

23fbca14ad9393355f9ec586cc2d8934.ppt
- Количество слайдов: 27
 Usher syndrome and other retinal dystrophy-hearing impairment associations Hussein Morfeq, MD King Abdulaziz University Jeddah, Saudi Arabia
Usher syndrome and other retinal dystrophy-hearing impairment associations Hussein Morfeq, MD King Abdulaziz University Jeddah, Saudi Arabia
 Usher syndrome v British ophthalmologist Charles Usher: who in 1914 wrote a paper describing several cases in which the link between congenital deafness and retinitis pigmentosa. Other names for Usher syndrome include : Hallgren syndrome Usher-Hallgren syndrome rp-dysacusis syndrome dystrophia retinae dysacusis syndrome.
Usher syndrome v British ophthalmologist Charles Usher: who in 1914 wrote a paper describing several cases in which the link between congenital deafness and retinitis pigmentosa. Other names for Usher syndrome include : Hallgren syndrome Usher-Hallgren syndrome rp-dysacusis syndrome dystrophia retinae dysacusis syndrome.
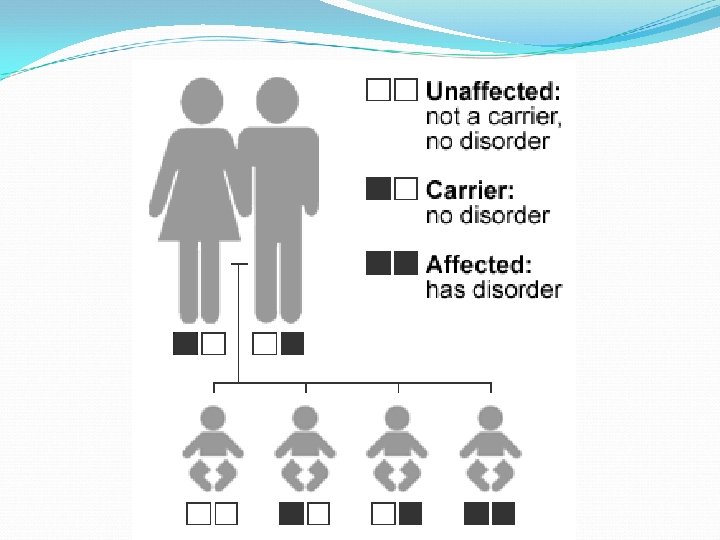
 Usher syndrome = progressive recessive retinitis pigmentosa + congenital moderate-to-severe SN hearing loss. It is inherited in an autosomal recessive pattern and is estimated to occur in 1 -3 in 10, 000 people. The genes also play a role in the development and stability of the retina by influencing the structure and function of both the rod photoreceptor cells and supporting cells called the retinal pigmented epithelium. Responsible for up to 10% of cases of congenital deafness. and accounts for about 50% of the deaf blind in the United States. it represents the major cause of syndromic deafness with blindness.
Usher syndrome = progressive recessive retinitis pigmentosa + congenital moderate-to-severe SN hearing loss. It is inherited in an autosomal recessive pattern and is estimated to occur in 1 -3 in 10, 000 people. The genes also play a role in the development and stability of the retina by influencing the structure and function of both the rod photoreceptor cells and supporting cells called the retinal pigmented epithelium. Responsible for up to 10% of cases of congenital deafness. and accounts for about 50% of the deaf blind in the United States. it represents the major cause of syndromic deafness with blindness.
 Usher syndrome is divided into three types: I, II and III. In the United States, types 1 and 2 are the most common types Children with type I syndrome are born profoundly deaf, and eyesight usually begins degrading after the first decade of life, beginning with night-blindness. They also experience degrading tunnel vision. Many use Sign language. When vision loss is severe or when one is blind one must use tactile signing. Problems with balance are present in people with Usher I and sometimes Usher III. Night vision loss begins first blind spots develop in the side (peripheral) vision doughnut shape tunnel vision Central vision is reduced and blurs Cataracts may develop in the teenage years or in adulthood.
