Лекция для магистров.ppt
- Количество слайдов: 130



Вопросы: 1. Генетическая основа наследственных заболеваний человека. 2. Молекулярно-генетические методы выявления мутаций, вызывающих наследственные заболевания человека. 3. Краткая характеристика моно- и полигенных наследственных заболеваний человека.
 ФЕНОКОПИИ ГЕНОТИПИЧЕСКАЯ (наследуемая) СОМАТИЧЕСКАЯ ГЕНЕРАТИВНАЯ МУТАЦИОННАЯ КОМБИНАТИВНАЯ")
ВИДЫ ИЗМЕНЧИВОСТИ ФЕНОТИПИЧЕСКАЯ (ненаследуемая, модификационная) ФЕНОКОПИИ ГЕНОТИПИЧЕСКАЯ (наследуемая) СОМАТИЧЕСКАЯ ГЕНЕРАТИВНАЯ МУТАЦИОННАЯ КОМБИНАТИВНАЯ

ВИДЫ МУТАЦИЙ ПО ПРИЧИНЕ “СПОНТАННЫЕ ” ИНДУЦИРОВАННЫЕ ПО ВИДУ КЛЕТОК, В КОТОРЫХ ПРОИЗОШЛА МУТАЦИЯ СОМАТИЧЕСКИЕ ГАМЕТИЧЕСКИЕ ПО ЗНАЧЕНИЮ ПАТОГЕННЫЕ НЕЙТРАЛЬНЫЕ БЛАГОПРИЯТНЫЕ ПО “УРОВНЮ” (“МАСШТАБУ”) ГЕННЫЕ ХРОМОСОМНЫЕ ГЕНОМНЫЕ

ВИДЫ МУТАЦИЙ “ПО МАСШТАБУ ” ГЕННЫЕ * изменения ДНК ХРОМОСОМНЫЕ * изменения структуры отдельных хромосом ГЕНОМНЫЕ * изменения числа хромосом ПРИМЕРЫ * гемоглобиноз S * гемофилии * муковисцидоз * нейрофиброматоз * фенилкетонурия * делеция хромосом (5 р – синдром “кошачьего крика”) * дупликация короткого плеча хромосомы 9 (множественные ВПР) * полиплоидии * анеуплоидии (моносомии, трисомии)

ОСНОВНЫЕ МЕХАНИЗМЫ ГЕННЫХ МУТАЦИЙ ДЕЛЕЦИЯ СЕГМЕНТА ДНК ДУПЛИКАЦИЯ УЧАСТКА ДНК ИНВЕРСИЯ СЕГМЕНТА ДНК ИНСЕРЦИЯ ФРАГМЕНТА ДНК ТРАНСВЕРСИЯ ТРАНЗИЦИЯ ОСНОВАНИЙ

ВИДЫ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ ГЕННЫЕ ХРОМОСОМНЫЕ БОЛЕЗНИ СОБСТВЕННО ХРОМОСОМНЫЕ ГЕНОМНЫЕ БОЛЕЗНИ ГЕНЕТИЧЕСКОЙ НЕСОВМЕСТИМОСТИ МАТЕРИ И ПЛОДА ГЕНЕТИЧЕСКИЕ СОМАТИЧЕСКИЕ БОЛЕЗНИ С ГЕНЕТИЧЕСКОЙ ПРЕДРАСПОЛОЖЕННОСТЬЮ (МНОГОФАКТОРНЫЕ)
 АУТОСОМНОДОМИНАНТНЫЕ РЕЦЕССИВНЫЕ ДОМИНАНТНЫЕ, СЦЕПЛЕННЫЕ")
ВИДЫ ГЕННЫХ БОЛЕЗНЕЙ В ЗАВИСИМОСТИ ОТ ТИПА ИХ НАСЛЕДОВАНИЯ (1) АУТОСОМНОДОМИНАНТНЫЕ РЕЦЕССИВНЫЕ ДОМИНАНТНЫЕ, СЦЕПЛЕННЫЕ АУТОСОМНОС Х ХРОМОСОМОЙ РЕЦЕССИВНЫЕ, СЦЕПЛЕННЫЕ С Х ХРОМОСОМОЙ П Р И М Е Р Ы ПОЛИДАКТИЛИЯ СИНДРОМ МАРФАНА СЕМЕЙНАЯ ГИПЕРХОЛЕСТЕРИНЕМИЯ НЕЙРОФИБРОМАТОЗ ХОРЕЯ ГЕНТИНГТОНА ПОЛИПОЗ ТОЛСТОГО КИШЕЧНИКА ГАЛАКТОЗЕМИЯ ФЕНИЛКЕТОНУРИЯ АЛЬБИНИЗМ ГЛИКОГЕНОЗЫ МУКОВИСЦИДОЗ АДРЕНОГЕНИТАЛЬНЫЙ СИНДРОМ ГИПЕРЛИПОПРОТЕИНЕМИЯ РАХИТ, УСТОЙЧИВЫЙ К ВИТАМИНУ D КАТАРАКТА ГЕМОФИЛИИ А, В ДАЛЬТОНИЗМ ГИПОГАМО ГЛОБУЛИНЕМИЯ МЫШЕЧНАЯ ДИСТРОФИЯ ДЮШЕНА
 ГОЛАНДРИЧЕСКИЕ МИТОХОНДРИАЛЬНЫЕ ПРИМЕРЫ ИЗБЫТОЧНОЕ")
ВИДЫ ГЕННЫХ БОЛЕЗНЕЙ В ЗАВИСИМОСТИ ОТ ТИПА ИХ НАСЛЕДОВАНИЯ (2) ГОЛАНДРИЧЕСКИЕ МИТОХОНДРИАЛЬНЫЕ ПРИМЕРЫ ИЗБЫТОЧНОЕ ОВОЛОСЕНИЕ УШНЫХ РАКОВИН АЗООСПЕРМИЯ АТРОФИЯ ЗРИТЕЛЬНОГО НЕРВА ЛЕБЕРА ЭНЦЕФАЛОПАТИЯ МИТОХОНДРИАЛЬНАЯ ЭПИЛЕПСИЯ МИОКЛОНАЛЬНАЯ КАРДИОМИОПАТИЯ

ВИДЫ ХРОМОСОМНЫХ МУТАЦИЙ ВНУТРИХРОМОСОМНЫЕ МЕЖХРОМОСОМНЫЕ РЕЦИПРОКНЫЕ ДЕЛЕЦИИ ИНВЕРСИИ ДУПЛИКАЦИИ НЕРЕЦИПРОКНЫЕ “ЦЕНТРИЧЕСКОЕ ” СЛИЯНИЕ

ВИДЫ ХРОМОСОМНЫХ БОЛЕЗНЕЙ В ЗАВИСИМОСТИ ОТ НАРУШЕНИЯ СТРУКТУРЫ ИЛИ ЧИСЛА ХРОМОСОМ НАРУШЕНИЕ СТРУКТУРЫ ХРОМОСОМ ИЗМЕНЕНИЕ ЧИСЛА ХРОМОСОМ ИЛИ ПЛОИДНОСТИ В ЗАВИСИМОСТИ ОТ ВИДА КЛЕТОК, В КОТОРЫХ ПРОИЗОШЛА МУТАЦИЯ ПОЛНЫЕ ФОРМЫ С ИЗМЕНЕНИЕМ ЧИСЛА ХРОМОСОМ СТРУКТУРЫ ХРОМОСОМ МОЗАИЧНЫЕ ФОРМЫ С МУТАЦИЯМИ ХРОМОСОМНЫМИ ГЕНОМНЫМИ
 АНЭУПЛОИДИЯ (2 n ± 1)")
ВИДЫ ГЕНОМНЫХ МУТАЦИЙ ПОЛИПЛОИДИЯ (3 n, 4 n, …) АНЭУПЛОИДИЯ (2 n ± 1)

