обычно имеются широкие извилины при отсутствии вторичных извилин. • Как и лиссэнцефалия, пахигирия может быть одно- или двусторонней и желудочки обычно расширены.
Б. Пахигирия менее тяжелая форма лиссэнцефалии. • Эти две формы часто смешивают, что неправильно. • При пахигирии обычно имеются широкие извилины при отсутствии вторичных извилин. • Как и лиссэнцефалия, пахигирия может быть одно- или двусторонней и желудочки обычно расширены.
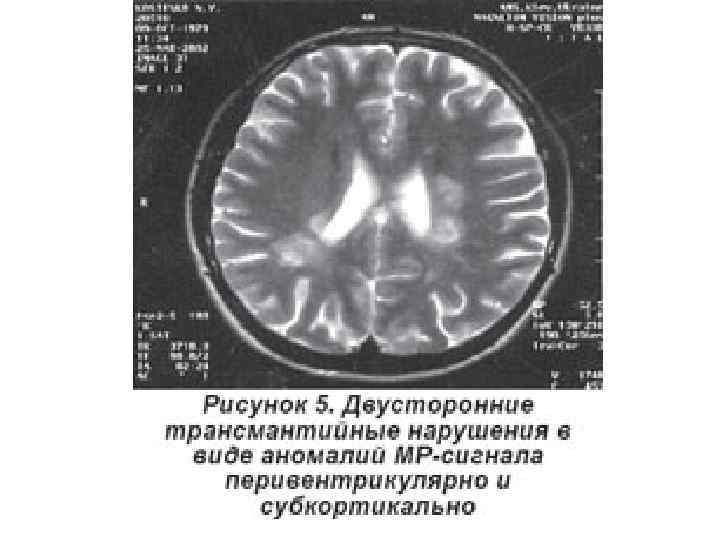
 • характеризуется наличием в пределах извилин множественных псевдоизвилин, что придает мозговой") В. Полимикрогирия (микрополигирия) • характеризуется наличием в пределах извилин множественных псевдоизвилин, что придает мозговой поверхности характерный шишкообразный вид. • Может сочетаться с микрогирией мозжечка.
В. Полимикрогирия (микрополигирия) • характеризуется наличием в пределах извилин множественных псевдоизвилин, что придает мозговой поверхности характерный шишкообразный вид. • Может сочетаться с микрогирией мозжечка.
 Микрогирия
Микрогирия
 Г. Нейрональная гетеротопия • представляет собой нарушение или остановку миграции нейробластов, что приводит к аномальному скоплению нервных клеток в атипичных местах. • Они могут появляться в виде узелков вдоль стенок желудочков либо диффузно, в виде аномальных клеток в белом веществе мозга.
Г. Нейрональная гетеротопия • представляет собой нарушение или остановку миграции нейробластов, что приводит к аномальному скоплению нервных клеток в атипичных местах. • Они могут появляться в виде узелков вдоль стенок желудочков либо диффузно, в виде аномальных клеток в белом веществе мозга.
 Нейрональная гетеротопия
Нейрональная гетеротопия
 III. НАРУШЕНИЯ ПРОЛИФЕРАЦИИ A. Шизенцефалия Б. Микрэнцефалия B. Мегалэнцефалия Г. Гемимегалэнцефалия
III. НАРУШЕНИЯ ПРОЛИФЕРАЦИИ A. Шизенцефалия Б. Микрэнцефалия B. Мегалэнцефалия Г. Гемимегалэнцефалия
 A. Шизенцефалия • характеризуется наличием симметричных аномальных полостей или расщелин, которые могут быть покрыты мягкой и арахноидальной или твердой мозговой оболочкой. • Может иметься гидроцефалия. • Это нарушение обычно сочетается с грубой умственной отсталостью, припадками и спастической тетраплегией.
A. Шизенцефалия • характеризуется наличием симметричных аномальных полостей или расщелин, которые могут быть покрыты мягкой и арахноидальной или твердой мозговой оболочкой. • Может иметься гидроцефалия. • Это нарушение обычно сочетается с грубой умственной отсталостью, припадками и спастической тетраплегией.
 - этот диагноз устанавливается при массе мозга у взрослых") Б. Микрэнцефалия • (маленький мозг) - этот диагноз устанавливается при массе мозга у взрослых менее 900 г. • Обычно имеется микроцефалия. • Часто возникает в результате аноксии или инфекции в пренатальном периоде и в таких случаях не относится к истинным мальформациям. • Может наблюдаться также рецессивно наследуемая семейная микрэнцефалия, при которой масса мозга составляет 500 -600 г.
Б. Микрэнцефалия • (маленький мозг) - этот диагноз устанавливается при массе мозга у взрослых менее 900 г. • Обычно имеется микроцефалия. • Часто возникает в результате аноксии или инфекции в пренатальном периоде и в таких случаях не относится к истинным мальформациям. • Может наблюдаться также рецессивно наследуемая семейная микрэнцефалия, при которой масса мозга составляет 500 -600 г.
 Микрэнцефалия
Микрэнцефалия
 определяется в случаях, когда масса мозга составляет 1600 -2850") B. Мегалэнцефалия • (большой мозг) определяется в случаях, когда масса мозга составляет 1600 -2850 г. • Сочетается с эпилепсией и умственной отсталостью. • Может наблюдаться также у лиц с нормальным и высоким интеллектом.
B. Мегалэнцефалия • (большой мозг) определяется в случаях, когда масса мозга составляет 1600 -2850 г. • Сочетается с эпилепсией и умственной отсталостью. • Может наблюдаться также у лиц с нормальным и высоким интеллектом.
 микроцефалия, мегалэнцефалия
микроцефалия, мегалэнцефалия
 Г. Гемимегалэнцефалия • наблюдается реже и может сочетаться с гемигипертрофией. • Может являться причиной резистентных судорожных припадков.
Г. Гемимегалэнцефалия • наблюдается реже и может сочетаться с гемигипертрофией. • Может являться причиной резистентных судорожных припадков.