Usher syndrome is divided into three types: I, II and III. In the United States, types 1 and 2 are the most common types Children with type I syndrome are born profoundly deaf, and eyesight usually begins degrading after the first decade of life, beginning with night-blindness. They also experience degrading tunnel vision. Many use Sign language. When vision loss is severe or when one is blind one must use tactile signing. Problems with balance are present in people with Usher I and sometimes Usher III. Night vision loss begins first blind spots develop in the side (peripheral) vision doughnut shape tunnel vision Central vision is reduced and blurs Cataracts may develop in the teenage years or in adulthood.
 Type II children are hearing impairment with stable hearing throughout their lives. Changes in sight in type II cases usually begin later, sometimes only becoming noticeable after the second decade of life. In the type III syndrome, hearing loss as well as retinitis pigmentosa can occur later in life. Hearing loss in Usher III is progressive.
Type II children are hearing impairment with stable hearing throughout their lives. Changes in sight in type II cases usually begin later, sometimes only becoming noticeable after the second decade of life. In the type III syndrome, hearing loss as well as retinitis pigmentosa can occur later in life. Hearing loss in Usher III is progressive.
 Type") Type 1: 3 to 6 per 100, 000 Type 2 (50% most common) Type 3 (Finland) Hearing Profound deafness in both Moderate to severe ears from birth hearing loss from birth Normal at birth; progressive loss in childhood or early teens Vision Decreased night vision before age 10 Decreased night vision Varies in severity; night begins in late childhood vision problems often begin or teens in teens Balance problems from birth Normal Vestibular function (balance) Normal to near-normal, chance of later problems
Type 1: 3 to 6 per 100, 000 Type 2 (50% most common) Type 3 (Finland) Hearing Profound deafness in both Moderate to severe ears from birth hearing loss from birth Normal at birth; progressive loss in childhood or early teens Vision Decreased night vision before age 10 Decreased night vision Varies in severity; night begins in late childhood vision problems often begin or teens in teens Balance problems from birth Normal Vestibular function (balance) Normal to near-normal, chance of later problems
 Type 1 Usher syndrome: MY 07 A, USH 1 C, CDH 23, PCDH 15, SANS Type 2 Usher syndrome: USH 2 A(Usherin protein), VLGR 1, WHRN Type 3 Usher syndrome: USH 3 A
Type 1 Usher syndrome: MY 07 A, USH 1 C, CDH 23, PCDH 15, SANS Type 2 Usher syndrome: USH 2 A(Usherin protein), VLGR 1, WHRN Type 3 Usher syndrome: USH 3 A
. Difficulty in adapting to bright light") symptoms Rods involvement: Night Blindness (1 st symptoms). Difficulty in adapting to bright light or rapidly changing light conditions. Tunnel Vision: Rods dysfunctioning in the periphery. Cones involvement: Early: "doughnut vision“. Late: Loss of central acuity: early degeneration of the cone cells in the macula. 50% of individuals develop complete blindness before age 50 years. Imbalance: Vestibulocerebellar ataxia (stereocilia)
symptoms Rods involvement: Night Blindness (1 st symptoms). Difficulty in adapting to bright light or rapidly changing light conditions. Tunnel Vision: Rods dysfunctioning in the periphery. Cones involvement: Early: "doughnut vision“. Late: Loss of central acuity: early degeneration of the cone cells in the macula. 50% of individuals develop complete blindness before age 50 years. Imbalance: Vestibulocerebellar ataxia (stereocilia)