ВИДЫ НАСЛЕДСТВЕННЫХ БОЛЕЗНЕЙ ПО ВИДУ МУТАНТНЫХ КЛЕТОК ГАМЕТИЧЕСКИЕ СОМАТИЧЕСКИЕ ПО РОЛИ ФАКТОРОВ НАСЛЕДСТВЕННОСТИ И СРЕДЫ В ИХ ВОЗНИКНОВЕНИИ СОБСТВЕННО НАСЛЕДСТВЕННЫЕ ВОЗНИКАЮЩИЕ ПРИ ДЕЙСТВИИ ОПРЕДЕЛЕННОГО ФАКТОРА СРЕДЫ ВЫЗЫВАЕМЫЕ ФАКТОРАМИ СРЕДЫ КОМБИНИРОВАННЫЕ ВОЗНИКАЮЩИЕ ПРИ ДЕЙСТВИИ ОПРЕДЕЛЕННОГО ФАКТОРА СРЕДЫ НА “ПРЕДРАСПОЛОЖЕННЫЙ” ОРГАНИЗМ

© П. Ф. Литвицкий, 2004 © ГЭОТАР-МЕД, 2004 ОСНОВНЫЕ МЕТОДЫ ДИАГНОСТИКИ И АНАЛИЗА ПАТОГЕНЕЗА НАСЛЕДСТВЕННЫХ ФОРМ ПАТОЛОГИИ клиникосиндромологический клиникогенеалогический генетики соматических клеток клонирования селекции гибридизации цитогенетический биологического моделирования близнецовый биохимический молекулярно- генетический гибридизация ДНК клонирование ДНК полимеразная цепная реакция блотгибридизации ДНК

ПРИНЦИПЫ И МЕТОДЫ ЛЕЧЕНИЯ НАСЛЕДСТВЕННЫХ ФОРМ ПАТОЛОГИИ ПРИНЦИПЫ МЕТОДЫ ЭТИОТРОПНЫЙ * коррекция дефекта генома: - введение в геном нормального гена, - подавление репликации патогенного гена * изменение генома: - введение в него гена, кодирующего синтез чужеродного антигена ПАТОГЕНЕТИЧЕСКИЙ * заместительная терапия (введение “дефицитного” вещества) СИМПТОМАТИЧЕСКИЙ * устранение тягостных, усугубляющих состояние симптомов * коррекция метаболизма: - ограничения попадания в организм неметаболизируемых веществ (лактозы, фенилаланина), - выведение избытка метаболитов (холестерина, фенилпировиноградной кислоты), - регуляция активности ферментов (липопротеинлипазы крови, КФКазы) * хирургическое устранение дефектов (шунтов, сращений, создание шунтов)

Характеристика моногенных болезней Наследуются в соответствии с законами Менделя. Классифицируются на: аутосомно-доминантные, Аутосомно-рецессивные, сцепленные с полом. Это наиболее широкая группа наследственных заболеваний. В настоящее время описано более 4000 вариантов моногенных наследственных болезней, подавляющее большинство которых встречается довольно редко (например, частота серповидноклеточной анемии - 1/6 000). Широкий круг моногенных болезней образуют наследственные нарушения обмена веществ, возникновение которых связано с мутацией генов, контролирующий синтез ферментов и обусловливающих их дефицит или дефект их строения.

Моногенные болезни • 1. Нарушение обмена аминокислот: фенилкетонурия. • 2. Нарушение обмена углеводов: галактоземия, фруктоземия. • 3. Нарушение обмена липидов: семейная гиперхолестеринемия. • 4. Нарушение биосинтеза гормонов: адреногенитальный синдром. • 5. Нарушение обмена витаминов: нарушение всасывания витамина В 12. • 6. Нарушение синтеза гемоглобина: серповидноклеточная анемия, талассемии.

Полигенные болезни наследуются по сложной схеме. Для них вопрос о наследовании не может быть решён на основании законов Менделя. Ранее такие наследственные заболевания характеризовались как болезни с наследственной предрасположенностью. Однако сейчас о них идёт речь как о мультифакториальных заболеваниях с аддитивнополигенным наследованием с пороговым эффектом. К этим заболеваниям относятся такие болезни как рак, сахарный диабет, шизофрения, эпилепсия, ишемическая болезнь сердца, гипертензия и др.

СВОЙСТВА МУЛЬТИФАКТОРИАЛЬНЫХ ЗАБОЛЕВАНИЙ в этиологии важна роль изменений в геноме предрасположенность к болезни зависит от большого числа генов (феномен аддитивности) характер наследования не объясняется только менделевскими законами предрасположенность реализуется под влиянием большого числа факторов среды П р и м е р ы: * Ишемическая болезнь сердца (ИБС) * Гипертоническая болезнь * Бронхиальная астма * Сахарный диабет * Язвенная болезнь желудка и кишечника * Псориаз * Эпилепсия * Системная красная волчанка *. . .

Хромосомные болезни обусловлены грубыми нарушениями наследственного аппарата — изменением числа или структуры хромосом. Сюда относятся синдром Дауна, синдром Кляйнфельтера, Шерешевского-Тернера, Эдвардса, синдром «кошачьего крика» и др.
 наследуется по материнской линии. Сюда")
Болезни, обусловленные дефектами митохондриальной ДНК Митохондриальная ДНК (мт. ДНК) наследуется по материнской линии. Сюда относят патологические нарушения клеточного энергетического обмена, обусловленные мутациями мт. ДНК. Они могут быть обусловлены дефектами различных звеньев в цикле Кребса в дыхательной цепи, процессах окисления. Вследствие гетероплазмии проявление и степень тяжести болезней, обусловленных сходными нарушениями мт. ДНК, могут быть неодинаковыми у разных людей, в зависимости от соотношения в цитоплазме клеток мутантных и нормальных митохондрий.

Механизм возникновения митохондриальных мутаций

Механизм экспансии тринуклеотидов

Незаконный кроссинговер

Принципы диагностики хромосомных заболеваний человека
 (фитогемагглютинина) 72 часа")
Отделение лейкоцитов Добавление Кровь (или стимулятора митоза – ФГА другой материал) (фитогемагглютинина) 72 часа Добавление колхицина – блокатора микротрубочек Деление лимфоцитов тормозится на стадии метафазы

Добавление гипотонического раствора – клетки разбухают Х ХХ Х Х х х при раскапывании от удара о стекло хромосомы разлетаются в стороны – образуется метафазная пластинка Затем препарат фиксируют и окрашивают

Виды окраски хромосом • Рутинная, появилась в 50 -х годах ХХ века. (Денверская классификация поделила все хромосомы человека на 7 групп по размеру и форме). • Дифференциальная, появилась в конце 60 -х годов (G, R, Q и С методы). Парижская конференция закрепила за каждой хромосомой номер, ввела обозначения для мутаций. • FISH - метод, был разработан в 90 -х годах и дал еще больше возможностей для диагностики.
")
Парижская классификация основана на дифференциальной окраске (чаще всего G-окраска)
 и суббенды")
Плечи делят на районы (бенды) и суббенды

FISH -метод – Fluorescent in situ hybridization дал еще больше возможностей

Гибридизация нуклеиновых кислот in situ • Метод основан на гибридизации изучаемой ДНК на цитологическом препарате с ДНК-пробой. Для выявления участков гибридизации ДНК-пробу метят либо радиоактивной меткой, либо с помощью флуорохромов. Для гибридизации предварительно проводят денатурацию ДНК пробы и ДНК цитологического препарата, затем осуществляют ренатурацию. В результате образуется дуплекс меченой ДНК и ДНК препарата. Не связавшаяся меченая ДНК отмывается, после чего производят выявление районов гибридизации. В настоящее время мечение ДНК делят на прямое и непрямое. • Прямое мечение – введение в ДНК репортерных элементов – флуорохромов (родамина, диэтиламинокумарина, техасского красного и др. ). • Непрямой метод мечения - ДНК-пробу предварительно конъюгируют с промежуточными лигандами (биотином, диоксигенином, 2, 4 -динитрофенолом), присутствие которых на цитологическом препарате выявляется затем с помощью флуорохромов. В этом случае чувствительность метода намного возрастает, так как лиганд может содержать несколько сайтов взаимодействия с флуорохромом. Гибридизация in situ позволяет выявлять хромосомные аномалии (числа хромосом), хромосомные перестройки – делеции, дупликации, частичные трисомии, транслокации.