 IV. НАРУШЕНИЯ КОМИССУРАЦИИ • А. Агенезия / гипогенезия мозолистого тела • Б. Агенезия / гипоплазия прозрачной перегородки
IV. НАРУШЕНИЯ КОМИССУРАЦИИ • А. Агенезия / гипогенезия мозолистого тела • Б. Агенезия / гипоплазия прозрачной перегородки
 А. Агенезия / гипогенезия мозолистого тела • заключается в полном либо частичном отсутствии мозолистого тела. • Часто встречается в сочетании с другими пороками развития, такими как миеломенингоцеле, синдромом Дэнди. Уолкера, дефект межжелудочковой перегородки, различные соматическе аномалии, нарушения дробления, миграции или сулькации. • Клинически может не проявляться либо выражаться припадками и умственной отсталостью.
А. Агенезия / гипогенезия мозолистого тела • заключается в полном либо частичном отсутствии мозолистого тела. • Часто встречается в сочетании с другими пороками развития, такими как миеломенингоцеле, синдромом Дэнди. Уолкера, дефект межжелудочковой перегородки, различные соматическе аномалии, нарушения дробления, миграции или сулькации. • Клинически может не проявляться либо выражаться припадками и умственной отсталостью.
 Б. Агенезия / гипоплазия прозрачной перегородки часто встречается в сочетании с пороками развития мозолистого тела.
Б. Агенезия / гипоплазия прозрачной перегородки часто встречается в сочетании с пороками развития мозолистого тела.
 V. ГИДРАНЭНЦЕФАЛИЯ • является следствием перинатального поражения, приводящего к отсутствию полушарий мозга. • В качестве "мозгового плаща" может присутствовать тонкая глиальная мембрана. • Промежуточный, средний мозг, мост и продолговатый мозг сохранены. • Считают, что гидранэнцефалия возникает вследствие билатерального церебрального некроза при окклюзии обеих внутренних сонных артерий в ранние сроки беременности. • Полагают, что окклюзия возникает в результате инфекции либо сдавления пуповины амниотической тканью (амниотические ленты). • Закупорка путей ликворотока, вызывая прогрессирующую гидроцефалию, может приводить к прогрессирующей макроцефалии. • Методом выбора является имплантация шунта для облегчения ухода за больным. • Большинство больных с гидранэнцефалией умирают в первые два года жизни.
V. ГИДРАНЭНЦЕФАЛИЯ • является следствием перинатального поражения, приводящего к отсутствию полушарий мозга. • В качестве "мозгового плаща" может присутствовать тонкая глиальная мембрана. • Промежуточный, средний мозг, мост и продолговатый мозг сохранены. • Считают, что гидранэнцефалия возникает вследствие билатерального церебрального некроза при окклюзии обеих внутренних сонных артерий в ранние сроки беременности. • Полагают, что окклюзия возникает в результате инфекции либо сдавления пуповины амниотической тканью (амниотические ленты). • Закупорка путей ликворотока, вызывая прогрессирующую гидроцефалию, может приводить к прогрессирующей макроцефалии. • Методом выбора является имплантация шунта для облегчения ухода за больным. • Большинство больных с гидранэнцефалией умирают в первые два года жизни.
 VI. СИНДРОМ ДЭНДИУОЛКЕРА • - мальформация структуры задней черепной ямки с кистозным расширением четвертого желудочка и гипоплазией либо аплазией мозжечка. • Может иметься атрезия отверстий Мажанди и Люшки. • Вторичный стеноз водопровода может стать причиной сопутствующей гидроцефалии. • Одновременно могут наблюдаться и другие аномалии: агенезия мозолистого тела, полидактилия, дефекты почек, спинальная дисплазия и нейрональная гетеротопия. • Лечение заключается в дренировании желудочков и/ или кисты.
VI. СИНДРОМ ДЭНДИУОЛКЕРА • - мальформация структуры задней черепной ямки с кистозным расширением четвертого желудочка и гипоплазией либо аплазией мозжечка. • Может иметься атрезия отверстий Мажанди и Люшки. • Вторичный стеноз водопровода может стать причиной сопутствующей гидроцефалии. • Одновременно могут наблюдаться и другие аномалии: агенезия мозолистого тела, полидактилия, дефекты почек, спинальная дисплазия и нейрональная гетеротопия. • Лечение заключается в дренировании желудочков и/ или кисты.
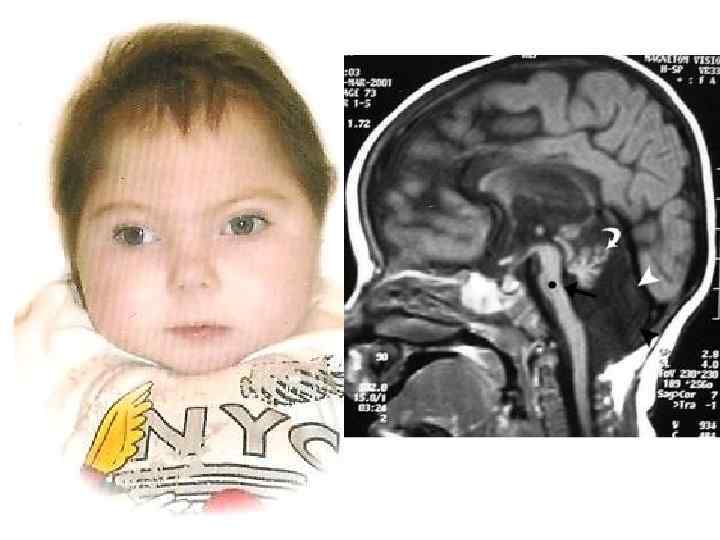
 VII. АРАХНОИДАЛЬНЫЕ КИСТЫ A. Доброкачественные врожденные аномалии, проявляющиеся в виде выстланных паутинной оболочкой полостей, обычно заполненных чистой бесцветной жидкостью, похожей на ликвор.
VII. АРАХНОИДАЛЬНЫЕ КИСТЫ A. Доброкачественные врожденные аномалии, проявляющиеся в виде выстланных паутинной оболочкой полостей, обычно заполненных чистой бесцветной жидкостью, похожей на ликвор.
 VII. АРАХНОИДАЛЬНЫЕ КИСТЫ • Б. Эти кисты не всегда напрямую сообщаются с субарахноидальным пространством или желудочковой системой. • Невидимый клапанный тип сообщения может объяснить иногда наблюдаемое в таких случаях прогрессирующее увеличение полости кисты.