 perimetry") Evaluation: VA: 20/20 to LP Fundus exam: 'bone corpuscle' lumps. VF Goldmann (kinetic) perimetry : most useful measure for ongoing follow-up care of patients with RP. Midperipheral scotomas ring scotoma tunnel vision IVFG: cystoid macular edema Optical coherence tomography (OCT): cystoid macular edema Electroretinography (the most crucial): measures the electrical responses of various cell types in the retina, including the photoreceptors (rods and cones), inner retinal cells (bipolar and amacrine cells), and the ganglion cells. Electrooculography: Central macular changes, normal ERG findings, and abnormal EOG findings suggest Best vitelliform macular dystrophy. Color testing: Mild blue-yellow axis color defects are common Audiometric tests : sensorineural hearing impairment Genetic testing: chromosomal mutations. the most common locus for syndromic RP
Evaluation: VA: 20/20 to LP Fundus exam: 'bone corpuscle' lumps. VF Goldmann (kinetic) perimetry : most useful measure for ongoing follow-up care of patients with RP. Midperipheral scotomas ring scotoma tunnel vision IVFG: cystoid macular edema Optical coherence tomography (OCT): cystoid macular edema Electroretinography (the most crucial): measures the electrical responses of various cell types in the retina, including the photoreceptors (rods and cones), inner retinal cells (bipolar and amacrine cells), and the ganglion cells. Electrooculography: Central macular changes, normal ERG findings, and abnormal EOG findings suggest Best vitelliform macular dystrophy. Color testing: Mild blue-yellow axis color defects are common Audiometric tests : sensorineural hearing impairment Genetic testing: chromosomal mutations. the most common locus for syndromic RP
 Evaluation: Inherited/syndromic disease lab tests Refsum disease - Serum phytanic acid in the presence of other neurologic abnormalities Gyrate atrophy - Ornithine levels Kearns-Sayre syndrome - ECG to help rule out heart block Abetalipoproteinemia - Lipid profile with possible protein electrophoresis
Evaluation: Inherited/syndromic disease lab tests Refsum disease - Serum phytanic acid in the presence of other neurologic abnormalities Gyrate atrophy - Ornithine levels Kearns-Sayre syndrome - ECG to help rule out heart block Abetalipoproteinemia - Lipid profile with possible protein electrophoresis
 Differential diagnosis Alport syndrome: nephropathy, sensorineural hearing loss, myopia, cataract, retinal detachment Alstrom syndrome: atypical retinal pigmentary degeneration, sensorineural hearing loss, obesity, diabetes mellitus, nephropathy, acanthosis nigricans Bardet-Biedl syndrome: retinitis pigmentosa, hypogenitalism, polysyndactyly, mental retardation, obesity, sensorineural deafness Cockayne syndrome: cachectic dwarfism, pigmenta ry retinal degeneration, optic atrophy, sensorineural hearing loss, characteristic faces, mental retardation spondyloepiphyseal dysplasia congenita: dwarfism, mild deafness, cleft palate, congenital high myopia with associated retinal degeneration, cataracts, glaucoma
Differential diagnosis Alport syndrome: nephropathy, sensorineural hearing loss, myopia, cataract, retinal detachment Alstrom syndrome: atypical retinal pigmentary degeneration, sensorineural hearing loss, obesity, diabetes mellitus, nephropathy, acanthosis nigricans Bardet-Biedl syndrome: retinitis pigmentosa, hypogenitalism, polysyndactyly, mental retardation, obesity, sensorineural deafness Cockayne syndrome: cachectic dwarfism, pigmenta ry retinal degeneration, optic atrophy, sensorineural hearing loss, characteristic faces, mental retardation spondyloepiphyseal dysplasia congenita: dwarfism, mild deafness, cleft palate, congenital high myopia with associated retinal degeneration, cataracts, glaucoma
 Differential diagnosis Flynn-Aird syndrome: severe myopia, atypical retinitis pigmentosa, sensorineural hearing loss, shooting pain, joint stiffness, muscular wasting kyphoscoliosis, skin atrophy, baldness, cystic bone changes Friedreich ataxia: spinocerebellar degeneration, limb incoordination, nerve deafness, retinal degeneration, optic atrophy Hurler syndrome (MPS-1): coarse facial features, short stature, progressive mental deterioration, corneal clouding, pigmentary retinopathy, optic atrophy Kearns-Sayre syndrome (CPEO): progressive external ophthalmoplegia, retinitis pigmentosa, heart block, sensorineural hearing loss Norrie syndrome: bilateral congenital retinal detachment, sensorineural hearing loss, mental retardation.
