
Topic: Methods of Human Genetics. Cytogenetic method and



 Studying the karyotypes of an organisms;")











,")



 can")








 For example, phenylketonuria (PKU) Cause: 1.")
 3. Phenylalanine, some of it changes into phenylpyruvic")
 5. This substance drastically affects the cells of the central")








 About 1 in 1. 000 females born have three")
 It is")
 Down syndrome occurs when")


")

")








 Phenylketonuria (PKU) Is an inborn error of")
 The homozygous recessive individual lacks the enzyme phenylalanine hydroxylase")








")





lecture_14_-_cytogenetic,_biochemical_methods,_chromo.diseases.ppt
- Размер: 17.7 Мб
- Автор:
- Количество слайдов: 71
Описание презентации Topic: Methods of Human Genetics. Cytogenetic method and по слайдам
Topic: Methods of Human Genetics. Cytogenetic method and Biochemical method. Chromosomal diseases of man which may result from changes in number of either the sex chromosomes and autosomes. Gene human diseases. Plan of the lecture: 1) Methods of Human Genetics. Cytogenetic methods: a) Preparation of karyotype or Method of culture of cells and tissues; b) Method of determination sex chromatin; 2) Method of amniocentesis. Method of chorionic villus sampling, significance in medicine. 3) Chromosome diseases of man which may result from changes in number of either the sex chromosomes and autosomes. 4) Biochemical method of diagnosis of the heredity pathology. Definition of “gene” diseases. Diagnosis of enzymopathia, significance.
Cytogenesis Term “cytogenesis” was introduced by Sutton in 1903. Cytogenesis is a branch of Biology, is very helpful in the study of human genetics. It is study of number, structure and functions of chromosomes with help of light microscopy.
Cytogenetic methods helps in: 1) Studying the karyotypes of an organisms; 2) Making more precise the human number of chromosomal sets, number of chromosomes and morphology of separate chromosomes for diagnosting the chromosomal diseases; 3) Making up the genetic cards of chromosomes; 4) Studying the genome and chromosomal mutations.
Culture of Cells Culture of cells can be received from piece of skin (fibroblasts), bone marrow, embryonic tissues, chorion, limphs nodes, cells of amniotic fluid, lymphocytes of blood (WBC).
Preparation of Karyotype A karyotype is the graphic representation of the physical appearance of the chromosomes of a particular organism. It is the chromosome complement of a cell or organism depicting the number, size and form of the chromosomes as seen in mitosis.
Preparation of Karyotypes are now prepared routinely for genetic Counseling of couples at risk for having children with Down’s Syndrome or other defects associated with chromosome abnormalities. The chromosomes show in a human karyotype are characteristically metaphase consisting of two sister chromatids held together at their centromere. The prepare a karyotype, white blood cells in the process of dividing are interrupted at metaphase by the addition of colchicine. The prevents the subsequent steps of mitosis from taking place.
Methaphase plate
Preparation of Karyotype By other words, division of the human cells in culture is arrested at the metaphase with the help of colchicine. After treating and staining such cells, when fixed with alcochol and stained, the chromosomes are photographed, enlarged, cut out, and arranged to size. The photographs or diagrams of chromosomes are arterially arranged in homologous pairs, which are arranged in order of their size, except the sex chromosomes that are placed at the end. Such a pictorial representation of all the chromosomes of an organism is called an idiogram. Certain abnormalities, such an extra chromosomes or piece of a chromosome, can be detected.
Preparation of human karyotype
Normal karyotype of male
Method of determination of sex-chromatin or Barr body Human and mammals somatic cells consist of two X-chromosomes, one of out form block of chromatin which is well visible in special cultivation in interkinetic nucleus. It is a condensed inactive X-chromosome of females , which was discovered in 1949 by Barr. He examined nucleus of neurons of female cat’s brain.
Method of determination of sex-chromatin or Barr body Later similar bodies was discovered in human. It is called Barr body or sex – chromatin (X-chromatin). Foetal skin cells are always sloughed off into the amniotic fluid. Microscopic study of these cells or check cells can reveal the sex of the unborn child. A dark-staining mass, called sex chromatin, or Barr body , is present in contact with the nuclear envelope in the cells of a female. The cells of male lack Barr body.