VII. АРАХНОИДАЛЬНЫЕ КИСТЫ • Б. Эти кисты не всегда напрямую сообщаются с субарахноидальным пространством или желудочковой системой. • Невидимый клапанный тип сообщения может объяснить иногда наблюдаемое в таких случаях прогрессирующее увеличение полости кисты.
 в") VII. АРАХНОИДАЛЬНЫЕ КИСТЫ • B. Кисты могут быть расположены (в порядке убывания частоты) в • сильвиевой щели, • мостомозжечковом углу, • надбугорковой области, • области червя и • в других местах.
VII. АРАХНОИДАЛЬНЫЕ КИСТЫ • B. Кисты могут быть расположены (в порядке убывания частоты) в • сильвиевой щели, • мостомозжечковом углу, • надбугорковой области, • области червя и • в других местах.
 VII. АРАХНОИДАЛЬНЫЕ КИСТЫ Г. Проявления различны. Часто арахноидальные кисты обнаруживаются случайно при рентгенографии, проводимой по другим показаниям. Они могут оказывать или не оказывать воздействие по типу обьемного процесса. В случае масс-эффекта могут отмечаться признаки повышения ВЧД, головные боли, припадки, очаговые неврологические нарушения или умственная отсталость.
VII. АРАХНОИДАЛЬНЫЕ КИСТЫ Г. Проявления различны. Часто арахноидальные кисты обнаруживаются случайно при рентгенографии, проводимой по другим показаниям. Они могут оказывать или не оказывать воздействие по типу обьемного процесса. В случае масс-эффекта могут отмечаться признаки повышения ВЧД, головные боли, припадки, очаговые неврологические нарушения или умственная отсталость.
 Д. Лечение симптоматических арахноидальных кист • заключается в краниотомии и марсупиализации кисты, либо её шунтировании или стентинге (образовании сообщения между кистой и ликворной системой). • Выжидательная тактика в случае развивающегося мозга может быть не в интересах ребенка, поскольку с течением времени нередко наблюдается медленный рост кист. • Во многих случаях, если операция проведена в раннем возрасте, после адекватной декомпрессии мозговая паренхима расправляется. • Лечение должно быть индивидуальным.
Д. Лечение симптоматических арахноидальных кист • заключается в краниотомии и марсупиализации кисты, либо её шунтировании или стентинге (образовании сообщения между кистой и ликворной системой). • Выжидательная тактика в случае развивающегося мозга может быть не в интересах ребенка, поскольку с течением времени нередко наблюдается медленный рост кист. • Во многих случаях, если операция проведена в раннем возрасте, после адекватной декомпрессии мозговая паренхима расправляется. • Лечение должно быть индивидуальным.
 представляют собой группу заболеваний, характеризующихся неврологическими, зрительными и кожными") Факоматозы • Нейрокожные поражения (факоматозы) представляют собой группу заболеваний, характеризующихся неврологическими, зрительными и кожными нарушениями. • Это наследственные заболевания, при которых с возрастом происходит прогрессирование симптомов. • Раннее выявление факоматозов позволяет осуществлять медико-генетическое консультирование больного, его братьев и сестер, его детей, а также обеспечивать оптимальное медицинское наблюдение и лечение. • В настоящее время обнаруживаются гены, ответственные за многие из этих заболеваний.
Факоматозы • Нейрокожные поражения (факоматозы) представляют собой группу заболеваний, характеризующихся неврологическими, зрительными и кожными нарушениями. • Это наследственные заболевания, при которых с возрастом происходит прогрессирование симптомов. • Раннее выявление факоматозов позволяет осуществлять медико-генетическое консультирование больного, его братьев и сестер, его детей, а также обеспечивать оптимальное медицинское наблюдение и лечение. • В настоящее время обнаруживаются гены, ответственные за многие из этих заболеваний.

 НЕЙРОФИБРОМАТОЗ • 1. НФ -наиболее частая форма факоматозов, которая встречается с частотой 1: 3000 живорожденных. • 2. Тип наследования аутосомнодоминантный с различной экспрессивностью. • Пенетрантность достигает 100%. • Пятьдесят (50%) процентов новых случаев представляют собой мутации.
НЕЙРОФИБРОМАТОЗ • 1. НФ -наиболее частая форма факоматозов, которая встречается с частотой 1: 3000 живорожденных. • 2. Тип наследования аутосомнодоминантный с различной экспрессивностью. • Пенетрантность достигает 100%. • Пятьдесят (50%) процентов новых случаев представляют собой мутации.
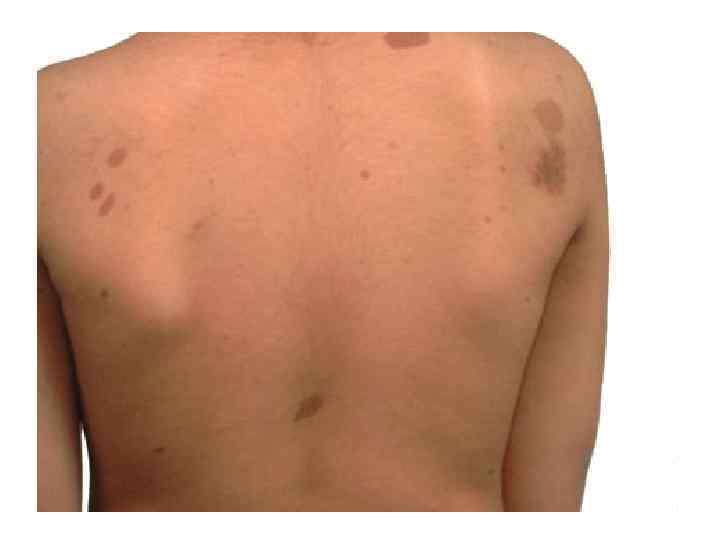
 НЕЙРОФИБРОМАТОЗ • 3. НФ может быть представлен двумя формами: периферическая форма НФ (классический НФ Реклингаузена) и центральная форма НФ (двусторонние невриномы слуховых нервов). • 4. Типичными признаками заболевания являются множественные пигментные пятна на коже (пятна cafe au lait ("кофе с молоком")) и множественные нейрофибромы.
