Врожденно-наследственные заболевания почек.ppt
- Количество слайдов: 67



Врожденнонаследственные заболевания почек

n Врожденные болезни органов мочевой системы ¨ это врожденные пороки развития, т. е. стойкие морфологические изменения органа (органов), выходящие за пределы возможных вариаций строения, возникающие внутриутробно. Могут быть генетически обусловлены или связаны с тератогенными влияниями на плод, включая внутриутробное инфицирование n Наследственными болезнями почек ¨ являются заболевания, связанные с аномалиями генов или хромосом

Генетика n n Международная цитогенетическая номенклатура, основанная на дифференциальном окрашивании хромосом и позволяющая подробно описывать отдельные хромосомы и их участки. Запись имеет следующий формат: ¨ n n [номер хромосомы] [символ плеча] [номер района (участка)]. [номер сегмента (полосы)] длинное плечо хромосомы обозначают буквой q, короткое — буквой p. Таким образом, 2 q 21. 3 – вторая хромосома, длинное плечо, район 21, сегмент 3.
 Нефротический синдром финского типа Аутосомнодоминантная поликистозная болезнь")
Моногенно-наследуемые нефропатии Болезнь синдром Альпорта (наследственный нефрит) Нефротический синдром финского типа Аутосомнодоминантная поликистозная болезнь (АДПКБ) Аутосомнорецессивная поликистозная болезнь (АРПКБ) Тип передачи Локализация мутантного гена Продукт гена Х-сцепленный АД АР COL 4 A 5 на Xq 21 -22 COL 4 A 3 на 2 chrom COL 4 A 4 на 2 chrom Цепи кол -лагена 4 типа АР 19 ql 3. 1 нефрин АД 16 р13. 3 4 q 21 полицистин 1 и 2 АР 6 р21. 1 -р22 Фиброцистин

Моногенно-наследуемые нефропатии Болезнь Нефронофтиз Первичная гипероксалурия 1 Первичная гипероксалурия 2 ФСГС* с ГРНС** Туберозный склероз 1 Туберозный склероз 2 Локализация Тип передачи мутантного гена АР 2 q 12 -q 13 Продукт гена нефроцистин АР 2 q 37. 3 9 р11 аланинглиоксилатаминотрансфераза Д-глицеро- фосфатдегидрогеназа АР 1 q 25 -q 31 подоцин 9 р34 АД туберин 16 р13 * фокалыю-сегментарный гломерулосклероз ** гормонорезистентный гломерулонефрит
 • аутосомно-рецессивный (АР) • доминантный, сцепленный с")
Пути передачи моногеннонаследуемых нефропатий • аутосомно-доминантный (АД) • аутосомно-рецессивный (АР) • доминантный, сцепленный с Х –хромосомой

Наследственные нефропатии в клинической практике Гематурия – Синдром Альпорта? n Кисты в почках – АДПКБ или АРПКБ? n Рахитоподобные изменения скелета – рахитоподобные заболевания? n Стойкие электролитные нарушения – псевдогипоальдостеронизм? n
 неимунная генетически детерминированная гломерулопатия, обусловленная мутацией генов, кодирующих коллаген 4")
Наследственный нефрит (синдром Альпорта) неимунная генетически детерминированная гломерулопатия, обусловленная мутацией генов, кодирующих коллаген 4 типа базальных мембран, проявляющаяся гематурией (+/- протеинурией), прогрессирующим снижением почечных функций (преимущественно у лиц мужского пола), нередко сочетающаяся с патологией зрения и слуха
 Частота в популяции – 1 : 5000 n 1% всех")
Наследственный нефрит (синдром Альпорта) Частота в популяции – 1 : 5000 n 1% всех случаев ХПН в Европе n 2, 3% случаев почечной трансплантации n n Встречается чаще, чем описывается
 Классификация Доминантный Х-сцепленный (классический, около 80% случаев) q Аутосомно-рецессивный (около")
Наследственный нефрит (синдром Альпорта) Классификация Доминантный Х-сцепленный (классический, около 80% случаев) q Аутосомно-рецессивный (около 20% случаев) q Аутосомно-доминантный (встречается редко) q q По данным НИИ педиатрии и детской хирургии из 200 детей Российской популяции – классический вариант – 78% , аутосомнодоминантный – 16%, аутосомно-рецессивный – 6%