Differential diagnosis Flynn-Aird syndrome: severe myopia, atypical retinitis pigmentosa, sensorineural hearing loss, shooting pain, joint stiffness, muscular wasting kyphoscoliosis, skin atrophy, baldness, cystic bone changes Friedreich ataxia: spinocerebellar degeneration, limb incoordination, nerve deafness, retinal degeneration, optic atrophy Hurler syndrome (MPS-1): coarse facial features, short stature, progressive mental deterioration, corneal clouding, pigmentary retinopathy, optic atrophy Kearns-Sayre syndrome (CPEO): progressive external ophthalmoplegia, retinitis pigmentosa, heart block, sensorineural hearing loss Norrie syndrome: bilateral congenital retinal detachment, sensorineural hearing loss, mental retardation.
: marble bone and spontaneous fractures, macrocephaly, optic atrophy, heptosplenomegaly,") Differential diagnosis Osteopetrosis (Albers-Schonberg disease): marble bone and spontaneous fractures, macrocephaly, optic atrophy, heptosplenomegaly, anemia, retinal degeneration, conductive hearing loss Refsum's disease (phytanic acid storage disease): dystopia canthorum, large root of nose, confluent eyebrows, heterochromia irides, sensorineural hearing loss, poliosis, pigment disturbance of RPE normal to subnormal ERG Zellweger syndrome (cerebro-hepato-renal syndrome): neonatal hypotonia, severe neurodevelopmental delay, hepatomegaly, renal cysts, sensorineural deafness, retinal dysfunction (abnormal ERG), facial dysmorphism
Differential diagnosis Osteopetrosis (Albers-Schonberg disease): marble bone and spontaneous fractures, macrocephaly, optic atrophy, heptosplenomegaly, anemia, retinal degeneration, conductive hearing loss Refsum's disease (phytanic acid storage disease): dystopia canthorum, large root of nose, confluent eyebrows, heterochromia irides, sensorineural hearing loss, poliosis, pigment disturbance of RPE normal to subnormal ERG Zellweger syndrome (cerebro-hepato-renal syndrome): neonatal hypotonia, severe neurodevelopmental delay, hepatomegaly, renal cysts, sensorineural deafness, retinal dysfunction (abnormal ERG), facial dysmorphism
 Acetazolamide Calcium channel blockers Lutein/zeaxanthin Medications with potential") Treatment Vitamin A/beta-carotene Docosahexaenoic acid (DHA) Acetazolamide Calcium channel blockers Lutein/zeaxanthin Medications with potential adverse effects in RP: Isotretinoin (Accutane)- Sildenafil (Viagra)- Vitamin E Surgery: - cataract extraction: Bastek et al studied 30 patients with RP; 83% of them improved by 2 lines on the Snellen visual acuity chart with cataract surgery. Perioperative use of corticosteroids is recommended to prevent postoperative cystoid macular edema. - Ciliary neurotrophic factor (CNTF) has been shown to slow retinal degeneration in a number of animal models. - RPE cell transplants - retinal prosthesis or phototransducing chip - Gene therapy to replace the defective protein by using DNA vector the ophthalmologist may act as the consultant to an internist. Audiology consultation Annual patient examinations usually are sufficient to measure Goldmann visual field and visual acuity. If medical treatment is initiated, more frequent visits and laboratory blood work may be indicated. Patients with systemic conditions that are associated with retinitis pigmentosa (RP) may require closer follow-up care.
Treatment Vitamin A/beta-carotene Docosahexaenoic acid (DHA) Acetazolamide Calcium channel blockers Lutein/zeaxanthin Medications with potential adverse effects in RP: Isotretinoin (Accutane)- Sildenafil (Viagra)- Vitamin E Surgery: - cataract extraction: Bastek et al studied 30 patients with RP; 83% of them improved by 2 lines on the Snellen visual acuity chart with cataract surgery. Perioperative use of corticosteroids is recommended to prevent postoperative cystoid macular edema. - Ciliary neurotrophic factor (CNTF) has been shown to slow retinal degeneration in a number of animal models. - RPE cell transplants - retinal prosthesis or phototransducing chip - Gene therapy to replace the defective protein by using DNA vector the ophthalmologist may act as the consultant to an internist. Audiology consultation Annual patient examinations usually are sufficient to measure Goldmann visual field and visual acuity. If medical treatment is initiated, more frequent visits and laboratory blood work may be indicated. Patients with systemic conditions that are associated with retinitis pigmentosa (RP) may require closer follow-up care.