FISH-метод Фиксированные клетки на препарате Ген Надрез участков эндонууклеазой Меченые биотином нуклеотиды Денатурация при 42⁰ С Ник- трансляция Денатурация Гибридизация на препарате Антитела и связывание с флуорофором Установление локализации

FISH-метод позволяет лучше распознавать хромосомны е перестройки, чем одноцветная окраска

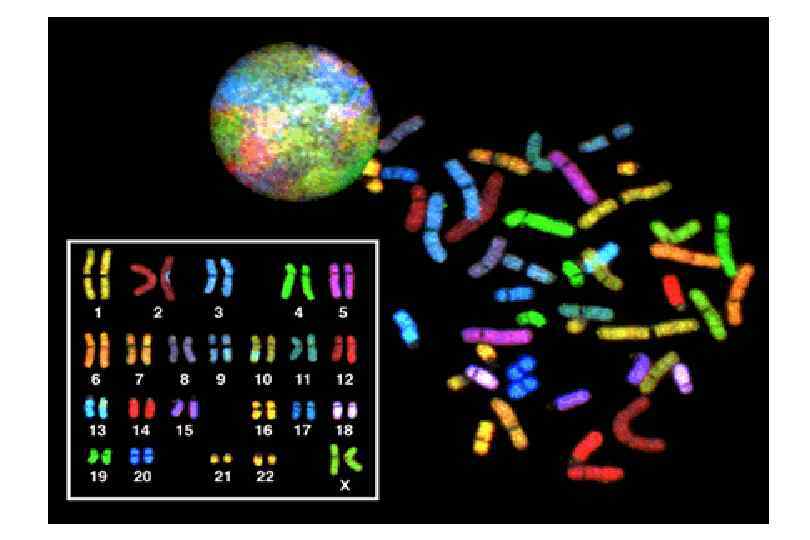
Методы выявление хромосомных мутаций с помощью спектрального кариотипирования Для визуализации нарушений структуры хромосом в последнее время используется методика так называемого спектрального кариотипирования, состоящая в окрашивании хромосом набором флуресцентных красителей, связывающихся со специфическими областями хромосом. В результате такого окрашивания гомологичные пары хромосом приобретают идентичные спектральные характеристики, что не только существенно облегчает выявление таких пар, но и облегчает обнаружение межхромосомных транслокаций, т. е. перемещений участков между хромосомами.

Методы выявление структурных генных мутаций Прямая диагностика. • Рестрикционный анализ. • Секвенирование. • Мультиплексная (мультипраймерная ПЦР). ●

ДНК-диагностика длин рестрикционных фрагментов ДНК. На примере спиноцеребеллярной атаксии-1. Экспансия тринуклеотидов в гетерозиготном состоянии. Дорожка 1 — маркер, дорожки 2, 3, 4, 5, 7 и 8 — больные, дорожка 6 — здоровый индивид. Длинной стрелкой указан мутантный аллель (экспансия CAG-повторов гена SCA 1), короткой стрелкой — нормальный аллель

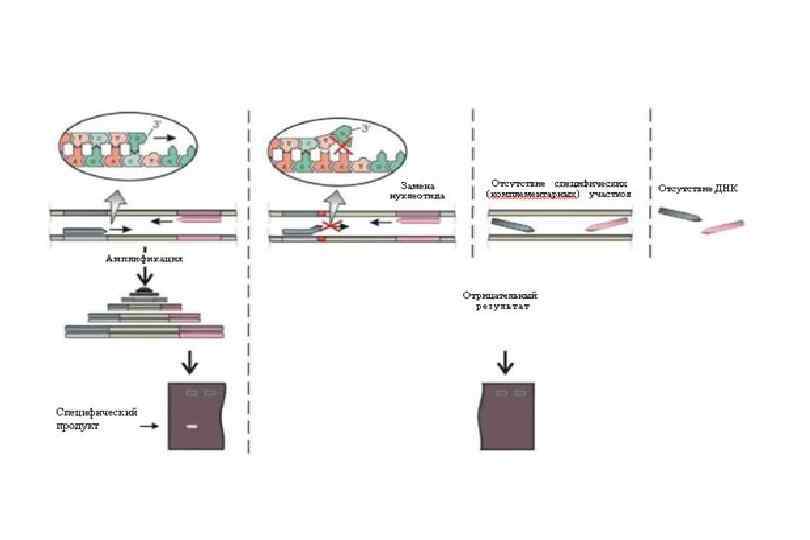
Метод выявления мутаций с помощью рестрикционного анализа А — амплифицируемый участок гена, содержащий сайт рестрикции AGCT для рестрикционной эндонуклеазы Alu I. Мутация G-» A изменяет данную нуклеотидную последовательность, в результате чего рестрикция ферментом Alu I блокируется; Б — электрофореграмма продуктов рестрикции: дорожка 1 — гомозиготность по нормальному аллелю; дорожка 2 — гомозиготность по мутации; дорожка 3 — гетерозиготное состояние (нормальный аллель + мутация)

Секвенирование Для выявления точковых мутаций, небольших делеций и инверсий в исследуемых генах используют методы, при помощи которых можно проанализировать уникальную последовательность ДНК. Примером может служить метод секвенирования ДНК. Любые типы мутаций могут быть обнаружены путем прямого секвенирования мутантной ДНК. Для некоторых генов, имеющих небольшие размеры, этот метод с успехом применяется как основной метод сканирования мутаций. Главное преимущество прямых методов диагностики - почти 100 % эффективность.
 для выявления делеций внутри гена • Прямая ДНК-диагностика мышечной дистрофии Дюшенна")
Мультиплексная (мультипраймерная ПЦР) для выявления делеций внутри гена • Прямая ДНК-диагностика мышечной дистрофии Дюшенна с помощью мультиплексной полимеразной цепной реакции (электрофорез в агарозном геле). У каждого из обследуемых лиц одновременно амплифицированы четыре экзона гена дистрофина (экзоны 17, 19, 44 и 45; стрелки указывают на соответствующие продукты амплификации). • Дорожка 1 — контроль. • Дорожки 2 -5 — больные мышечной дистрофией Дюшена с различными делециями гена дистрофина (дорожки 2 и 5 — делеция экзона 45, дорожка 3 — делеция экзона 44, дорожка 4 — делеция экзонов 17 и 19)

Диагностика генных мутаций

Точковые генные мутации не всегда имеют фенотипическое проявление Последовательность ДНК Аминокислотная последовательность Тип мутации ATG CAG GTG ACC TCA GTG M Q V T S V нет ATG CAG GTT ACC TCA GTG M Q V T S V Молчащая (синонимическая 0 ATG CAA GTG ACC TCA GTG M Q L T S V Консервативная ATG CCG GTG ACC TCA GTG M P V T S V Неконсервативная ATG CAG GTG ACC TGA GTG M Q V T ter Нонсенс ATG CAG GTG AAC CTC AGT G M Q V N L S Фреймшифт Консервативной заменой аминокислоты называется мутационная замена не приводящая к значительным изменениям структуры и функции белка

Методы идентификации мутаций делятся на две группы I. Идентификация известных мутаций: ► блот-гибридизация по Саузерну; ► полимеразная цепная реакция (ПЦР); ► рестрикционный анализ. ► анализ с помощью биочипов. II. Идентификация неизвестных мутаций: ► прямое секвенирование; ► метод конформационного полиморфизма одноцепочечных фрагментов SSCP; ► метод электрофореза в денатурирующем градиентном геле; ► метод химического расщепления некомплементарных сайтов; ► гететродуплексный анализ и др.