НЕЙРОФИБРОМАТОЗ • 3. НФ может быть представлен двумя формами: периферическая форма НФ (классический НФ Реклингаузена) и центральная форма НФ (двусторонние невриномы слуховых нервов). • 4. Типичными признаками заболевания являются множественные пигментные пятна на коже (пятна cafe au lait ("кофе с молоком")) и множественные нейрофибромы.
 Б. Периферическая форма НФ. 1. Признаки. • а. Кожные нейрофибромы могут варьировать в числе и размерах. • Нейрофибромы развиваются на периферических нервах, нервных корешках и симпатическом стволе. • Морфологически нейрофибромы состоят из шванновских клеток, фибробластов, тучных клеток и соединительной ткани.
Б. Периферическая форма НФ. 1. Признаки. • а. Кожные нейрофибромы могут варьировать в числе и размерах. • Нейрофибромы развиваются на периферических нервах, нервных корешках и симпатическом стволе. • Морфологически нейрофибромы состоят из шванновских клеток, фибробластов, тучных клеток и соединительной ткани.
 Б. Периферическая форма НФ. 1. Признаки. • б. Пятна cafe au lait представляют собой гиперпигментированные пятна, обычно наблюдающиеся на коже туловища. • в. Пигментные гамартомы радужки (узелки Лиха) наблюдаются у большинства больных старше 6 лет; с возрастом их число увеличивается. • г. Веснушчатые пятна в областях опрелостей (подмышечные впадины, ягодицы).
Б. Периферическая форма НФ. 1. Признаки. • б. Пятна cafe au lait представляют собой гиперпигментированные пятна, обычно наблюдающиеся на коже туловища. • в. Пигментные гамартомы радужки (узелки Лиха) наблюдаются у большинства больных старше 6 лет; с возрастом их число увеличивается. • г. Веснушчатые пятна в областях опрелостей (подмышечные впадины, ягодицы).
 Б. Периферическая форма НФ. 1. Признаки. • д. Плексиформные нейрофибромы могут быть спаяны с периферическими волокнами симпатического ствола. • Они могут значительно разрастаться, приводя к гемигипертрофии (elephantiasis neuromatosa). • Нейрофибромы могут также перерождаться в нейрофибросаркомы, что проявляется бурным ростом опухоли.
Б. Периферическая форма НФ. 1. Признаки. • д. Плексиформные нейрофибромы могут быть спаяны с периферическими волокнами симпатического ствола. • Они могут значительно разрастаться, приводя к гемигипертрофии (elephantiasis neuromatosa). • Нейрофибромы могут также перерождаться в нейрофибросаркомы, что проявляется бурным ростом опухоли.
 Б. Периферическая форма НФ. 1. Признаки. • е. Буфтальм ("бычий глаз") развивается вследствие глаукомы, вызывающей увеличение (чаще одностороннее) глазного яблока. • ж. Сколиоз является наиболее частой скелетной аномалией, связанной с НФ. Хирургическое вмешательство может предотвратить тяжелую деформацию позвоночника.
Б. Периферическая форма НФ. 1. Признаки. • е. Буфтальм ("бычий глаз") развивается вследствие глаукомы, вызывающей увеличение (чаще одностороннее) глазного яблока. • ж. Сколиоз является наиболее частой скелетной аномалией, связанной с НФ. Хирургическое вмешательство может предотвратить тяжелую деформацию позвоночника.
 Б. Периферическая форма НФ. 2. Диагностика. • Диагноз основывается на клинических критериях и семейном анамнезе. • У детей наличие пяти пятен, диаметр которых превышает 5 мм, предполагаетт диагноз НФ. • Подсчет пятен удобнее всего производить с помощью лампы Вуда. • Однако 20% больных НФ не удовлетворяют этому критерию, поэтому он не может считаться абсолютным. • Диагноз подтверждает наличие нейрофибром и/или узелков Лиха. • Семейный анамнез с указанием на НФ важен, однако не является неотъемлемым диагностическим критерием, поскольку нередки случаи возникновения НФ как мутации.
Б. Периферическая форма НФ. 2. Диагностика. • Диагноз основывается на клинических критериях и семейном анамнезе. • У детей наличие пяти пятен, диаметр которых превышает 5 мм, предполагаетт диагноз НФ. • Подсчет пятен удобнее всего производить с помощью лампы Вуда. • Однако 20% больных НФ не удовлетворяют этому критерию, поэтому он не может считаться абсолютным. • Диагноз подтверждает наличие нейрофибром и/или узелков Лиха. • Семейный анамнез с указанием на НФ важен, однако не является неотъемлемым диагностическим критерием, поскольку нередки случаи возникновения НФ как мутации.
 Б. Периферическая форма НФ. 3. Тактика лечения • Тактика лечения выжидательная. • Нейрофибромы, вызывающие клиническую симптоматику, можно удалить хирургическим путем, несмотря на некоторый риск нарушения функции при таких операциях. • Неотъемлемой частью лечения является генетическое консультирование.
Б. Периферическая форма НФ. 3. Тактика лечения • Тактика лечения выжидательная. • Нейрофибромы, вызывающие клиническую симптоматику, можно удалить хирургическим путем, несмотря на некоторый риск нарушения функции при таких операциях. • Неотъемлемой частью лечения является генетическое консультирование.
 Б. Периферическая форма НФ. 4. Прогноз. • Периферический НФ представляет собой прогрессирующее заболевание. • Прогноз зависит от степени выраженности признаков. • Имеется тенденция к нарастанию симптоматики. • При саркоматозном перерождении нейрофибром лишь 23% больных живут более 5 лет.
Б. Периферическая форма НФ. 4. Прогноз. • Периферический НФ представляет собой прогрессирующее заболевание. • Прогноз зависит от степени выраженности признаков. • Имеется тенденция к нарастанию симптоматики. • При саркоматозном перерождении нейрофибром лишь 23% больных живут более 5 лет.
 В. Центральный НФ. 1. Признаки • а. Двусторонние невриномы слуховых нервов, которые проявляются обычно в период юношества. • б. Кожные нейрофибромы (меньше, чем при периферическом НФ). • в. Пятна cafe au lait (меньше, чем при периферическом НФ). • г. Врожденные катаракты.