 Treatment: genetic counseling and early diagnosis. 10% of congenitally deaf children may have Usher syndrome Currently, there is no cure for Usher syndrome. The best treatment involves early identification so that educational programs can begin as soon as possible. Typically, treatment will include hearing aids, assistive listening devices, cochlear implants, or other communication methods such as American Sign Language; orientation and mobility training; and communication services and independent-living training that may include Braille instruction, low-vision services, or auditory training. Some ophthalmologists believe that a high dose of vitamin A palmitate may slow, but not halt, the progression of retinitis pigmentosa. This belief stems from the results of a long-term clinical trial supported by the National Eye Institute and the Foundation for Fighting Blindness. Based on these findings, the researchers recommend that most adult patients with the common forms of RP take a daily supplement of 15, 000 IU (international units) of vitamin A in the palmitate form under the supervision of their eye care professional.
Treatment: genetic counseling and early diagnosis. 10% of congenitally deaf children may have Usher syndrome Currently, there is no cure for Usher syndrome. The best treatment involves early identification so that educational programs can begin as soon as possible. Typically, treatment will include hearing aids, assistive listening devices, cochlear implants, or other communication methods such as American Sign Language; orientation and mobility training; and communication services and independent-living training that may include Braille instruction, low-vision services, or auditory training. Some ophthalmologists believe that a high dose of vitamin A palmitate may slow, but not halt, the progression of retinitis pigmentosa. This belief stems from the results of a long-term clinical trial supported by the National Eye Institute and the Foundation for Fighting Blindness. Based on these findings, the researchers recommend that most adult patients with the common forms of RP take a daily supplement of 15, 000 IU (international units) of vitamin A in the palmitate form under the supervision of their eye care professional.
 Other guidelines regarding this treatment option include: Do not substitute vitamin A palmitate with a beta-carotene supplement. Do not take vitamin A supplements greater than the recommended dose of 15, 000 IU or modify your diet to select foods with high levels of vitamin A. Women who are considering pregnancy should stop taking the high-dose supplement of vitamin A three months before trying to conceive due to the increased risk of birth defects. Women who are pregnant should stop taking the high-dose supplement of vitamin A due to the increased risk of birth defects. In addition, according to the same study, people with RP should avoid using supplements of more than 400 IU of vitamin E per day. NIDCD researchers, along with collaborators from universities in New York and Israel, pinpointed a mutation, named R 245 X, of the PCDH 15 gene that accounts for a large percentage of type 1 Usher syndrome in today’s Ashkenazi Jewish population.
Other guidelines regarding this treatment option include: Do not substitute vitamin A palmitate with a beta-carotene supplement. Do not take vitamin A supplements greater than the recommended dose of 15, 000 IU or modify your diet to select foods with high levels of vitamin A. Women who are considering pregnancy should stop taking the high-dose supplement of vitamin A three months before trying to conceive due to the increased risk of birth defects. Women who are pregnant should stop taking the high-dose supplement of vitamin A due to the increased risk of birth defects. In addition, according to the same study, people with RP should avoid using supplements of more than 400 IU of vitamin E per day. NIDCD researchers, along with collaborators from universities in New York and Israel, pinpointed a mutation, named R 245 X, of the PCDH 15 gene that accounts for a large percentage of type 1 Usher syndrome in today’s Ashkenazi Jewish population.