Важнейший этап идентификации мутантного гена – его выделение. ДНК может быть изолирована из любого типа тканей и клеток, содержащих ядра. Этапы выделения ДНК включают: быстрый лизис клеток, удаление с помощью центрифугирования фрагментов клеточных органелл и мембран, ферментативное разрушение белков и их экстрагирование из раствора с помощью фенола и хлороформа, концентрирование молекул ДНК путём преципитации в этаноле.

Следующий этап подготовки материала к исследованию – «разрезание» ДНК на фрагменты в участках со строго специфической последовательностью оснований, которое осуществляют с помощью бактериальных ферментов - рестрикционных эндонуклеаз (рестриктаз).

Идентификацию конкретных фрагментов в геле среди геномной ДНК провести труднее. Из-за больших размеров генома человека после рестрикции образуется так много фрагментов, что агарозный гель после электрофореза и окраски этидум бромидом выглядит в ультрафиолетовых лучах более или менее равномерно окрашенным. Поэтому для выявления специфических фрагментов ДНК используют следующие подходы: блот-гибридизацию по Саузерну, полимеразную цепную реакцию (ПЦР), рестрикционный анализ, а также гибридизацию с помощью биочипов.
.")
Блот-гибридизация • • ДНК-диагностика мутации в гене дистрофина с помощью блот-гибридизации по Саузерну (авторадиография). В дорожке 2 у больного отсутствуют два облигатных фрагмента ДНК (указаны стрелками), что свидетельствует о протяженной делеции соответствующего участка гена дистрофина. В дорожке 4 (мать больного) указанные стрелками фрагменты визуализируются в виде полос пониженной интенсивности сигнала {по сравнению с контролем в дорожках 1 и 3), что свидетельствует о сниженной вдвое "дозе гена" (гетерозиготное носительство делеции)

Блот-гибридизация по Саузерну

Рестрикционный анализ Выявление мутаций с помощью рестрикционного анализа

Прямая ДНК-диагностика спиноцеребеллярной атаксии-1 с помощью электрофореза в агарозном геле Дорожка 1 – маркер, дорожки 2, 3, 4, 5, 7 и 8 – больные, дорожка 6 – здоровый индивид. Длинной стрелкой указан мутантный аллель (экспансия CAG-повторов гена SCA 1), короткой стрелкой – нормальный аллель
 Прямая ДНК-диагностика мышечной дистрофии Дюшена с помощью мультиплексной ПЦР. Примечание:")
Полимеразная цепная реакция (ПЦР) Прямая ДНК-диагностика мышечной дистрофии Дюшена с помощью мультиплексной ПЦР. Примечание: у каждого из обследуемых лиц одновременно амплифицированы четыре экзона гена дистрофина (экзоны 17, 19, 44 и 45; стрелки указывают на соответствующие продукты амплификации). Дорожка 1 – контроль, дорожки 2 -5 – больные мышечной дистрофией Дюшена с различными делециями гена дистрофина (дорожки 2 и 5 – делеция экзона 45, дорожка 3 – делеция экзона 44, дорожка 4 – делеция экзонов 17 и 19)

Метод биочипов

Метод биочипов в последние годы заняли существенное место в разнонаправленных молекулярно-биологических исследованиях
 Дает возможность генотипирования всех видов точечных нуклеотидных замен С Для")
Метод аллель-специфической амплификации (ПЦР) Дает возможность генотипирования всех видов точечных нуклеотидных замен С Для эффективного осуществления ПЦР 3'-концевой нуклеотид и предшествующий ему нуклеотид праймера должны быть комплементарны cоответствующему нуклеотиду матричной ДНК. В противном случае эффективность удлинения праймера во время ПЦР резко снижается и при определенных сочетаниях ошибочно спаренных нуклеотидов может отсутствовать вообще.

Отжиг праймера Амплификация Выщепление метки Амплификат Метод количественной ПЦР в реальном режиме времени Метод использует общие принципы ПЦР. Основное отличие состоит в том, что измеряется количество амплифицированной ДНК в реальном времени после каждого цикла амплификации. Для количественного определения используют два метода — флюоресцентные красители, интеркалирующие в двуцепочечные молекулы ДНК, и модифицированные олигонуклеотиды (ДНК-зонды), которые флюоресцируют после гибридизации с комплементарными участками ДНК. Часто ПЦР в реальном времени комбинируют с ОТ -ПЦР для измерения малых количеств м. РНК, что позволяет исследователю получать количественную информацию о содержании данной м. РНК в клетке и, соответственно, позволяет судить об уровне экспрессии данного гена в отдельной клетке или ткани.

. Метод обратно-транскриптазной ПЦР
 A – контроль (два нормальных аллеля); Б –")
Анализ конформационного полиморфизма однонитевой ДНК (SSCP-анализ) A – контроль (два нормальных аллеля); Б – гетерозиготность по нормальному аллелю и точковой мутации. 1 – исходные двойные цепи ДНК, подвергающиеся денатурации; 2 – разделенные нити ДНК. Черными кружками обозначены нормальные нуклеотиды в комплементарных цепях ДНК, черными треугольниками – мутантные нуклеотиды в тех же сайтах. 3 – короткими стрелками на электрофореграмме указаны нормальные фрагменты ДНК, длинными стрелками — мутантные фрагменты ДНК с измененной электрофоретической подвижностью

Гетеродуплексный анализ ДНК-дуплексы подвергаются электрофорезу в геле с градиентом денатурирующих условий Миграция продолжается до тех пор, пока ДНК-дуплексы не достигают в геле точки плавления и не разделяются, после чего миграция фрагментов останавливается. Однонуклеотидные различия в нормальной и тестируемой ДНК выявляются по различной электрофоретической подвижности в геле.

Метод биочипов В случае некомплементарности - темное пятно При комплементарности – светлое При комплементарности – пятно Иммобилизованный олигонуклеотидный зонд на биочипе
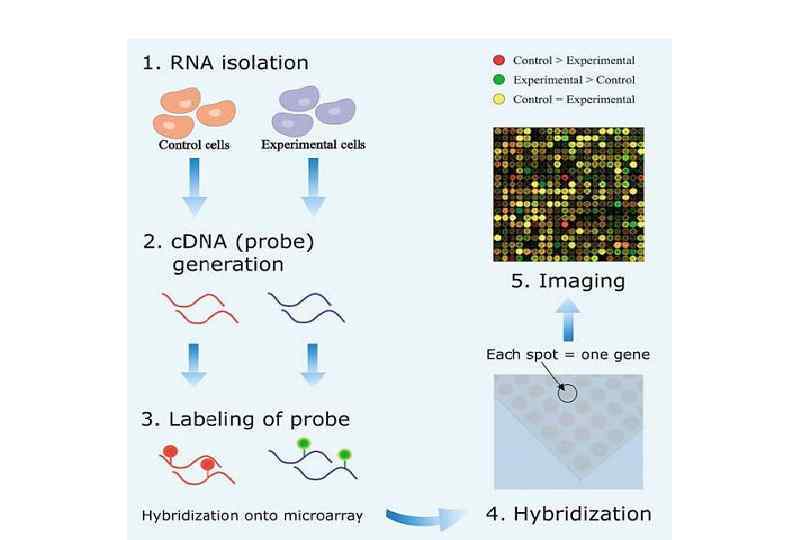

Микрочиповая технология позволяет проводить реакцию в микрообъемах, а, следовательно, требуется незначительное количество материала, и в то же время проводить многосторонний анализ многих чипов одного и того же объекта. • Возможность получения информации не об одном или нескольких, а о многих (тысячах) генов одновременно. • Возможность функциональной характеристики ранее не охарактеризованных генов.