Синдром Альпорта Фрагмент родословной Гематурия Патология зрения Тугоухость ХПН Смерть

Синдром Альпорта Этиология Мутация одного из генов: COL 4 A 5 на Xq 21 -22 COL 4 A 3 на 2 chrom COL 4 A 4 на 2 chrom

Схема гломерулярного барьера 1 – эпителий, 2 – эндотелий, 3 – lamina rara externa, 4 – lamina densa, 5 - lamina rara interna, 6 – фенестры эндотелия, 7 – подоциты эпителия
2, 2(IV) 3(IV),")
Этапы изменения гломерулярной базальной мембраны при синдроме Альпорта Нормальная ГБМ эндотелий 1(IV)2, 2(IV) 3(IV), 3(IV) и 5(IV) эпителий Поздний СА Ранний СА 1(IV)2, 2(IV) коллаген IV коллаген V эпителий Истончение БМ (ГБМ, капсула хрусталика) Гематурия и лентиконус эпителий Протеинурия и снижение функции почек Утолщение и дезорганизация ГБМ

Фрагмент стенки гломерулярного капилляра. Диагноз: синдром Альпорта Базальная мембрана расширена, вещество её просветлено, плотная пластинка выражена слабо. Электронограмма. Увел. 16000
 Орган зрения – базальная мембрана капсулы")
Коллаген IV типа (цепи α 3 -α 5) Орган зрения – базальная мембрана капсулы хрусталика, роговицы и др. n Орган слуха – внутреннее ухо (основная мембрана внутреннего уха, базальная мембрана спирального выступа, спирального гребешка, внутренней и наружной спиральных борозд) n

Синдром Альпорта Клинико-лабораторный симптомокомплекс n n n Первый симптом – гематурия При осмотре - бледность, снижение мышечного тонуса, стигмы дизэмбриогенеза, артериальная гипертензия Лабораторные показатели – гематурия, лейкоцитурия (лимфоцитарная), протеинурия Снижение почечных функций до развития терминальной ХПН (мужской пол) Нарушение слуха и зрения (снижение слуха по данным аудиометрии к 7 -8 годам, клинически выраженная тугоухость – к подростковому возрасту; аномалии органа зрения (лентиконус)

Синдром Альпорта

Синдром Альпорта Диагностика n n Клинико-лабораторный симптомокомплекс, наследственность, биопсия почки Для диагностики синдрома Альпорта необходимо 3 из 5 признаков: ¨ Гематурия или летальный исход от ХПН в семье; ¨ Гематурия и/или протеинурия в семье; ¨ Специфические изменения базальной мембраны клубочков (расслаивание и утолщение); ¨ Снижение слуха по данным аудиометрии; ¨ Врожденная патология зрения

Лечение синдрома Альпорта n n Специфическое консервативное лечение - не разработано. Основная цель терапии – замедление прогрессирования заболевания: ¨ Охранительный режим, диета, ограничение контакта с инфекциями, санация хронических очагов инфекции ¨ Длительная терапия ингибиторами АПФ n Трансплантация почек.
 или доброкачественная")
Дифференциальный диагноз n Болезнь тонких базальных мембран – доброкачественная гематурия (спорадические случаи) или доброкачественная семейная гематурия (если заболевание поражает несколько членов одной семьи) – наследственное поражение базальной мембраны клубочка, проявляющееся хронической гематурией.

Болезнь тонких базальных мембран Отличия от синдрома Альпорта n n n Нет внепочечных проявлений. Протеинурия, артериальная гипертензия и почечная недостаточность не характерны. Пол не влияет на течение заболевания. Наследуется аутосомно-доминантно. Предполагается генетическая гетерогенность заболевания. Иммуногистохимические исследования коллагена IV типа не выявили никаких нарушений в распределении ни одной из его 6 цепей.