В. Центральный НФ. 1. Признаки • а. Двусторонние невриномы слуховых нервов, которые проявляются обычно в период юношества. • б. Кожные нейрофибромы (меньше, чем при периферическом НФ). • в. Пятна cafe au lait (меньше, чем при периферическом НФ). • г. Врожденные катаракты.
 В. Центральный НФ. 2. Диагностика. а. КТ или МРТ головного мозга. б. ССВП могут выявить небольшие невриномы слухового нерва. в. Семейный анамнез.
В. Центральный НФ. 2. Диагностика. а. КТ или МРТ головного мозга. б. ССВП могут выявить небольшие невриномы слухового нерва. в. Семейный анамнез.
 В. Центральный НФ. 3. Лечение. • также выжидательное. • С помощью МРТ можно выявить опухоли слуховых нервов даже самых маленьких размеров. • В настоящее время преобладает тенденция к раннему удалению этих опухолей. • Наблюдение за этими больными в течение нескольких последующих десятилетий даст ответ на вопрос, имеет ли такая тактика преимущества. • Неотъемлемой частью лечения является генетическое консультирование.
В. Центральный НФ. 3. Лечение. • также выжидательное. • С помощью МРТ можно выявить опухоли слуховых нервов даже самых маленьких размеров. • В настоящее время преобладает тенденция к раннему удалению этих опухолей. • Наблюдение за этими больными в течение нескольких последующих десятилетий даст ответ на вопрос, имеет ли такая тактика преимущества. • Неотъемлемой частью лечения является генетическое консультирование.
 В. Центральный НФ. 4. Прогноз. • Центральный НФ представляет собой прогрессирующее заболевание. • Самым важным является удаление опухолей слуховых нервов с сохранением при этом слуха и функции лицевого нерва. • При саркоматозном перерождении опухолей прогноз неблагоприятный.
В. Центральный НФ. 4. Прогноз. • Центральный НФ представляет собой прогрессирующее заболевание. • Самым важным является удаление опухолей слуховых нервов с сохранением при этом слуха и функции лицевого нерва. • При саркоматозном перерождении опухолей прогноз неблагоприятный.
 1. При НФ наблюдается поражение головного и") Г. Поражение ЦНС (при ПНФ и ЦНФ) 1. При НФ наблюдается поражение головного и спинного мозга, черепных нервов, нервных корешков и мозговых оболочек. 2. В принципе, при наличии клинических проявлений очаг поражения должен быть оперирован. Тактика при асимптомати-ческих очагах поражения индивидуальна. Часто множественность опухолей препятствует их удалению хирургическим путем.
Г. Поражение ЦНС (при ПНФ и ЦНФ) 1. При НФ наблюдается поражение головного и спинного мозга, черепных нервов, нервных корешков и мозговых оболочек. 2. В принципе, при наличии клинических проявлений очаг поражения должен быть оперирован. Тактика при асимптомати-ческих очагах поражения индивидуальна. Часто множественность опухолей препятствует их удалению хирургическим путем.
 3. У детей с НФ часты глиомы") Г. Поражение ЦНС (при ПНФ и ЦНФ) 3. У детей с НФ часты глиомы зрительных нервов. 4. Прогноз этой опухоли при НФ лучше, по сравнению с глиомами зрительных нервов у "нормальных" детей. 5. Эта опухоль может служить первым проявлением НФ у ребенка.
Г. Поражение ЦНС (при ПНФ и ЦНФ) 3. У детей с НФ часты глиомы зрительных нервов. 4. Прогноз этой опухоли при НФ лучше, по сравнению с глиомами зрительных нервов у "нормальных" детей. 5. Эта опухоль может служить первым проявлением НФ у ребенка.
 4. Глиомы возникают также и в других") Г. Поражение ЦНС (при ПНФ и ЦНФ) 4. Глиомы возникают также и в других отделах ЦНС. Поражение гипоталамической области можно заподозрить при преждевременном половом созревании либо при половом инфантилизме. 5. Менингиомы (внутричерепные либо спинальные). 6. Часто встречаются спинальные нейрофибромы
Г. Поражение ЦНС (при ПНФ и ЦНФ) 4. Глиомы возникают также и в других отделах ЦНС. Поражение гипоталамической области можно заподозрить при преждевременном половом созревании либо при половом инфантилизме. 5. Менингиомы (внутричерепные либо спинальные). 6. Часто встречаются спинальные нейрофибромы
 7. Последствия а. Припадки. б. Макроцефалия (75%).") Г. Поражение ЦНС (при ПНФ и ЦНФ) 7. Последствия а. Припадки. б. Макроцефалия (75%). в. Задержка развития. г. Трудности обучения. д. Умственная отсталость.
Г. Поражение ЦНС (при ПНФ и ЦНФ) 7. Последствия а. Припадки. б. Макроцефалия (75%). в. Задержка развития. г. Трудности обучения. д. Умственная отсталость.
 Д. Сегментарный нейрофиброматоз. • Наличие пятен cafe au lait и нейрофибром в пределах ограниченного участка верхних отделов тела (например, на руке и половине туловища).
Д. Сегментарный нейрофиброматоз. • Наличие пятен cafe au lait и нейрофибром в пределах ограниченного участка верхних отделов тела (например, на руке и половине туловища).
 Е. Кожный нейрофиброматоз. • Наличие в клинике только пятен cafe au lait.
Е. Кожный нейрофиброматоз. • Наличие в клинике только пятен cafe au lait.
 II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. А. Общая характеристика. • 1. Аутосомно-доминантный тип наследования с большим числом спорадических случаев. • 2. Частота 1: 10 000 на живорожденных. Чаще встречается у мужчин; редко - у негров.
II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. А. Общая характеристика. • 1. Аутосомно-доминантный тип наследования с большим числом спорадических случаев. • 2. Частота 1: 10 000 на живорожденных. Чаще встречается у мужчин; редко - у негров.