Молекулярно-генетическая характеристика наследственных и генетических заболеваний

Аутосомно-доминантные заболевания • • • Причины: избыточная экспрессия мутантного гена изменение или появление новых функций мутантного белка недостаточность структурных белков генетический фон • Заболевания этого типа характеризуются: • полной или неполной пенетрантностью • варьирующей экспрессивностью • антиципацией (нарастание тяжести заболевания в последующих поколениях)
. Фамильная гиперхолестеринемия.")
Аутосомно-доминантные наследственные заболевания Нейрофиброматоз. Частота 1: 3 500 (1 : 40 000). Фамильная гиперхолестеринемия. Частота 1 : 500. Болезнь Альцгеймера. Частота от 0, 02 % до 10, 6 %. Синдром Марфана. Частота 2 -3 : 10 000 Хорея Гентингтона. Частота 1 : 2 500. Миотоническая дистрофия. Частота 1 : 7 500. Ахондроплазия. Частота от 1 : 26 000 до 1 : 100 000. Множественный склероз. Частота от 1 : 6 000 до 1 : 10 000. Мозжечковая атаксия. Частота 1 : 250 000. Панкреатическая аденокарцинома. Семейный рак молочной железы. Частота до 5 -10 % у жителей Европы. Некоторые виды рака щитовидной железы. Семейный неполипозный рак ободочной кишки. Заболевание Паркинсона. Частота 1 -2 человека на 1 000. Поликистоз почек. Частота от 1 : 1 000 до 1: 1250.
, которые")
Нейрофиброматоз – это наследственное заболевание, при котором в нервной ткани образуются опухоли (нейрофибромы), которые могут быть как доброкачественными, так и злокачественными.

Нейрофибромин представляет собой цитоплазматический белок, состоящий из 2818 аминокислот. Он участвует в инактивации белка RAS и его аналогов, обеспечивая динамический контроль клеточного роста. Ген НФ-1 является одним из основных геновсупрессоров опухолевого роста для примерно половины тканей организма, в первую очередь нейроэктодермального происхождения, пролиферация которых определяется системой белков RAS. Нейрофибромин также влияет на содержание в клетке аденозинмонофосфата (АМФ). АМФ в свою очередь опосредованно тормозит процессы клеточного деления. Ген НФ 1 локализован на длинном плече 17 хромосомы – 17 q 11. 2. Частота заболевания 1 : 3 500 человек.

Основными симптомами НФ 1 являются: наличие множества светло-коричневых пятен на коже (от 5 до 15 мм); наличие нескольких нейрофибром; гиперпигментация; наличие глиомы зрительного нерва; костные аномалии.
 - возникает в результате мутации гена")
Нейрофиброматоз 2 типа (также известный как "центральный нейрофиброматоз") - возникает в результате мутации гена merlin (другое название которого "schwannomin" или NР 2). Ген расположен на хромосоме 22 q 12. Продуктом гена является merlin - цитоскелетный белок, который играет роль супрессора опухолевого роста. Частота заболевания 1 : 40 000.

Основными симптомами НФ 2 являются: • двусторонняя невринома VIII нерва; • наличие нейрофибром; • менингом; • глиом; • шванном.
 – наследственное аутосомно-доминантное нарушение метаболизма липопротеинов, характеризующееся высоким содержанием в плазме")
Семейная гиперхолестеринемия (СГХС) – наследственное аутосомно-доминантное нарушение метаболизма липопротеинов, характеризующееся высоким содержанием в плазме крови липопротеинов низкой плотности (ЛНП), наличием кожных и сухожильных ксантом и высоким риском раннего развития ИБС.

Семейная гиперхолестеринемия может быть обусловлена мутациями нескольких генов: 1. Гена рецептора ЛНП – LDLR. Локус LDLR находится на 19 хромосоме p 13. 1– 13. 3. Ген содержит около 45 т. п. н. , 18 кодирующих областей (экзонов) и 17 некодирующих областей (интронов). В настоящее время в мире известно более 1000 мутаций в гене ЛНП, приводящих к развитию семейной гиперхолестеринемии. 2. Гена аполипопротеина В-100 – APOB. Ген APOB локализован на 2 хромосоме, его длина 43 т. п. н. , содержит 29 экзонов и 28 интронов. Описаны всего четыре мутации гена APOB вызывающие семейную гиперхолестеринемию. Все эти мутации расположены в одном 26 экзоне гена, что значительно упрощает диагностику данных мутаций. Мутации гена АРОВ встречаются у 1 -6% пациентов с клиническим диагнозом СГХС.

Идентифицировано 4 разных класса мутаций рецептора ЛПНП. Вследствие этих мутаций нарушается синтез, транспорт, связывание и кластеризация ЛПНП в клетке. Мутации 1 -го класса делают соответствующие аллели "недействительными", что выражается в полном отсутствии рецепторов. При наличии мутантных аллелей 2 -го класса нарушается транспорт любых синтезируемых рецепторов ЛПНП. Мутантные аллели 3 -го класса вызывают образование функционально дефектных рецепторов, способных связывать ЛПНП. При наличии мутаций в аллелях 4 -го класса формируются рецепторы, но они не могут собираться в группы (кластеризуются) в окаймленных ямках, а это препятствует их "втягиванию" в клетку (интернализация) после связывания ЛПНП.

Синдром Марфана Наследственная болезнь соединительной ткани, вызванная мутацией гена, кодирующего структуру белка фибриллина.

Известные люди с синдромом Марфана Ш. де Голль Эхнатон Н. Паганини А. Линкольн

Это наследственное заболевание соединительной ткани , проявляющееся изменениями скелета: высоким ростом с относительно коротким туловищем , длинными паукообразными пальцами (арахнодактилия), разболтанностью суставов, часто сколиозом, кифозом , деформациями грудной клетки, Арахнодактилия аркообразным небом. Характерны также поражения глаз. В связи с аномалиями сердечно-сосудистой системы средняя продолжительность жизни сокращена.

Высокий выброс адреналина , характерный для заболевания, способствует не только развитию сердечно-сосудистых осложнений, но и появлению у некоторых лиц особой силы духа и умственной одаренности. Способы лечения неизвестны. Считают, что ею болели Паганини, Андерсен, Чуковский.
 – хроническое прогрессирующее дегенеративное заболевание ЦНС, что клинически проявляется нарушением произвольных")
Болезнь Паркинсона (БП) – хроническое прогрессирующее дегенеративное заболевание ЦНС, что клинически проявляется нарушением произвольных движений. Частота заболевания 2 -3 случаев на 1000 человек, а у пожилых людей от 1 % до 3 % и более.

Идиопатическая болезнь Паркинсона, как и другие нейродегенеративные заболевания, например болезнь Альцгеймера, относится к протеинопатиям, т. е. болезням с патологическим накоплением белка α-синуклеина. Мутации гена, кодирующего этот белок, были обнаружены у больных аутосомно-доминантной формой болезни Паркинсона. При данном заболевании альфа-синуклеин откладывается в нейронах в виде телец Леви.

В странах Европы и Северной Америки болезнь Паркинсона поражает в среднем 100— 200 человек на 100 000 населения. В Азии и Африке это заболевание встречается реже. Источник: http: //www. medn. ru/statyi/Boleznparkinsona. html

Хорея Гентингтона • Одно из тяжелейших и постоянно прогрессирующих наследственных заболеваний нервной системы, обусловленной системной дегенерацией экстрапирамидных двигательных структур и коры головного мозга. • Тип наследования: аутосомно-доминантный с высокой пенетрантностью мутантного гена (хромосома 4). Это заболевание “тринуклеотидной экспансии”. Заболевание вызывается увеличением числа кодонов ЦАГ в гене IT-15. Этот ген кодирует 350 -k. Da белок хантингтин.
, присутствующий у всех людей, кодирует белок хантингтин (Htt). Ген HTT расположен")
Ген хантингтин (HTT), присутствующий у всех людей, кодирует белок хантингтин (Htt). Ген HTT расположен на коротком плече 4 -й хромосомы (4 p 16. 3). Этот ген включает последовательности трёх азотистых оснований — цитозин-аденин-гуанин, которые повторяются множество раз (т. е. . ЦАГЦАГЦАГ. . . ) и известны как тринуклеотидные повторы. Триплет ЦАГ кодирует аминокислоту глутамин, поэтому синтезируемый белок хантингтин состоит из цепочки глутаминовых остатков, называемых полиглутаминовый тракт.
 и гене")
Семейный рак молочной железы Причина – мутация в гене BRCA-1 (17 q-хромосома) и гене BRCA-2 (13 q-хромосома). Сложность диагностики заключается в том, что заболевание возникает при различной локализации мутации в гене, нет «горячих точек» . Для гена BRCA-2 уже описано 150 мутаций, а для BRCA-1 – более 300. Только у евреев «ашкенази» обнаружены горячие зоны, что говорит о том, что мутация «гуляет» внутри популяции людей этой национальности. Частота в мутации в популяции женщин 1/800, из которых 85 % заболевает до 70 лет.