Болезнь тонких базальных мембран n Если в семейном анамнезе гематурия без ХПН, наследуемая по аутосомнодоминантному типу, и не выявлено других явных причин гематурии – диагноз доброкачественной семейной гематурии может быть поставлен и без биопсии почек. n Семейная доброкачественная гематурии (и спорадические случаи болезни тонких мембран) не прогрессирует, не требует лечения.

Поликистозная болезнь почек n Наследственная нефропатия, связанная с мутацией генов, определяющих структуру почечных канальцев в их эмбриональном развитии, проявляющаяся образованием кист в почечной паренхиме, увеличение которых ведет к склерозированию ткани почек и развитию ХПН
 n Аутосомно-рецессивная поликистозная болезнь почек (АРПКБ)")
Поликистозная болезнь почек Аутосомно-доминантная поликистозная болезнь почек (АДПКБ) n Аутосомно-рецессивная поликистозная болезнь почек (АРПКБ) n

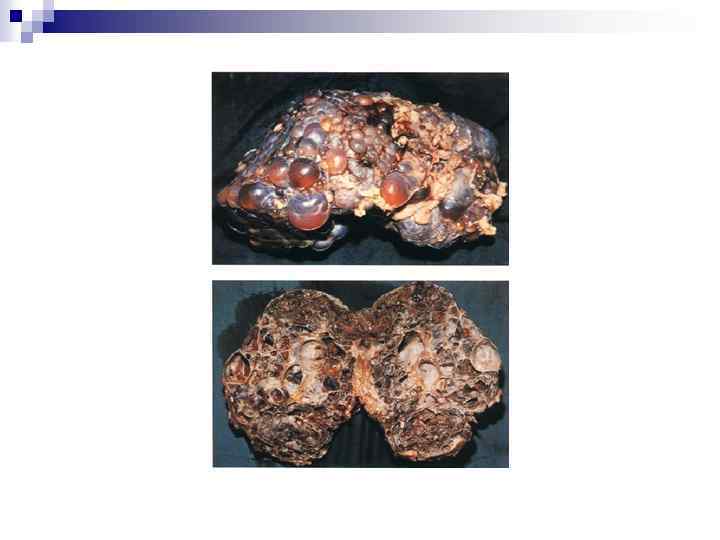
Аутосомно-доминантная поликистозная болезнь почек n n n Одно из наиболее распространенных генетически обусловленных заболеваний в популяции Частота - 1: 400 (европейская популяция) 1: 1000 (в среднем в мире) Устаревшее название – взрослый тип поликистозной болезни

Нормальное строение нефрона Формирование кист при АДПКБ

Почки при АДПКБ q q q Значительно увеличены С множественными кистами в коре и мозговом слое Кисты: Видны на поверхности почек Сферические Различной величины (мм – см) Содержат соломенного цвета жидкость (может быть геморрагической при кровоизлияниях) q Развиваются в 1 -5% нефронов q Всегда билатеральное расположение (в начальной стадии возможно асимметричное развитие) q q q Оставшаяся паренхима имеет различную степень канальцевой атрофии и интерстициальный фиброз

Клиническая картина АДПКБ q q q q Признаки болезни могут появляться в любом возрасте. У детей часто бессимптомное течение с выявлением при УЗИ почек Хроническая боль в пояснице, животе (чаще при физической нагрузке и во время интеркуррентных заболеваний) – вследствие растяжения капсулы почек Редко – пальпируемые увеличенные почки Макро- или микрогематурия, протеинурия, кристаллурия Клинико-лабораторные симптомы инфекции мочевой системы Нарушение функции концентрирования Никтурия
. Образованию способствуют:")
Клиническая картина АДПБ МКБ у 15 -20% больных (камни кальциевооксалатные и уратные). Образованию способствуют: q ртериальная гипертензия у 20 -30% детей и у 75% взрослых, развивается из-за изменения архитектуры почек, интраренальной ишемии, активации ренинангиотензиновой системы q Терминальная хроническая недостаточность к 50 -60 годам (наиболее быстрый темп развития ХПН при 1 типе АДПКБ) q
: В печени у")
Клиническая картина АДПКБ q Кисты могут выявляться (чаще после пубертатного периода): В печени у 30 -70% больных (не влияют на функцию) q В селезенке у 5% больных q В поджелудочной железе у 10% больных q В яичниках q q q Дивертикулез кишечника, У 5 -10% больных аневризмы внутричерепных артерий Аневризмы грудного и брюшного отделов аорты У 25% пролапс митрального клапана и недостаточность аортального клапана