 Ophthalmic diseases associated with hearing loss: Cataracts - Congenital rubella Coloboma - Coloboma of iris, heart deformities, choanal atresia, retarded growth, genital and ear deformities (CHARGE) association Dystopia canthorum - Waardenburg syndrome (WS) Heterochromia irides - WS Keratitis - Cogan syndrome Ocular palsy - Duane syndrome Retinal atrophy - Cockayne syndrome Retinitis pigmentosum - Usher syndrome Retinal degeneration - Alström syndrome Congenital blindness, pseudotumor retinae - Norrie syndrome
Ophthalmic diseases associated with hearing loss: Cataracts - Congenital rubella Coloboma - Coloboma of iris, heart deformities, choanal atresia, retarded growth, genital and ear deformities (CHARGE) association Dystopia canthorum - Waardenburg syndrome (WS) Heterochromia irides - WS Keratitis - Cogan syndrome Ocular palsy - Duane syndrome Retinal atrophy - Cockayne syndrome Retinitis pigmentosum - Usher syndrome Retinal degeneration - Alström syndrome Congenital blindness, pseudotumor retinae - Norrie syndrome
 references EARLY DIAGNOSIS OF USHER SYNDROME IN CHILDREN BY Marilyn B. Mets, MD, Nancy M. Young, MD (BY INVITATION), Arlene Pass, BS (BY INVITATION), AND Janice B. Lasky, MD (BY INVITATION) http: //www. ncbi. nlm. nih. gov/pmc/articles/PMC 1298229/pdf/taos 00001 -0240. pdf Novel USH 2 A compound heterozygous mutations cause RP/USH 2 in a Chinese family Xiaowen Liu, 1, 2 Zhaohui Tang, 2 Chang Li, 2 Kangjuan Yang, 3 Guanqi Gan, 2 Zibo Zhang, 3 Jingyu Liu, 2 Fagang Jiang, 1 Qing Wang, 2 and Mugen Liu 2 http: //www. ncbi. nlm. nih. gov/pmc/articles/PMC 2842093/ Cottet S, Schorderet DF. Mechanisms of apoptosis in retinitis pigmentosa. Curr Mol Med. Apr 2009; 9(3): 37583. [Medline]. Emedicine: Author: David G Telander, MD, Ph. D, Assistant Professor, Department of Ophthalmology and Vision Science, Division of Vitreo-Retinal Diseases and Surgery, University of California Davis School of Medicine Coauthor(s): Anthony de Beus, MD, Ph. D, Consulting Staff, Southwest Eye Centers; Kent W Small, MD, Director/President, Macular and Retinal Disease Center; President, Molecular Insight LLC; Consulting Surgeon, Glendale Eye Medical Group Usher Syndrome(National Institute on Deafness and Other Communication Disorders) Usher Syndrome(National Eye Institute, National Institute on Deafness and Other Communication Disorders) retina-international. org nidcd. nih. gov
references EARLY DIAGNOSIS OF USHER SYNDROME IN CHILDREN BY Marilyn B. Mets, MD, Nancy M. Young, MD (BY INVITATION), Arlene Pass, BS (BY INVITATION), AND Janice B. Lasky, MD (BY INVITATION) http: //www. ncbi. nlm. nih. gov/pmc/articles/PMC 1298229/pdf/taos 00001 -0240. pdf Novel USH 2 A compound heterozygous mutations cause RP/USH 2 in a Chinese family Xiaowen Liu, 1, 2 Zhaohui Tang, 2 Chang Li, 2 Kangjuan Yang, 3 Guanqi Gan, 2 Zibo Zhang, 3 Jingyu Liu, 2 Fagang Jiang, 1 Qing Wang, 2 and Mugen Liu 2 http: //www. ncbi. nlm. nih. gov/pmc/articles/PMC 2842093/ Cottet S, Schorderet DF. Mechanisms of apoptosis in retinitis pigmentosa. Curr Mol Med. Apr 2009; 9(3): 37583. [Medline]. Emedicine: Author: David G Telander, MD, Ph. D, Assistant Professor, Department of Ophthalmology and Vision Science, Division of Vitreo-Retinal Diseases and Surgery, University of California Davis School of Medicine Coauthor(s): Anthony de Beus, MD, Ph. D, Consulting Staff, Southwest Eye Centers; Kent W Small, MD, Director/President, Macular and Retinal Disease Center; President, Molecular Insight LLC; Consulting Surgeon, Glendale Eye Medical Group Usher Syndrome(National Institute on Deafness and Other Communication Disorders) Usher Syndrome(National Eye Institute, National Institute on Deafness and Other Communication Disorders) retina-international. org nidcd. nih. gov
 RP group of inherited disorders characterized by progressive peripheral vision loss and night vision difficulties (nyctalopia) that can lead to central vision loss. RP is a misnomer, as the word retinitis implies an inflammatory response, which has not been found to be a predominant feature of this condition. RP can be passed on by all types of inheritance: 20 -25% is autosomal dominant, 15 -20% is autosomal recessive, and 5 -10% is X linked, while the remaining 45 -50% is found in patients without any known affected relatives. RP is most commonly found in isolation, but it can be associated with systemic disease. The most common systemic association is hearing loss (up to 30% of patients). Many of these patients are diagnosed with Usher syndrome.