Аутосомно-рецессивные заболевания Серповидноклеточная анемия 1 : 625 Муковисцидоз 1 : 2 500 Болезнь Тея-Сакса 1 : 5 000 Фенилкетонурия (ср. 1 : 8 000) от 1 : 2 600 до 1 : 199 000 Мукополисахаридозы 1 : 25 000 Галактоземия 1 : 57 000 Проксимальная спинальная амниотрофия 1 : 3 000 Адреногенитальный синдром 1 : 12 000 Нарушение обмена моно- и дисахаридов Талассемия и др.

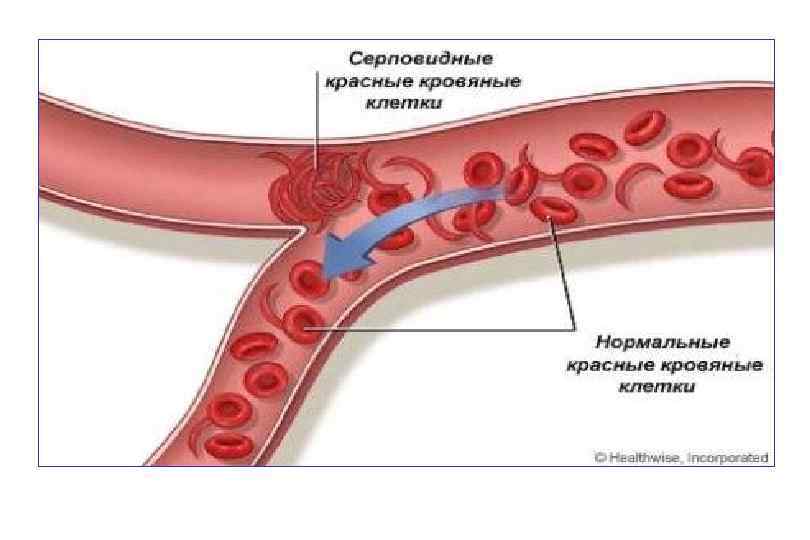
Серповидноклеточная анемия Серповидно-клеточная анемия это аутосомно-рецессивное заболевание крови, характеризующееся присутствием неправильных красных кровяных клеток (эритроцитов), устойчивой, серповидной формы.

Серповидность клеток уменьшает их гибкость и эластичность, что увеличивает риск возникновения различных осложнений. Причиной появления клеток серповидной формы являются мутации в гене глобинового белка. Как следствие сокращается ожидаемая продолжительность жизни, в среднем она составляет 42 года у мужчин и 48 у женщин.
 — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным")
Фенилкетонурия (фенилпировиноградная олигофрения) — наследственное заболевание группы ферментопатий, связанное с нарушением метаболизма аминокислот, главным образом фенилаланина. Сопровождается накоплением фенилаланина и его токсических продуктов, что приводит к тяжёлому поражению ЦНС, проявляющемуся, в частности, в виде нарушения умственного развития.

Фенилаланингидроксилаза катализирует реакцию преобразования фенилаланин + О 2 + тетрагидробиоптерин ↓ тирозин + вода + окисленный биоптерин Ген, кодирующий ФАГ человека, расположен на участке q 22 -24 12 ой хромосомы и имеет размер более 100 т. п. н. Он включает 13 экзонов и сложную 5΄-нетранслируемую область, содержащую трансактивируемые регуляторные цис-элементы. В настоящее время в гене идентифицировано более 400 мутаций, связанных с возникновением заболевания.

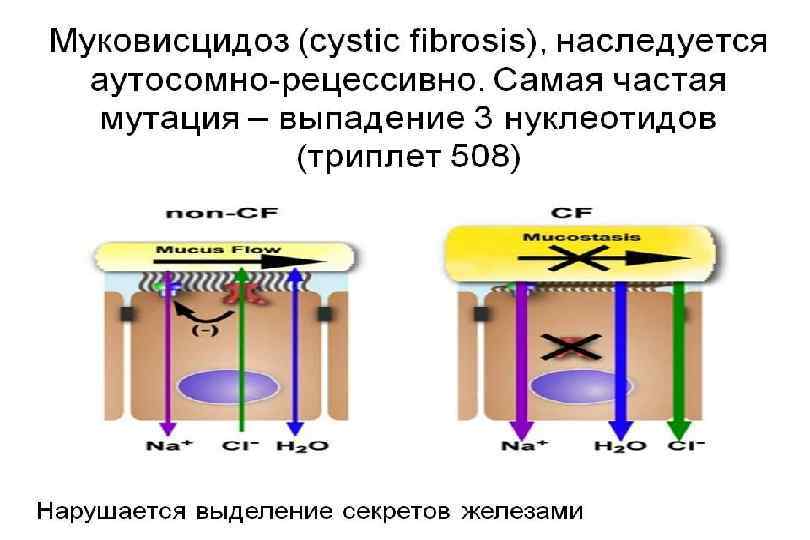
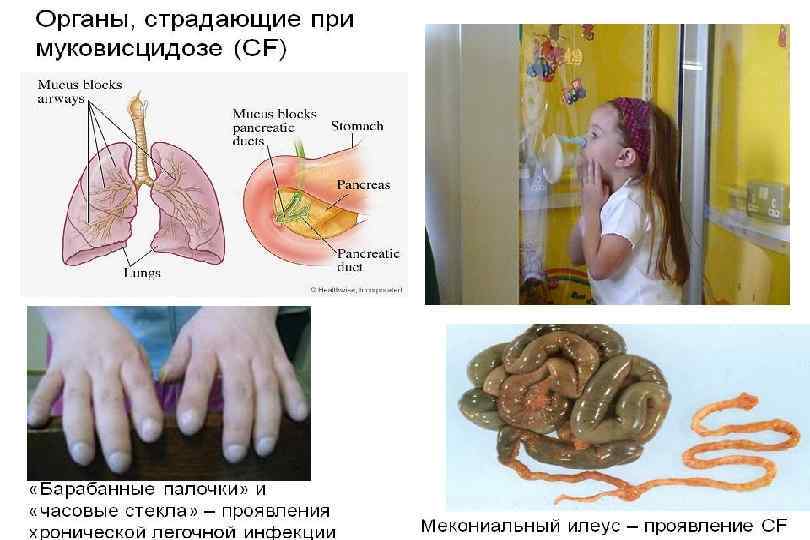
Муковисцидоз Заболевание, при котором поражаются экзокринные железы. Наследуется по аутосомно-рецессивному типу. Системное наследственное заболевание, обусловленное мутацией гена трансмембранного регулятора муковисцидоза и характеризующееся поражением желёз внешней секреции, тяжёлыми нарушениями функций органов дыхания и желудочно-кишечного тракта.