Ультразвуковые признаки аутосомнодоминантного поликистоза почек q Увеличение почек в размерах q Неровность контуров почек q Наличие множества эхонегативных образований в паренхиме обеих почек q Нечеткая дифференцировка коркового и мозгового слоев почек q Деформация чашечно-лоханочной системы

УЗИ левой почки пациента 13 лет Диагноз – АДПКБ


Лечение АДПКБ n n Цель – замедление развития терминальной ХПН Симптоматическая терапия: ¨ Лечение артериальной гипертензии ¨ Лечение инфекции мочевой системы n Трансплантация почек n Экспериментальные исследование – противоопухолевые препараты

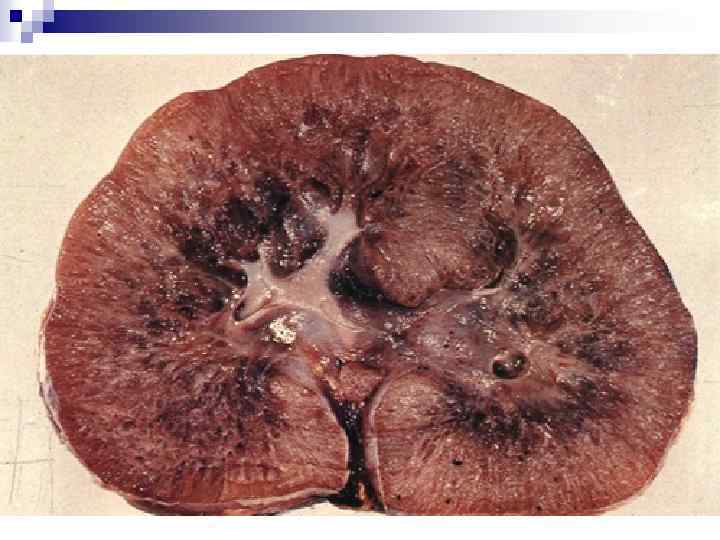
Аутосомно-рецессивная поликистозная болезнь почек n n n Поликистоз почек детского типа, «младенческий» Частота 1: 6000 -1: 40000 живых новорожденных Высокая смертность в перинатальном и раннем неонатальном периоде Этиология Болезнь Тип передачи Локализация мутантного гена Продукт гена АРПКБ (ген – PKHD 1) АР 6 р21. 1 -р22 Фиброцистин

Фрагмент родословной. Диагноз - АРПКБ I 50 65 67 50 II 29 III Поликистозная болезнь Сердечнососудистая патология Мочекаменная болезнь 34

Нормальный нефрон Нефрон при АРПКБ. Собирательные трубочки расширены в виде веретенообразных кист

Аутосомно-рецессивная поликистозная болезнь q q Ишемия в антенатальном периоде, маловодие Множественные аномалии развития (синдром Поттер): q q q q Гипоплазия легких Аномалии лицевого скелета (низкорасположенные уши, плоский нос, недоразвитый подбородок – лицо Поттер) Большие почки с гладкой поверхностью Артериальная гипертензия Рецидивирующая инфекция мочевой системы Гепатоспленомегалия (формирование печеночного фиброза, портальной гипертензии, возможно развитие печеночной недостаточности) Задержка развития

Основные диагностические признаки АРПКБ Клинические признаки n Отсутствие семейного анамнеза n Ранняя манифестация болезни n Прогрессирующее снижение функции почек n Гепатоспленомегалия n Портальная гипертензия

Основные диагностические признаки АРПКБ УЗИ-диагностика n УЗИ почек: увеличение почек в размерах, паренхима гиперэхогенная, крупнозернистая, с наличием мелких эхонегативных зон ( «соль перцем» ) - микрокистоз, нарушение кортикомедулярной дифференциации n УЗИ печени: повышенная эхогенность, кисты желчных протоков