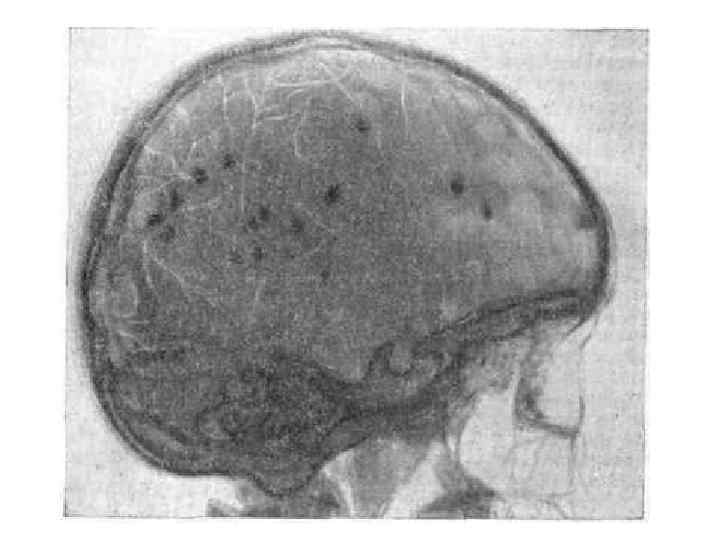
 II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. А. Общая характеристика. 3. ТС поражает центральную нервную систему, кожу, внутренние органы, сетчатку, почки, кости. Классическая триада включает в себя 1. аденомы сальных желез, 2. умственную отсталость и 3. припадки.
II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. А. Общая характеристика. 3. ТС поражает центральную нервную систему, кожу, внутренние органы, сетчатку, почки, кости. Классическая триада включает в себя 1. аденомы сальных желез, 2. умственную отсталость и 3. припадки.
 II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. А. Общая характеристика. 5. Болезнь Бурневилля. ТС с поражением ЦНС. 6. Болезнь Прингля. ТС с исключительно кожными проявлениями. 7. Синдром Веста. Кожные поражения в сочетании с умственной отсталостью, инфантильными спазмами и гипсаритмией.
II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. А. Общая характеристика. 5. Болезнь Бурневилля. ТС с поражением ЦНС. 6. Болезнь Прингля. ТС с исключительно кожными проявлениями. 7. Синдром Веста. Кожные поражения в сочетании с умственной отсталостью, инфантильными спазмами и гипсаритмией.
 II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. Б. Признаки. 1. Пятна типа "листьев ясеня" представляют собой депигменированные кожные пятна, заостренные с одного конца и закруглённые с другого. Они могут присутствовать с рождения либо появляться позднее в детском возрасте. Для обнаружения этих пятен, в особенности у светлокожих, используется лампа Вуда.
II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. Б. Признаки. 1. Пятна типа "листьев ясеня" представляют собой депигменированные кожные пятна, заостренные с одного конца и закруглённые с другого. Они могут присутствовать с рождения либо появляться позднее в детском возрасте. Для обнаружения этих пятен, в особенности у светлокожих, используется лампа Вуда.
 представляют собой красные") II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. Б. Признаки. 2. Аденомы сальных желез (лицевые ангиофибромы) представляют собой красные возвышающиеся гамартомы, развивающиеся из нервных элементов кожи. Они имеются у 90% больных ТС. 3. Подногтевые ангиофибромы на пальцах кистей и стоп. 4. Подкожный фиброз (акулья кожа)
II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. Б. Признаки. 2. Аденомы сальных желез (лицевые ангиофибромы) представляют собой красные возвышающиеся гамартомы, развивающиеся из нервных элементов кожи. Они имеются у 90% больных ТС. 3. Подногтевые ангиофибромы на пальцах кистей и стоп. 4. Подкожный фиброз (акулья кожа)
 II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. Б. Признаки. 5. Поражение сетчатки; очаги поражения состоят из глии, ганглиональных клеток и фибробластов. 6. Почечные гамартомы, ангиолипомы и кисты. 7. Рабдомиома сердца. 8. Гамартомы и ангиомы печени. 9. Лимфоангиоматоз легких. 10. Скелетные аномалии (кистозные полости, остеосклероз)
II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. Б. Признаки. 5. Поражение сетчатки; очаги поражения состоят из глии, ганглиональных клеток и фибробластов. 6. Почечные гамартомы, ангиолипомы и кисты. 7. Рабдомиома сердца. 8. Гамартомы и ангиомы печени. 9. Лимфоангиоматоз легких. 10. Скелетные аномалии (кистозные полости, остеосклероз)
 II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. В. Поражение ЦНС 1. Субэпендимальные узелки выступают в полость третьего и боковых желудочках и могут частично внедряться в таламус или хвостатое ядро. Гистологически эти узлы состоят из вытянутых астроцитов с частым наличием кальцификатов. 2. Корковые узелки ("бугорки") возникают преимущественно в коре головного мозга. Патоморфологически они представляют собой участки ткани коры с патологически измененными нейронами, астроцитами и цитоархитектоникой. 3. Субэпендимальные гигантоклеточные астроцитомы возникают у 7 -23% больных с ТС. Чаще всего они локализуются в области отверстия Монро и проявляются признаками повышения ВЧД в результате обструктивной гидроцефалии. 4. Пахигирия, микрогирия и различные варианты гетеротопии. 5. Сосудистая гиперплазия.
II. ТУБЕРОЗНЫЙ СКЛЕРОЗ. В. Поражение ЦНС 1. Субэпендимальные узелки выступают в полость третьего и боковых желудочках и могут частично внедряться в таламус или хвостатое ядро. Гистологически эти узлы состоят из вытянутых астроцитов с частым наличием кальцификатов. 2. Корковые узелки ("бугорки") возникают преимущественно в коре головного мозга. Патоморфологически они представляют собой участки ткани коры с патологически измененными нейронами, астроцитами и цитоархитектоникой. 3. Субэпендимальные гигантоклеточные астроцитомы возникают у 7 -23% больных с ТС. Чаще всего они локализуются в области отверстия Монро и проявляются признаками повышения ВЧД в результате обструктивной гидроцефалии. 4. Пахигирия, микрогирия и различные варианты гетеротопии. 5. Сосудистая гиперплазия.
 Г. Диагностика. • Больные обычно попадают в поле зрения врача в связи с судорожными припадками. • КТ или МРТ выявляют патогномоничные изменения мозговой ткани. • Тщательное обследование помогает обнаружить некоторые другие признаки заболевания. • Изредка больные попадают ко врачу по поводу нарушения функции почек (азотемии, гипертензии, альбуминурии).