Method of determination of sex-chromatin or Barr body Knowledge of the sex of very young foetus can help in predicting the occurrence of sex-linked genetic disorders. Sometimes it necessary for judicial – medical examination or experts in questionable cases of determination of sex, if defect in structure of external sex organs occurs.
Barr bodies
Amniocentesis At an early stage of pregnancy (16 -17 weeks), a small amount of amniotic fluid about 10 ml is drawn by passing a special surgical syringe needle (without causing any damage to the foetus) through the abdominal wall and uterine wall into the amniotic sac containing amniotic fluid.
Amniocentesis The amniotic fluid contains cells that have sloughed off the foetus skin; these cells can be cultured in the laboratory. They may be examined for protein or enzyme alterations or deficiencies, metabolic disorders such as PKU, sickle-cell anemia, DNA changes, and chromosomal abnormalities.
Amniocentesis
Amniocentesis In case of any serious congenital defect termination of pregnancy is advised. Amniocentesis is often misused to determine the sex of the child so as to abort the female foetus. Because amniocentesis is complicated and costly, it is primarily used in high-risk cases.
Chorionic-villus sampling A new technique, named chorionic-villus sampling (CVS) can be done during the light to eighth to tenth week (8 -10 week) of pregnancy when abortion is safe for the women. That is earlier than amniocentesis. The chorion is a membrane layer surrounding the foetus, and it consist entirely of embryonic tissues.
Chorionic-villus sampling A chorionic villus tissue sample may be taken from the developing placenta through the abdomen (as in amniocenteses) or via the vagina using biopsy forceps or a flexible catheter, aided by ultrasound. Once the tissues sample is obtained, the analysis is similar to that used in amniocentesis.
Chorionic-villus sampling
Biochemical method is a study of biological liquids such as blood, urine, amniotic fluid, liquor or spinal fluid, that allow to reveal the breakage of metabolism with help of chemical analysis.
Biochemical method There are 300 inborn metabolism or gene disease was discovered by biochemical methods. Biochemical method is useful for genetic counseling of couples at risk for having children with genetic disorders, gene diseases. Many human genetic diseases are caused by deficiencies in enzyme activities. Most of these diseases are inherited as recessive traits.
Beadle and Tatum’s “one gene — one enzyme” hypothesis Molecular genetics is a specific relationship between genes and enzymes, historically embodied in Beadle and Tatum’s “one gene — one enzyme” hypothesis , which states that each gene controls the synthesis or activity of a single enzyme. Since we know that enzymes may consist of more than one polypeptide and that genes code for individual polypeptide chains, a more modern title for this hypotheses is “ one gene-one polypeptide”.
Cause of gene diseases is breakage of metabolism. As known proteins may be structural; proteins, having specific functions like transfer of oxygen, protein-hormones and protein enzymes. Protein- enzymes are catalysators and participants of all biochemical reactions in the cell. Therefore the absence or inactivity of the enzyme is due to the absence of the normal form of the gene that controls the synthesis of the enzyme. Accumulation of derivatives or intermediate products of metabolism in blood, urine, amniotic fluid and other occurs, which damages the brain and other organs and causes the disease.
Enzymopathy Lack of the enzyme is due to the abnormal recessive gene. This mutant gene controls the synthesis of the “wrong” enzyme. It is also called enzymopathy.
General scheme of enzymopathy Mutant gene → changing nonfunctional enzyme → breakage of chemical reaction → absence of any substrates (impossible synthesis) → accumulation of derivatives in the urine, blood, secret of bronches, amniotic fluid and other biological liquids ↓ non-function of organs, damage the brain, liver, kidney… ↓ disease (suffers whole organism)