Основные диагностические признаки АРПКБ Морфологические признаки Почки: диффузная дилатация и кистоз собирательных трубочек n Печень: диффузная билиарная дисгенезия с пролиферацией и эктазией билиарных протоков, портальный фиброз n

Дифференциальный диагноз поликистоза почек АРПКБ АДПКБ Увеличение почек ++ + Повышение АД + + Новорожденный + - Увеличение печени, селезенки + - Наследственность + ++ Кисты в других органах - + Гематурия ± + Инфекция мочевых путей + + Неблагоприятный прогноз + -

Аутосомно-рецессивная поликистозная болезнь Лечение Цель – замедление прогрессирования почечной и печеночной недостаточности q Симптоматическое: q q Артериальной гипертензии, q Инфекции мочевой системы q Почечной недостаточности, q Патологии печени – портальная гипертензия, варикозное расширение вен пищевода q Трансплантация

Рахит при наследственных нефропатиях

Витамин Д-резистентный рахит q Доминантное сцепленное с Х-хромосомой заболевание с глубокими нарушениями Р-Са обмена, которые не удается восстановить обычными дозами витамина D q Другое название – гипофосфатемический рахит, фосфат-диабет, синдром Олбрайта. Батлера-Блюмберга, относится к тубулопатиям

Витамин Д-резистентный рахит n n n n Самое частое из группы рахитоподобных заболеваний Частота 1 : 20000 детского населения Ген Рех-ген Хр22. 2 -р22. 1 Продукт гена – фосфатрегулирующий белок, который обеспечивает почечную реабсорбцию фосфатов и метаболизм витамина Д Выявлено более 130 мутаций гена фосфат-диабета Существуют аутосомно-доминантные моногенные формы фосфат-диабета (12 р13. 3, 5 q 35) 4 клинико-биохимических варианта заболевания с различными сроками манифестации и эффективностью терапии

Патогенез фосфат-диабета Снижение реабсорбции фосфатов в почечных канальцах q Нарушение всасывания фосфора и кальция в кишечнике q Нарушение метаболизма витамина Д q Вторичный гиперпаратиреоз q

Фосфат-диабет

Клиническая картина фосфат-диабета n n n n Период новорожденности без особенностей Манифестация на 2 -м году (возможна ранняя манифестация – 1 год, поздняя манифестация – 7 -9 лет) Первый симптом – искривление нижних конечностей (по типу варусных деформаций) Задержка физического развития, нарушение походки Прогрессирующий характер поражения скелета Интеллект не страдает Позднее прорезывание зубов, дефекты эмали, множественный кариес

Диагностическая программа выявления витамин Д-резистентного рахита n n n n Сбор генеалогических данных Оценка параметров физического развития Характеристика костных деформаций Общий анализ крови, общий анализ мочи Проба Сулковича Определение показателей кальция и фосфора в крови Определение суточной экскреции кальция и фосфора с мочой Определение активности щелочной фосфатазы сыворотки крови Исследование показателей 25(ОН)D 3, 1, 25(ОН)2 D 3 Определение концентрации паратгормона, кальцитонина, остеокальцина в крови Исследование минеральной плотности костной ткани (денситометрия) Рентгенография трубчатых костей верхних и нижних конечностей (активность процесса) УЗИ почек

Лабораторные изменения n n n Снижение Р в сыворотке крови Увеличение выделения Р с мочой Снижение реабсорбции фосфатов в почечных канальцах (менее 80%) Повышенная активность щелочной фосфатазы Нормальный уровень Са в сыворотке крови Повышение паратгормона, кальцитонина, остеокальцина