RP group of inherited disorders characterized by progressive peripheral vision loss and night vision difficulties (nyctalopia) that can lead to central vision loss. RP is a misnomer, as the word retinitis implies an inflammatory response, which has not been found to be a predominant feature of this condition. RP can be passed on by all types of inheritance: 20 -25% is autosomal dominant, 15 -20% is autosomal recessive, and 5 -10% is X linked, while the remaining 45 -50% is found in patients without any known affected relatives. RP is most commonly found in isolation, but it can be associated with systemic disease. The most common systemic association is hearing loss (up to 30% of patients). Many of these patients are diagnosed with Usher syndrome.
 Pathophysiology RP is typically thought of as a rod-cone dystrophy in which the genetic defects cause cell death (apoptosis), predominantly in the rod photoreceptors; less commonly, the genetic defects affect the RPE and cone photoreceptors. RP has significant phenotypic variation, as there are many different genes that lead to a diagnosis of RP, and patients with the same genetic mutation can present with very different retinal findings. The first histologic change found in the photoreceptors is shortening of the rod outer segments. The outer segments progressively shorten, followed by loss of the rod photoreceptor. This occurs most significantly in the mid periphery of the retina. These regions of the retina reflect the cell apoptosis by having decreased nuclei in the outer nuclear layer. In many cases, the degeneration tends to be worse in the inferior retina, thereby suggesting a role for light exposure. Cone photoreceptor death occurs in a similar manner to rod apoptosis with shortening of the outer segments followed by cell loss. This can occur early or late in the various forms of RP.
Pathophysiology RP is typically thought of as a rod-cone dystrophy in which the genetic defects cause cell death (apoptosis), predominantly in the rod photoreceptors; less commonly, the genetic defects affect the RPE and cone photoreceptors. RP has significant phenotypic variation, as there are many different genes that lead to a diagnosis of RP, and patients with the same genetic mutation can present with very different retinal findings. The first histologic change found in the photoreceptors is shortening of the rod outer segments. The outer segments progressively shorten, followed by loss of the rod photoreceptor. This occurs most significantly in the mid periphery of the retina. These regions of the retina reflect the cell apoptosis by having decreased nuclei in the outer nuclear layer. In many cases, the degeneration tends to be worse in the inferior retina, thereby suggesting a role for light exposure. Cone photoreceptor death occurs in a similar manner to rod apoptosis with shortening of the outer segments followed by cell loss. This can occur early or late in the various forms of RP.
 Frequency The prevalence of typical RP is reported to be approximately 1 in 4000 in the United States. The carrier state is believed to be approximately 1 in 100. Worldwide prevalence of RP is approximately 1 in 5000. because of these X-linked varieties, men may be affected slightly more than women. The age of onset can vary. RP usually is diagnosed in young adulthood, although it can present anywhere from infancy to the mid 30 s to 50 s.
Frequency The prevalence of typical RP is reported to be approximately 1 in 4000 in the United States. The carrier state is believed to be approximately 1 in 100. Worldwide prevalence of RP is approximately 1 in 5000. because of these X-linked varieties, men may be affected slightly more than women. The age of onset can vary. RP usually is diagnosed in young adulthood, although it can present anywhere from infancy to the mid 30 s to 50 s.