Phenylketonuria (PKU) For example, phenylketonuria (PKU) Cause: 1. Recessive mutation of gene on chromosome-1 which control the synthesis of enzyme phenylalanine hydroxylase. 2. The absence of that enzyme activity prevents the conversion of the amino acid phenylalanine to the amino acid tyrosine. Phenylalanine is one of the essential amino acid.
Phenylketonuria (PKU) 3. Phenylalanine, some of it changes into phenylpyruvic acid and metabolic block in the pathway occurs. 4. Phenylalanine accumulates in the tissues, accumulation of phenylketones and other derivatives in urine, blood and other fluids of an organism.
Phenylketonuria (PKU) 5. This substance drastically affects the cells of the central nervous system (CNS) and produces serious symptoms: severe mental retardation, a slow growth rate, and early death. 6. Disease is called phenylketonuria (PKU). PKU homozygotes develop normally on low phenylalanine diets.
Turner syndrome is caused by XO genotype. The individual has 45 chromosomes (2 n-1) and is a sterile female with underdeveloped breasts, reduced ovaries, small uterus. Turner syndrome individuals occur with a frequency of 1 in 10. 000 female births. It is estimated that up to 99 percent of all 45, X embryos die before birth. Surviving Turner syndrome individuals have few noticeable major defects until puberty, when they fail to develop secondary sexual characteristics.
Symptoms of Turner syndrome Female tend to be shorter than average, and they have web-like necks, poorly developed breast, and immature internal sexual organs. They have a reduced ability to interpret spatial relationships and are usually infertile. Females are often sterile as they may not menstruate or ovulate, subnormal intelligence, with many male characteristics such as heavy neck muscles and narrow hips.
Turner syndrome
Y O — individual does not survive, is not viable
Klinefelter syndrome About 1 in 1. 000 males born have Klinefelter syndrome. The male has 47 chromosome (2 n+1). These 47 , XXY males have underdeveloped testes, and are often taller then the average male. Some degree of breast development is seen in about 50 percent of affected individual, and some show subnormal intelligence. Individuals with similar phenotypes are also found with higher numbers of X and /or Y chromosomes, e. g. , 48, XXXY, 49, XXXXY… male individuals that are greatly mentally retarded. Some individuals have one X and two Y chromosomes; they have XYY syndrome.
Symptoms of Klinefelter syndrome They have one Barr body. Male individual with following features: underdeveloped testes, enlarged breasts, little hair on the body, infertility due to abnormal sominiferous tubules, mentally retarded, small testes, unusually long legs, obesity, with many female characteristics. The more the X chromosomes, the greater is the mental defects.
Klinefelter syndrome
47, XYY These 47, XYY individuals are male because of the presence of the Y. The XYY karyotype results from nondisjunction of the Y- chromosome in meiosis. About 1 in 1. 000 males born have XYY syndrome with following features: they are taller than normal people, has reduced intelligence, more aggressive than normal males, over produces male hormones like testosterone, has criminal and brutal tendencies.
47, XXX (triplo-X) About 1 in 1. 000 females born have three X-chromosomes instead of the normal two. These 47, XXX (triplo-X) females are mostly completely normal, although they are slightly less fertile and a small number have less than average intelligence. Barr bodies are two.
Autosomal abnormalities Down syndrome (mongolism or trisomy 21 ) It is due to the presence of an extra chromosome 21. These individual have 47 chromosomes (not 46) in all their body cells. Trisomy -21 occurs with a frequency of about 3. 510 per 1 million conceptions and about and 1. 430 per 1 million live births.
Down syndrome ( trisomy 21 ) Down syndrome occurs when there are three copies of chromosome 21 as shown in the karyotype in figure.
Down syndrome Down’s syndrome was first reported in in 1866 by Langdon Down but its cause was found in 1959 by Lejeune and his coworkers
Symptoms of Down syndrome severe mental retardation number of abnormalities in the facial structure are: prominent fore head, flattened nasal bridge, habitually open mouth, protruding lower lips, large generally protruding tongue, a characteristic fold of skin at the corner of the eyes, short and broad neck, flat hands, oval mongoloid face having superficial similarity to mongolians. malformation of heart underdevelopment of gonads