Рентгенологические изменения при фосфат-диабете q Рахитоподобные изменения скелета: Через 3 -4 месяца после манифестации заболевания – небольшое разрыхление зон предварительного обызвествления (на границе между эпифизом и метафизом) q В период активного рахитического процесса – генерализованный остеопороз, увеличение метафизов, стирание нормальной границы, отделяющей метафиз от эпифиза, грубоволокнистая структура губчатого вещества кости, в метафизах и эпифизах кистоподобные разряжения, грубые деформации длинных трубчатых костей q На фоне лечения – обратное развитие – появление зон предварительного обызвествления, выравнивается линия препараторного роста, восстанавливаются контуры костной ткани, более четко контурируются ядра окостенения q

Фосфат-диабет Лечение Учет индивидуальной переносимости препаратов, активности рахитического процесса и клинико-биохимических вариантов заболевания n Базисные препараты – витамин Д и его активные метаболиты n

Фосфат-диабет Лечение n Показания для консервативного лечения витамином Д: ¨ Активный рахитический процесс в костной ткани по рентгенологическим данным (системный остеопороз, неровность эпифизов, нечеткость зон препараторного роста) ¨ Повышение активности щелочной фосфатазы сыворотки крови ¨ Повышенная экскреция фосфатов с мочой ¨ В период подготовки детей к проведению хирургической коррекции костных деформаций

Фосфат-диабет Лечение n n n Начальная доза витамина Д – 10000 -15000 МЕ в сутки Увеличение дозы под контролем уровня кальция, фосфора крови и мочи, показателей щелочной фосфатазы (каждые 10 -14 дней). При положительной динамики – дозу не увеличивать Максимальные суточные дозы витамина Д с учетом вариантов: 100000 – 300000 Ед
 n n n")
Фосфат-диабет Лечение n Активные метаболиты Витамина Д: ¨ Альфакальцидол (1 -гидроксихолекальциферол) n n n Оксидевит Этальфа Альфа D 3 -Тева ¨ Кальцитриол (1, 25 дигидроксихолекальциферол) n n n Рокальтрол Остеотриол Индивидуальное дозирование (0, 25 -3 мкг/сут)
")
Фосфат-диабет Лечение q q q Препараты кальция (кальция карбонат 0, 5 -1, 5 г/сут) Неорганические фосфаты (1, 5 – 3 г/сут) Цитратные смеси (лимонная кислота 24 г + натрия цитрат 48 г + дистиллированная вода до 500 мл) по 20 -50 мл/сут Перспективы лечения: соматропин, кальцитонин, дериваты паратиреоидного гормона, бисфосфонаты Лечение продолжается в течение всего периода роста Во время лечения контроль состояния костной ткани и содержания Са и Р в крови и моче

Симптомы интоксикации витамином D Жажда q Ухудшение аппетита q Полиурия q Запор q Экскреция Са с мочой >5 мг/кг за 24 ч q Гиперкальциемия q Положительная реакция Сулковича q

Исходы лечения q q q Дети растут, но отставание в росте сохраняется Витамин D усиливает минерализацию костей, но не приводит к полной нормализации тонкой костной структуры После пубертатного периода возможна стойкая ремиссия без активной терапии Рецидивы возможны в периоды напряженного минерального обмена (половое созревание, беременность, лактация) Фосфат-диабет не осложняется органическим поражением почек, не развивается ХПН

Витамин Д-зависимый рахит Рахитоподобное заболевание с аутосомно-рецессивным типом наследования, в основе которого лежит дефект 1α-гидроксилазы, что приводит к недостаточному образованию 1, 25(ОН)2 D 3

Болезнь де Тони-Дебре-Фанкони Наиболее тяжелая генерализованная проксимальная тубулопатия с аутосомно-рецессивным типом наследования. Является канальцевой полиэнзимопатией, характеризующейся нарушением синтеза ферментов, ответственных за транспорт аминокислот, фосфатов, глюкозы и бикарбонатов

Болезнь де Тони-Дебре-Фанкони Исходы заболевания

Почечный канальцевый ацидоз Генетически гетерогенное заболевание, имеющее различные типы наследования, в основе которого лежит неспособность почечных канальцев подкислять мочу (дефект реабсорбции бикарбонатов в проксимальных канальцах или нарушение ацидогенеза в дистальных канальцах), что приводит к постоянному метаболическому ацидозу

Спасибо за внимание!
Врожденно-наследственные заболевания почек.ppt