Г. Диагностика. • Больные обычно попадают в поле зрения врача в связи с судорожными припадками. • КТ или МРТ выявляют патогномоничные изменения мозговой ткани. • Тщательное обследование помогает обнаружить некоторые другие признаки заболевания. • Изредка больные попадают ко врачу по поводу нарушения функции почек (азотемии, гипертензии, альбуминурии).
 Д. Лечение. 1. Противосудорожные средства. 2. Нейрохирургическое вмешательство по поводу опухолей, вызывающих некупируемые припадки либо гидроцефалию. 3. Лазеротерапия либо дермабразия при поражениях лица. 3. Генетическое консультирование.
Д. Лечение. 1. Противосудорожные средства. 2. Нейрохирургическое вмешательство по поводу опухолей, вызывающих некупируемые припадки либо гидроцефалию. 3. Лазеротерапия либо дермабразия при поражениях лица. 3. Генетическое консультирование.
 Е. Прогноз. • Припадки, начинающиеся в возрасте до 2 -х лет, свидетельствуют о неблагоприятном прогнозе; • такие дети обычно отличаются выраженной умственной отсталостью. • У больных с отсутствием припадков умственное развитие может быть нормальным.
Е. Прогноз. • Припадки, начинающиеся в возрасте до 2 -х лет, свидетельствуют о неблагоприятном прогнозе; • такие дети обычно отличаются выраженной умственной отсталостью. • У больных с отсутствием припадков умственное развитие может быть нормальным.
.") III. БОЛЕЗНЬ ФОН ГИППЕЛЯЛИНДАУ • (ретиноцебеллярный ангиоматоз).
III. БОЛЕЗНЬ ФОН ГИППЕЛЯЛИНДАУ • (ретиноцебеллярный ангиоматоз).
 А. Общая характеристика. 1. Тип наследования аутосомнодоминантный с различной экспрессивностью и проявляющийся обычно у взрослых. 2. Одиночные гемангиобластомы могут наблюдаться у детей, не страдающих ФГЛ.
А. Общая характеристика. 1. Тип наследования аутосомнодоминантный с различной экспрессивностью и проявляющийся обычно у взрослых. 2. Одиночные гемангиобластомы могут наблюдаться у детей, не страдающих ФГЛ.
 являются наиболее типичными") Б. Признаки. 1. Капиллярные гемангиобластомы (опухоли, состоящие из эмбриональных сосудистых элементов) являются наиболее типичными поражениями ри ФГЛ. Наиболее часто они локализуются в мозжечке, однако могут возникать вдоль всей кра-ниоспинальной оси. 2. Ангиомы сетчатки. 3. Феохромоцитома. 4. Кисты, опухоли и ангиомы почек. 5. Ангиомы поджелудочной железы, печени, селезенки, легких и эпидидимиса.
Б. Признаки. 1. Капиллярные гемангиобластомы (опухоли, состоящие из эмбриональных сосудистых элементов) являются наиболее типичными поражениями ри ФГЛ. Наиболее часто они локализуются в мозжечке, однако могут возникать вдоль всей кра-ниоспинальной оси. 2. Ангиомы сетчатки. 3. Феохромоцитома. 4. Кисты, опухоли и ангиомы почек. 5. Ангиомы поджелудочной железы, печени, селезенки, легких и эпидидимиса.
 В. Диагностика. • Диагноз ФГЛ устанавливается у больных, имеющих более одного очага поражения либо одиночный очаг поражения в сочетании с семейным анамнезом.
В. Диагностика. • Диагноз ФГЛ устанавливается у больных, имеющих более одного очага поражения либо одиночный очаг поражения в сочетании с семейным анамнезом.
 Г. Лечение симптоматических гемангиобластом хирургическое. Гемангиобластомы спинного мозга могут сочетаться с сирингомиелией.
Г. Лечение симптоматических гемангиобластом хирургическое. Гемангиобластомы спинного мозга могут сочетаться с сирингомиелией.
 Д. Прогноз. ФГЛ - прогрессирующее заболевание. Рекомендуется периодическое рентгенографическое обследование и исследование функции почек с хирургическим удалением симптоматических очагов поражения. Обязательно генетическое консультирование.
Д. Прогноз. ФГЛ - прогрессирующее заболевание. Рекомендуется периодическое рентгенографическое обследование и исследование функции почек с хирургическим удалением симптоматических очагов поражения. Обязательно генетическое консультирование.
. • 1. Спорадичность") IV. НЕЙРОКОЖНЫЕ АНГИОМАТОЗЫ • А. Синдром Штурге. Вебера (энцефалотригеминал ьный ангиома-тоз). • 1. Спорадичность случаев заболевания свидетельствует в пользу мутаций.
IV. НЕЙРОКОЖНЫЕ АНГИОМАТОЗЫ • А. Синдром Штурге. Вебера (энцефалотригеминал ьный ангиома-тоз). • 1. Спорадичность случаев заболевания свидетельствует в пользу мутаций.
 2. Признаки. • Невус цвета портвейна на лице в зоне иннервации первой или второй ветви тройничного нерва, сочетающийся с ипсилатеральным ангиоматозом лептоменинкса, обычно теменной и затылочной локализации. • Под пораженными оболочками может развиваться атрофия коры мозга и отложение кальцификатов. • В некоторых случаях ангиомы возникают в других частях тела.
2. Признаки. • Невус цвета портвейна на лице в зоне иннервации первой или второй ветви тройничного нерва, сочетающийся с ипсилатеральным ангиоматозом лептоменинкса, обычно теменной и затылочной локализации. • Под пораженными оболочками может развиваться атрофия коры мозга и отложение кальцификатов. • В некоторых случаях ангиомы возникают в других частях тела.
 3. Клинические проявления. • • Припадки, контрлатеральный гемипарез, ипсилатеральная глаукома, гомонимная гемианопсия и, в более тяжелых случаях, • умственная отсталость.
3. Клинические проявления. • • Припадки, контрлатеральный гемипарез, ипсилатеральная глаукома, гомонимная гемианопсия и, в более тяжелых случаях, • умственная отсталость.
 4. Ренгенографические исследования. • Обычная рентгенография выявляет кальцификаты вдоль извилин коры по типу "трамвайных путей". • КТ обнаруживает внутричерепные кальцификаты, ангиоматоз мозговых оболочек и унилатеральную атрофию коры.