Down syndrome ( trisomy 21 )
Relationship between age of mother risk of a Trisomy -21 in child Age of mother Risk of Trisomy -21 in child, % 16 -26 77/10000 27 -34 4/10000 35 -39 29/10000 40 -44 100/10000 45 -47 333/10000 All mothers combined 15/
Down syndrome ( trisomy 21 )
Patau syndrome or trisomy — 13 Trisomy — 13 produces Patau syndrome. About 2 in 10. 000 live births produce individuals with trisomy-13.
Symptoms of Patau syndrome cleft lip and palate, small eyes polydactyly (extra fingers and toes) mental and developmental retardation and cardiac anomalies, among many other abnormalities Most die before the age of three months.
Edwards syndrome Trisomy-18 produces Edwards syndrome. It occur in about 2. 5 in 10. 000 live births. For reasons that are not known, about 80 percent of Edwards syndrome infants are female. Individuals with trisomy-18 a small at birth and have multiple congenital malformations affecting almost every organ system in the body.
Edwards syndrome or trisomy-18 Clenched fists elongated skull low-set malformed ears mental and developmental retardation and many other abnormalities are associated with the syndrome. Ninety percent (90%) of infants with trisomy-18 die within six months, often from cardiac problems
Cri-du-chat syndrome One human disorder cased by a heterozygous deletion is Cri-du-chat syndrome which results from an observable deletion of part of the short arm of chromosome 5, one of the larger human chromosomes.
Cri-du-chat syndrome Children with cri-du-chat syndrome are severely mentally retarded, have a number of physical abnormalities, and cry with a sound like the mew of a cat (hence the name, which is French for “cry of the cat”).
Prader-Willi syndrome Another example is Prader-Willi syndrome, which results from heterozygosity for a deletion of part of the long arm of chromosome 15.
Prader-Willi syndrome Infants with this syndrome are weak because their sucking reflex is poor, making feeding difficult. As a result, growth is poor. Other phenotypes associated with the syndrome include poor sexual development in males, behavioral problems, and mental retardation. Many individual with the syndrome go undiagnosed, so its frequency of occurrence is not known.
Gene diseases (metabolism errors) Phenylketonuria (PKU) Is an inborn error of metabolism. PKU occur in about 1 in 12. 000 births. PKU is most commonly caused by a recessive mutation of a gene on chromosome 1.
Phenylketonuria (PKU) The homozygous recessive individual lacks the enzyme phenylalanine hydroxylase needed to change one amino acid, phenylalanine, to another, tyrosine. The absence of that enzyme activity prevents the conversion of the amino acid phenylalanine to the amino acid tyrosine. Phenylalanine, phenylketones and other derivatives in urine, in the tissues accumulate, and some of it into phenylpyruvic acid which damages the brain and causes the disease.
Albinism Is caused by an autosomal recessive mutation, and individuals must be homozygous for the mutation to exhibit the condition. Albinism is characterized by lack of dark pigment melanin in the skin , hair and iris. It is caused by the absence of the enzyme tyrosinase which is necessary for the synthesis of the pigment melanin from dihydroxyphenylalanine. The gene for albinism (a) does not produce enzyme tyrosinase , but its normal allele (A) does. Thus, only homozygous individual (aa) is affected.
Alkaptonuria Is an inborn metabolic disorder produced by the action of a single gene. Its symptoms include blackening of urine on exposure to air and darkening of certain cartilages. Urine of alkaptonuric person contains homogentisic acid, which spontaneously combines with oxygen to form a black pigment. This pigment slowly deposits in the cartilages of ears and nose. It also causes mild arthritis.
Tay-Sachs disease or Infantile Amaurotic Idiocy The child having this disease is born normal, but a few months later, due to an error in lipid metabolism, his brain and spinal cord are damaged. This results in mental retardation and paralysis. The victim dies in 3 or 4 years. These is no treatment for this disease. Tay-Sachs disease is caused by homozygosity for a rare recessive mutation that maps to chromosome 15. Lysosomes are membrane-bound organelles that contain 40 or more different digestive enzymes. These enzymes catalyze the breakdown of nucleic acids, proteins, polysaccharides, and lipids. A number of human diseases are caused by mutations in gene that code for lysosomal enzymes. These diseases called lysosomal-storage diseases , are generally caused by recessive mutations.