Прогерия детей – синдром Хатчинсона-Гилфорда § § § § Маленький рост Птичье лицо с клювообразным профилем Преобладание мозгового черепа над лицевым Выпадение бровей, ресниц Продожительность жизни -13 лет Причина: нарушение репарации, Сшивки ДНК
 семейная")
Синдром Альцгеймера В настоящее время известно две формы болезни Альцгеймера: 1. ранняя (пресинильная) семейная форма заболевания, возникающая до 60 -65 лет; 2. поздняя форма (спорадическая) (сенильная) возникает после 60 -65 лет.

Характеристика заболевания: 1. 2. 3. 4. Более часто возникает в пожилом возрасте. Как правило, имеет летальный исход. Заболевание развивается в течение 10 лет. На начальных этапах наблюдается потеря памяти. Затем начинают постепенно проявляться и другие симптомы деменции. 5. На поздних этапах полная потеря памяти.

Нейропатология и биохимия заболевания Заболевание характеризуется нарушением передачи нервных импульсов и функционирования синапсов в коре головного мозга и подкорковой области. Это приводит к дегенерационным процессам в височной области и передней доле головного мозга. Редукционные процессы в этих областях приводят к нарушению когнитивных процессов.

Образование β -амилоидного белка γ-секретаза β-секретаза Клеточная мембрана Амилоидный предшественник АРР βамилоид Бляшка
 b-секретазаразрезает APP белко, давая: (2)")
b-секретазный путь: APP белок: b a g g (1) b-секретазаразрезает APP белко, давая: (2) g-секретаза разрезает этот конец, давая: Ab 40 Фрагмент Растворимый или Ab 42 Фрасгент Нерастворимый, аггрегирущийся в бляшки

Генетика болезни Альцгеймера

К настоящему времени обнаружено, по крайней мере, четыре гена, мутации в которых вызывают БА или являются факторами генетической предрасположенности: ген белка амилоидного предшественника (АРР) на хромосоме 21. Ген аполипопротеина Е (Аро Е) на хромосоме 19. Ген пресенилина 1 на хромосоме 14. Ген пресенилина 2 на хромосоме 1.

Полигенные заболевания

Полигенные заболевания человека обусловлены взаимодействием определенных комбинаций аллелей разных локусов и внешних факторов. Не наследуются по законам Г. Менделя. Являются многофакторными. • Это наиболее распространенные болезни: ревматизм, врожденные пороки сердца, ишемическая болезнь сердца, гипертоническая и язвенная болезни, цирроз печени, сахарный диабет, бронхиальная астма, псориаз, шизофрения , некоторые формы онкологических заболеваний и др. • Так, шизофренией болеют около 1% населения, сахарным диабетом — 5%, аллергическими заболеваниями — более 10%, гипертонией — около 30%.

Три степени генетической предрасположенности • слабая (болезнь, обычно, не развивается, благодаря способности организма поддерживать гомеостаз) • умеренная • сильная В двух последних случаях появляется заболевание, выраженное в большей или меньшей степени

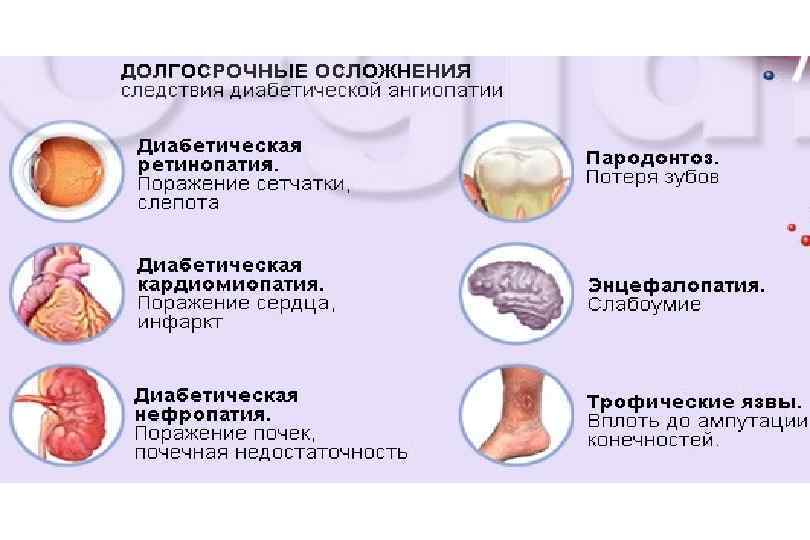
Сахарный диабет

В МИРЕ:

Классификация сахарного диабета Сахарный диабет 1 -го типа Сахарный диабет 2 -го типа Сахарный диабет 3 -го типа (Mody. Другие типы диабета при: тип) Гестационный сахарный диабет – диабет беременных
 Возраст Юношеский возраст После 40 лет Юношеский возраст Лечение")
Сахарный диабет Тип III (MODY-тип) Возраст Юношеский возраст После 40 лет Юношеский возраст Лечение Требует инсулинотерапии Обычно контролируется медикаментами Не всегда контролируется медикаментами Сопутствующие симптомы Не связан с ожирением Связан с ожирением Не связан с ожирением Генетическая компонента Наследуется Связь с HLAсистемой Связан с HLA генами Не связан с HLA генами
 6")
Генетическая основа сахарного диабета I типа Хромосома Гены Хромосома 6 (локус IDDM 1) 6 р21. 3 Гены главного комплекса гистосовместимости: Гуппа В: HLA-В 8, В 15 Группа D: HLA-DP, DR, DQ (главная роль в формировании иммунного ответа) HLA-DQA 1 и DQB 1 (наиболее существенный, частота встречаемости у больных ИЗД – 95%) HLA- DR 3 и DR 4 (частота встречаемости у больных 95%). Наследование предрасположенности связано гаплотипами: HLA-DRВ 1, DR 3, DR 4, DQA 1, DQB 1 и HLA-DPВ 1. Хромосома 11 (локус IDDM 2) 11 р15. 5 Хромосома 11 (локус IDDM 4) Ген INS - синтеза инсулина. На 5’-концевой части гена имеются тандемные VNTR-повторы (14 нуклеотидов), повторяющиеся от 26 до 200 раз. Ген FGF 3, кодирующий синтез фактора роста фибробластов (7% риска). Хромосома 7 Ген Ian 5. Мутация в гене приводит к тому, что иммунная система уничтожает В-клетки.
 2 q 33 Обнаружены гены предрасположенности. Риск")
Хромосома Ген Хромосома 2 (локус IDDM 12) 2 q 33 Обнаружены гены предрасположенности. Риск заболевания 10, 7%. Локус IDDM 12 определяет предрасположенность не только к сахарному диабету, но также диффузно-токсическому зобу, аутоиммунному тиреоидиту. Хромосома 18 (локус IDDM 6) 18 q 21 Ген TCFL 2, кодирующий синтез транскрипционного фактора, контролирующего экспрессию протоглюкагона. Ген DCC. Продукт этого гена участвует в апоптозе. Известно, что нарушения процесса апоптоза могут приводить к развитию аутоиммунных заболеваний. Хромосома 14 (локус IDDM 16) 14 q 32 Ген, кодирующий синтез тяжелой цепи иммуноглобулина. Хромосома 3 (локус. IDDM 9) 3 q 21 - q 25 Обнаружены гены предрасположенности. Хромосома 10 (локус IDDM 10) Обнаружены гены предрасположенности.

Сахарный диабет II типа • Группа генетически обусловленных гетерогенных метаболических нарушений , которые вызывают аномалии в усвоении глюкозы: – Основная причина - развитие инсулинрезистентности. Поэтому этот тип диабета называют инсулиннезависимым. Кроме того, возможно уменьшение числа панктеатических β-клеток или ослабленная секреция инсулина. • ~90% больных диабетом имеют диабет II типа. • Лечение: – Помогает диета, лекарственные препараты, физическая активность. – Период манифестации признаков может быть очень длительный. • Полигенная и мультифактораная природа заболевания: – Связана с нарушением функционирования и взаимодействия большого числа генов. – Наряду с генетической компонентой велико влияние внешних факторов и образа жизни.