4. Ренгенографические исследования. • Обычная рентгенография выявляет кальцификаты вдоль извилин коры по типу "трамвайных путей". • КТ обнаруживает внутричерепные кальцификаты, ангиоматоз мозговых оболочек и унилатеральную атрофию коры.
 5. Лечение • направлено на прекращение судорожных припадков. • Методом выбора является хирургическое вмешательство (гемисферэктомия, лобэктомия), в особенности при раннем начале припадков.
5. Лечение • направлено на прекращение судорожных припадков. • Методом выбора является хирургическое вмешательство (гемисферэктомия, лобэктомия), в особенности при раннем начале припадков.
 6. Прогноз • у разных больных различен. • О неблагоприятном прогнозе свидетельствуют обширные поражения головного мозга и раннее начало припадков.
6. Прогноз • у разных больных различен. • О неблагоприятном прогнозе свидетельствуют обширные поражения головного мозга и раннее начало припадков.
 Б. Синдром Ослера -Рандю Вебера. 1. Аутосомно-доминантный тип наследования. 2. Признаки. Ангиомы кожи, слизистых оболочек и нервной системы. 3. Ангиомы ЦНС могут кровоточить либо проявляться иными симптомами, требуя нейрохирургического вмешательства.
Б. Синдром Ослера -Рандю Вебера. 1. Аутосомно-доминантный тип наследования. 2. Признаки. Ангиомы кожи, слизистых оболочек и нервной системы. 3. Ангиомы ЦНС могут кровоточить либо проявляться иными симптомами, требуя нейрохирургического вмешательства.
. 1. Х-сцепленный тип наследования. Причиной заболевания служит дефект") В. Болезнь Фабри (диффузная ангиокератома тела). 1. Х-сцепленный тип наследования. Причиной заболевания служит дефект альфа-галактозидазы А, что приводит к накоплению в различных клетках и тканях тригексозил-церамида. 2. Признаки. Помутнение роговицы и множественные багровые макулярные и макулопапулярные очаги гиперкератоза на животе и нижних конечностях.
В. Болезнь Фабри (диффузная ангиокератома тела). 1. Х-сцепленный тип наследования. Причиной заболевания служит дефект альфа-галактозидазы А, что приводит к накоплению в различных клетках и тканях тригексозил-церамида. 2. Признаки. Помутнение роговицы и множественные багровые макулярные и макулопапулярные очаги гиперкератоза на животе и нижних конечностях.
. 3. Клинические проявления. Боль и чувство жжения в") В. Болезнь Фабри (диффузная ангиокератома тела). 3. Клинические проявления. Боль и чувство жжения в конечностях, ангидроз, нарушение функции почек. 4. Лечение. Есть данные о том, что фенитоин уменьшает болевой синдром. При возникновении почечной недостаточности показана о трансплантация почек. 5. Прогноз. Болезнь имеет прогрессирующий характер. Могут развиваться цереброваскулярные нарушения, почечная недостаточность и поражение миокарда.
В. Болезнь Фабри (диффузная ангиокератома тела). 3. Клинические проявления. Боль и чувство жжения в конечностях, ангидроз, нарушение функции почек. 4. Лечение. Есть данные о том, что фенитоин уменьшает болевой синдром. При возникновении почечной недостаточности показана о трансплантация почек. 5. Прогноз. Болезнь имеет прогрессирующий характер. Могут развиваться цереброваскулярные нарушения, почечная недостаточность и поражение миокарда.
. • 1. Заболевание с аутосомнорецессивным • типом наследования и частотой") Г. Атаксия-телеангиэктазия (синдром Луи-Бар). • 1. Заболевание с аутосомнорецессивным • типом наследования и частотой возникновения 2 -3 : 100. 000 живорожденных.
Г. Атаксия-телеангиэктазия (синдром Луи-Бар). • 1. Заболевание с аутосомнорецессивным • типом наследования и частотой возникновения 2 -3 : 100. 000 живорожденных.
 2. Признаки. : 1. 2. 3. 4. Прогрессирующая атаксия, множественные телеангиэктазии, иммунодефицитное состояние и предрасположенность к развитию опухолей
2. Признаки. : 1. 2. 3. 4. Прогрессирующая атаксия, множественные телеангиэктазии, иммунодефицитное состояние и предрасположенность к развитию опухолей
. 3. Клинические проявления. Развитие в детском возрасте прогрессирующей атаксии, дизартрии") Г. Атаксия-телеангиэктазия (синдром Луи-Бар). 3. Клинические проявления. Развитие в детском возрасте прогрессирующей атаксии, дизартрии и хореоатетоза. 4. Рентгенографические исследования выявляют атрофию мозжечка, преимущественно червя, и признаки демиелинизации в области ствола и спинного мозга.
Г. Атаксия-телеангиэктазия (синдром Луи-Бар). 3. Клинические проявления. Развитие в детском возрасте прогрессирующей атаксии, дизартрии и хореоатетоза. 4. Рентгенографические исследования выявляют атрофию мозжечка, преимущественно червя, и признаки демиелинизации в области ствола и спинного мозга.
. 5. Лечение включает борьбу с инфекционными осложнениями, развивающимися вследствие иммунологической") Г. Атаксия-телеангиэктазия (синдром Луи-Бар). 5. Лечение включает борьбу с инфекционными осложнениями, развивающимися вследствие иммунологической недостаточности, физиотерапию и другие формы поддерживающей терапии. 6. Прогноз неблагоприятен. Большинство больных по достижении юношеского возраста передвигаются в инвалидных колясках. Смерть обычно наступает до 20 -летнего возраста от опухолей либо инфекционных осложнений.
Г. Атаксия-телеангиэктазия (синдром Луи-Бар). 5. Лечение включает борьбу с инфекционными осложнениями, развивающимися вследствие иммунологической недостаточности, физиотерапию и другие формы поддерживающей терапии. 6. Прогноз неблагоприятен. Большинство больных по достижении юношеского возраста передвигаются в инвалидных колясках. Смерть обычно наступает до 20 -летнего возраста от опухолей либо инфекционных осложнений.
 Спасибо за внимание!
Спасибо за внимание!