 Sytmptoms: Nyctalopia: First symptoms and the hall mark Loss of vision: Painless, slowly progressive. Peripheral then tunnel vision. Photopsia +ve family history Drug history is essential to rule out phenothiazine/thioridazine toxicity.
Sytmptoms: Nyctalopia: First symptoms and the hall mark Loss of vision: Painless, slowly progressive. Peripheral then tunnel vision. Photopsia +ve family history Drug history is essential to rule out phenothiazine/thioridazine toxicity.
 Physical VA: vary from 20/20 to light perception, but usually preserved until late of the disease. Pupil: Normal +/- RAPD Anterior segment: 50% of RP patient will develop posterior subcapsular cataract.
Physical VA: vary from 20/20 to light perception, but usually preserved until late of the disease. Pupil: Normal +/- RAPD Anterior segment: 50% of RP patient will develop posterior subcapsular cataract.
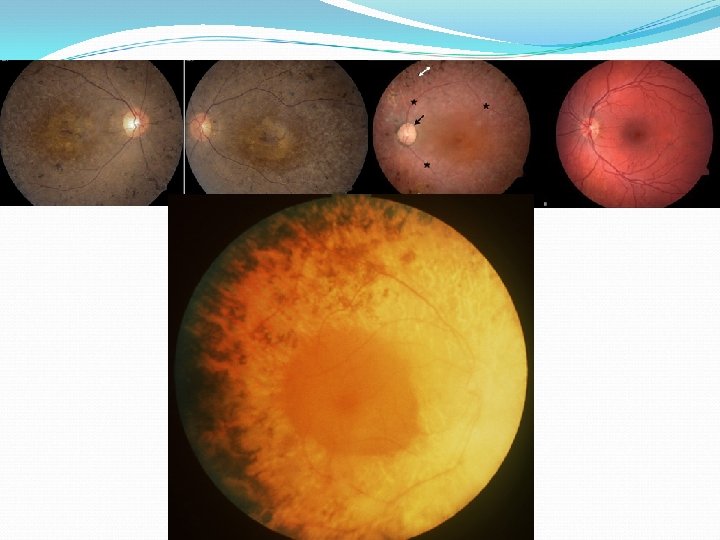
 Phsyical Fundus: The retina can appear unaffected early in the disease. Typical key findings include the following: Bone spicules - Midperipheral retinal hyperpigmentation in a characteristic pattern Optic nerve waxy pallor Atrophy of the RPE in the mid periphery of the retina Retinal arteriolar attenuation The presence of vitreous cells is common. Patients can have a loss of the foveolar reflex an abnormal vitreoretinal interface. A subset of patients with RP develops cystoid macular edema with an associated more rapid and potentially reversible loss of vision. Retinitis punctata albescens, a variant of RP, presents with yellow deposits deep in the retina rather the normal increased pigmentation of the peripheral retina.
Phsyical Fundus: The retina can appear unaffected early in the disease. Typical key findings include the following: Bone spicules - Midperipheral retinal hyperpigmentation in a characteristic pattern Optic nerve waxy pallor Atrophy of the RPE in the mid periphery of the retina Retinal arteriolar attenuation The presence of vitreous cells is common. Patients can have a loss of the foveolar reflex an abnormal vitreoretinal interface. A subset of patients with RP develops cystoid macular edema with an associated more rapid and potentially reversible loss of vision. Retinitis punctata albescens, a variant of RP, presents with yellow deposits deep in the retina rather the normal increased pigmentation of the peripheral retina.
.") Fundus: Cone-rod retinal degenerations present with central macular pigmentary changes (bull's eye maculopathy). Choroideremia and gyrate atrophy typically present with large scalloped areas of peripheral retinal atrophy.
Fundus: Cone-rod retinal degenerations present with central macular pigmentary changes (bull's eye maculopathy). Choroideremia and gyrate atrophy typically present with large scalloped areas of peripheral retinal atrophy.