Уровень Blood glucose глюкозы в крови levels Диабет II типа Действие генов Секреция инсулина и Резистентность к инсулину Факторы внешней среды Уровень жирных кислот acid levels

Что известно о генах, связанных с возникновением диабета II типа ? • Хромосома 2 q 37. 3. Ген, кодирующий внутриклеточную Cа-зависимую цитоплазматическую протеазу, активность которой контролируется убиквитином. • Хромосома 11 p 15. 1. Ген, кодирующий ATФ-связывающая кассета субсемейства С. • Хромосома 11 p 15. 1. Калиевый канал субсемейства J. Хромосома 10 q 25. Транскрипционный фактор 7. ● • Хромосома 3 p 25. Пероксисомный пролифератор, активируемый рецептором-ϒ. Ответственен за 25 % диабета II типа.

Недавно установлено, что ген SUR кодирует синтез рецептора сульфанилмочевины. Сульфанилмочевина входит в состав комплекса, который стимулирует секрецию инсулина. Этот ген – кандидат на индукцию гипергликемии и избыточного веса. Дефект гена в 22 экзоне (кодон 761) приводит к ожирению в 8 % случаев.
 • • Составляет 5 % от всех диабетов. Вызывается")
Диабет III тип (MODY –тип) • • Составляет 5 % от всех диабетов. Вызывается повреждение одного гена: – имеет аутосомно-доминантный тип наследования; – мутация генерирует множественные эффекты. • Возникает в молодом возрасте(< 25 лет). • Характеризуется отсутствием ожирения, образования кетоновых телец и изменений аутоиммунной системы. • Гипергликемия корректируется диетой.
. Описано три формы")
Типы сахарного диабета III типа • Саханрый диабет III типа (MODY-тип). Описано три формы – MODY 1 (связана с хромосомой 20, дефект синтеза инсулина, а также мутация в гене, контролирующем синтез транскрипционного фактора гепатоцитов HNF 4), приводит к дефекту синтеза и секреции инсулина. • MODY-2 – мутация в гене глюкокиназы (7 хромосома, наследуется по аутосомнодоминантному типу), приводит к хронической гипергликемии. • MODY-3 – мутация в гене транскрипционного фактора гепатоцитов HNF 1α (12 хромосома), приводит к дефекту секреции инсулина.

Резюме • Все гены, ассоциированные с заболеванием диабета III-типа экспрессируются в клетках поджелудочной железы и играют роль в: – Метаболизме глюкозы. – Являются регуляторами инсулиновых генов, а также других генов, участвующих в транспорте глюкозы.

Диагностика • Исследование крови: если глюкоза плазмы натощак: • менее 6, 1 ммоль/л - гипергликемии нет • в интервале от 6, 1 до 7, 0 ммоль/л - нарушенная гликемия натощак • показатель свыше 7, 0 ммоль/л может служить основанием для постановки предварительного диагноза сахарного диабета, но с дальнейшим обязательным подтверждением.

Острые осложнения • Гипогликемия — снижение уровня глюкозы в крови ниже нормального значения (обычно ниже 3, 3 ммоль/л).

Бронхиальная астма Хроническое воспалительное заболевание дыхательных путей, которое характеризуется обратимой обструкцией и гиперреактивностью бронхов.
. Распространенность астмы")
Эпидемиология астмы • Наиболее распостарненным типом астмы является атопическая астма (аллергическая астма). Распространенность астмы среди детей и взрослых составляет 10 -15% соответственно. Аспирин вызывает аллергию у 10 % астматиков. • Несмотря на оптимизацию подходов к лечению больных астмой, смертность от этого заболевания не снижается. • Причины повсеместного возрастания числа больных /смертности остаются не установленными.

Нормальный просвет бронха Хроническая астма Бронхоспазм Воспаление и отек Эпителий Сокращение Гладкомышечные гладко-мышечных клетки клеток Альвеолярные перегородки Секреция слизи, экссудаци я плазмы Повреждение/слущиван ие эпителия

Развитие астмы происходит с участием Ig. E- механизма. Аллергены, попадая с воздухом в дыхательные пути, взаимодействуют с дендритными клетками слизистой бронхов. Этот комплекс связывается с рецепторами CD 4 Тклеток, что стимулирует дифференцировку Т-лимфоцитов в форму Th 2, которые начинают секретировать IL-10 и цитокины, гены которых расположены на хромосоме 5, а функция связана с гуморальным ответом. Th 2 -лимфоциты высвобождают также IL-4 и IL-13, которые взаимодействуют со своими рецепторами на В-клетках, активируют образование Ig. E-типа иммуноглобулина. Ig. E связывается с тучными клетками, что индуцирует образование медиаторов воспаления и хемокинов: гистамина, простагландинов, фактора активации тромбоцитов, протеаз и др. Действуя в совокупности эти факторы приводят к изменениям сосудов стенок дыхательной системы, сокращению гладкой мускулатуры бронхов, гиперсекреции слизи. Цитокины связаны с индукцией воспалительного процесса.

Факторы среды, провоцирующие бронхиальную астму
 картирован на")
Генетика астмы ●Ген, кодирующий β-цепь высокоаффинного рецептора для Ig. E (FcεR 1β) картирован на длинном плече 11 хромосомы -11 q 13. ●Мутация в этом гене повышает активность Fc рецептора, потенцируя высвобождение тучными клетками IL 4, что в свою очередь, стимулирует Th 2 - тип реагирования и гиперпродукцию Ig. E. ●Именно с этой мутацией связывают атопию, передающуюся по материнской линии.

Ключевую роль в определении специфичности иммунного ответа отводят Ir-генам, находящимся в ассоциативной связи с некоторыми аллелями II класса главного комплекса гистосовместимости – DR, DQ, DP (6 q 21, 3), а также гену (или генам) длинного плеча 14 хромосомы (14 q), кодирующему α цепь Т-клеточного рецептора (TCR).
 идентифицирован кластер цитокиновых генов, играющих решающую")
На длинном плече 5 хромосомы (5 q 31) идентифицирован кластер цитокиновых генов, играющих решающую роль в формировании аллергического воспаления: ●IL 4 – ответственный за наследование высокого базального уровня Ig. E. ●IL 13 – эффекты совпадают с IL 4. ●IL 5 – активирует эозинофилы. ●IRF 1 – фактор регуляции интерферона. ●CSF 2 – ген, кодирующий гранулоцитарно-моноцитарный колониестимулирующий фактор. ●IL 3 - активатор пролиферации воспалительных клеток.

В области генома 5 q 32 -q 33, кроме кластера генов IL 4, расположены: ● ген IL 9, активирующего тучные клетки. ● ген, кодирующий β 2 -адренорецепторы. ● гены, осуществляющие контроль тонуса гладкой мускулатуры бронхов.

Описаны три варианта мутации гена, кодирующего β 2 -адренорецепторы, которые приводят к снижению регуляторной функции адренорецепторов и повышению чувствительности бронхов к метахолину, что ассоциируется с тяжелой неаллергической астмой и частыми ночными приступами удушья.

На длинном плече 11 хромосомы аллель 168 в локусе D 11 S 527 ассоциирован с гиперреактивностью бронхов. Гены (как иммунных, так и неиммунных механизмов) расположены в разных хромосомах и ПЕРЕДАЮТСЯ ПО НАСЛЕДСТВУ НЕЗАВИСИМО ДРУГ ОТ ДРУГА. РЕБЕНОК ПОЛУЧАЕТ РАЗЛИЧНЫЙ НАБОР ГЕНОВ ИЗ ГЕНОТИПОВ РОДИТЕЛЕЙ (ИММУННЫХ И НЕИММУННЫХ МЕХАНИЗМОВ), ЧТО И ОПРЕДЕЛЯЕТ ГЕТЕРОГЕННОСТЬ ФОРМИРОВАНИЯ И ТЕЧЕНИЯ БРОНХИАЛЬНОЙ АСТМЫ.
Лекция для магистров.ppt