Lesch-Nyhan syndrome Is fatal human trait caused by a recessive, single gene mutation on the X-chromosome. An estimate 1 in 10000 male exhibit Lesch-Nyhan syndrome. Because this genetic disease is often lethal before reproductive age. Lesch-Nyhan syndrome is the result of a deficiency in the enzyme hypoxanthine guanine phosphoribosyl transferase , an enzyme essential to the utilization of purines (HG PRT). In this case, excess purines accumulate, and are converted to uric acid. At birth, Lesch-Nyhan individuals are healthy and develop normally for several months. Between three and eight months, delays in motor development lead to weak muscles. Later, the muscle tone change, radically, producing uncontrollable movements and involuntary spasms, seriously affecting feeding activities.
Galactosemia Is defect of metabolism sugar — lactosae. In blood accumulation of galactosae occurs, mental and developmental retardation, liver damages, nervous system, eyes and other organs. It is due to recessive mutation of gene. Autosomal-recessive type of inheritance. It is about of 1 in 16. 000 births.
Glycogen disease Glycogen accumulated in liver and kidney, can not converted again to glycosae and hypoglycosemya is developed due to lack enzyme glucosae-6 -phosphatasae in liver. Type of inheritance is autosomal-recessive. Symptoms: large head, short neck, large abdomen, dolly face, no mental retarded, but low growth rate.
Haemoglobinopathy — Sickle-cell anaemia is hereditary disease found widely in tropical Africa and also in American blacks whose ancestors came from that part of Africa. It is characterize by sickle-shaped (crescentic) red blood cells formed under low oxygen conditions. Sickle-cell anemia is an genetically based human disease that results from an animo acid substitution in the hemoglobin protein that transports oxygen in the blood.
Sickle-cell anaemia Sickle-cell anemia was first described in 1910 by J. Herrick. He found that red blood cells from individuals with the disease lose the characteristics disk shape in condition of low oxygen tension and assume the shape of a sickle. The sickled red blood cells are more fragile than normal red blood cells and tend to break easily.
Methods of prenatal diagnosis (amniocentesis, chorionic-villus sampling)
Genetic counseling is the analysis of the probability that individuals have a genetic defect, or of the risk that prospective parents may produce a child with a genetic defect. In the latter case, genetic counseling involves presenting the available options for avoiding or minimizing those possible risks. Thus, genetic counseling provides people with an understanding of the genetic problems that are or may be inherent in their families or prospective families.
Genetic counseling generally starts with pedigree analysis of both families to determine the likelihood that a genetic disease is present in either group. Once evidence is found of a genetic disease in a family or families, prospective parents need to be informed of the probability that they will produce a child with that disease. Early detection of a genetic disease occurs at one or both of two levels. One is the detection of heterozygotes (carriers) of recessive mutations, and the other is the determination of whether or not the developing fetus shows the biochemical defect.
Genetic counseling In sum, genetic counseling is a lot more than the simple presentation of risk facts and figures to patients; it is the prevention of disease, the relief of pain, and the maintenance of health, all of which are goals of the medical profession in general.
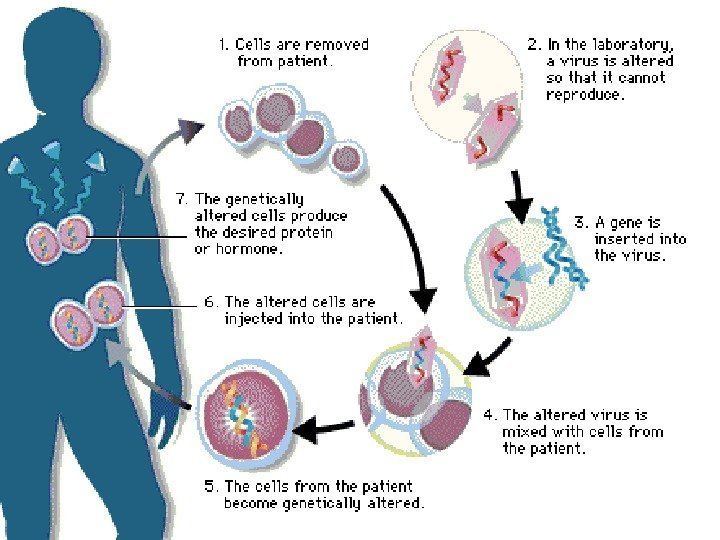
Gene therapy Diagnosis of Diseases Gene Therapy Gene therapy involves supplying a functional gene to cells lacking that function, with the aim of correcting a genetic disorder or acquired disease. It can be divided into two categories: 1)alteration of germ cells — a permanent genetic change for the whole organism and subsequent generations. 2)Somatic cell therapy, is analogous to an organ transplant.
Genetic therapy of Cancer 1. 1. Introduction in cell proteins – increasing immune system 2. 2. Introduction of virus genes – application of antivirus drugs 3. 3. Introduction of antioncogenes