
TOPIC: Human hereditary diseases. Chromosomal









 Down’s syndrome was first")
 It is caused by the presence of an extra")




 Reciprocal translocation B) Terminal deletion C) Ring chromosome D)")








 1: 1080 of birth Presence of")

 About 1 in 1. 000 females born have three")









 For example, phenylketonuria (PKU) Cause: 1. Recessive")
 3. Phenylalanine, some of it changes into phenylpyruvic")
 5. This substance drastically affects the cells of the central")

. Recessively inherited traits Phenylketonuria (PKU) Is")
 The homozygous recessive individual lacks the enzyme phenylalanine hydroxylase")
 Lack of the enzyme is due to the abnormal recessive")



 was first reported")










 - defeat of the connective tissue due")
 —")

")


")





lecture_15_-_genetic_disorders.ppt
- Размер: 6.2 Мб
- Автор:
- Количество слайдов: 71
Описание презентации TOPIC: Human hereditary diseases. Chromosomal по слайдам
TOPIC: Human hereditary diseases. Chromosomal diseases which are connected with genomic mutations and chromosomal aberrations. Gene human diseases Plan of the lecture: Chromosomal human diseases Gene diseases, diseases with hereditary predisposition. Prenatal diagnosis of hereditary diseases. Methods of prenatal diagnosis. Role of medico-genetic counseling in prophylaxis of human hereditary diseases.
Hereditary diseases The basis of hereditary diseases are caused by mutations. The Latin word mutatio means «change». Mutation is a change in the hereditary characteristics of an organism as a result of rearrangements in the structures responsible for the storage and transmission of genetic information.
Chromosomal diseases Diseases associated with pathological changes in chromosomes, usually well known as chromosomal disorders. Under hereditary diseases to understand the violations caused by gene mutations.
Hereditary diseases are divided into two groups: chromosomal and gene that is associated with «breakdowns» at the level of chromosomes or individual genes. Gene diseases , in turn, divided into monogenic and multifactorial. The origin of the first depends on the presence of mutations in a gene. Mutations can disrupt the structure, increase or reduce the quantitative content of the gene encoding the protein
Multifactorial diseases are caused by the combined action of adverse factors of environment and the genetic risk factors forming hereditary predisposition to disease. This group of diseases concern the majority of chronic diseases of the human with affect cardiovascular, respiratory, endocrine and other systems. In many cases genetically determined.
The reasons of occurrence ● The reasons of 40 -60 % of anomalies of development are unknown. To them apply the term «sporadic defects of a birth» , the term designating the unknown reason, casual occurrence and low risk of repeated occurrence at the future children. ● For 20 -25% of anomalies the «multifactorial» reason — complex interaction of many small genetic defects and risk factors of an environment is more probable. ● The others of 10 -13 % of anomalies are connected with influence of environment. ● Only 12 -25 % of anomalies have cleanly genetic reasons.
Appearance of Genetic disorders The genetics disorders arise in humans in the following ways – I. By numerical changes in chromosomes 1. in autosomes (Down’s syndrome) 2. in sex chromosomes (Turner’s and Klinefelter’s syndrome, trisomy-X)
Appearance of Genetic disorders II. By gene mutation in chromosomes 1. in autosomes (alkaptonuria, phenylketonuria, sickle-cell anaemia, albinism) 2. In sex chromosomes (haemophilia, red-green colour blindness, night blindness, muscular dystrophy) III. By incompatibility of genes (Rh-factor, blood groups)
Autosomal abnormalities Down syndrome (mongolism or trisomy 21 ) Down’s syndrome was first reported in in 1866 by Langdon Down but its cause was found in 1959 by Lejeune and his coworkers
Down syndrome (Trisomy 21) It is caused by the presence of an extra chromosome 21. These individual have 47 chromosomes (not 46) in all their body cells. 1: 800 of birth Mental deficiency, heart defects. Round face Short digits
Cause Both the chromosomes of the pair 21 pass into a single egg due to nondisjunction during oogenesis in the mother’s ovary. Thus, the egg has 24 chromosomes instead of 23, and the offspring has 47 chromosomes (45 + XY in male, 45 + XX in female). The presence of three chromosomes of the same kind is called trisomy (21 -trisomy)
Relationship between age of mother risk of a Trisomy -21 in child Age of mother Risk of Trisomy -21 in child, % 16 -26 77/10000 27 -34 4/10000 35 -39 29/10000 40 -44 100/10000 45 -47 333/10000 All mothers combined 15/
Female Trisomy 18 1: 8000 of birth Mental deficiency low set ear Growth retardation Second and fifth digit overlapping the third and fourth Short sternum Narrow pelvis Clenched fists elongated skull infants with trisomy-18 die within six months, often from cardiac problems
Female with trisomy 13 1: 25000 of birth Mental deficiency, CNS malformation, microphthalmia Bilateral cleft lip &palate Low set malformed ears Polydactyly and cardiac anomalies, among many other abnormalities Most die before the age of three months.
Structural chromosomal abnormalities A) Reciprocal translocation B) Terminal deletion C) Ring chromosome D) Duplication E) Paracentric inversion F) Isochromosome G) Robertsonian translocation
Cri-du-chat syndrome Terminal deletion of the short arm end of the chromosome no. 5 Child has a weak catlike cry Mental retardation , microcephaly , heart disease.
Cri-du-chat syndrome One human disorder cased by a heterozygous deletion is Cri-du-chat syndrome which results from an observable deletion of part of the short arm of chromosome 5 (5 p-), one of the larger human chromosomes. Frequency in population is 1:
Cri-du-chat syndrome Children with cri-du-chat syndrome are severely mentally retarded, have a number of physical abnormalities, muscular hypotony, moon-like face, microcephalia, low-set malformed ears, anti-mongoloid eyes and cry with a sound like the mew of a cat (hence the name, which is French for “cry of the cat”). Duration of life is only 14% of affected children die before the age of ten year.
Prader-Willi syndrome Another example is Prader-Willi syndrome , which results from heterozygosity for a deletion of part of the long arm of chromosome 15 (15 q-).
Prader-Willi syndrome Infants with this syndrome are weak because their sucking reflex is poor, making feeding difficult. As a result, growth is poor. Other phenotypes associated with the syndrome include poor sexual development in males, behavioral problems, and mental retardation. Many individual with the syndrome go undiagnosed, so its frequency of occurrence is not known.
Sex-chromosomal abnormalities Turner syndrome is caused by XO genotype. The individual has 45 chromosomes (2 n-1) and is a sterile female with underdeveloped breasts, reduced ovaries, small uterus. Turner syndrome individuals occur with a frequency of 1 in 3. 000 -5. 000 births. It is estimated that up to 99 percent of all 45, X embryos die before birth. Surviving Turner syndrome individuals have few noticeable major defects until puberty, when they fail to develop secondary sexual characteristics.
Symptoms of Turner syndrome Female tend to be shorter than average, and they have web-like necks, poorly developed breast, and immature internal sexual organs. They have a reduced ability to interpret spatial relationships and are usually infertile. Females are often sterile as they may not menstruate or ovulate, subnormal intelligence, with many male characteristics such as heavy neck muscles and narrow hips.
Y O — individual does not survive, is not viable
Young male with Klinefelter syndrome (XXY trisomy) 1: 1080 of birth Presence of breasts Gynecomastia (excessive development of male mammary glands) Small testes, aspermatogenesis due to hyalinization of seminiferous tubules Less intelligent
47, XYY These 47, XYY individuals are male because of the presence of the Y. The XYY karyotype results from nondisjunction of the Y- chromosome in meiosis. About 1 in 400 males born have XYY syndrome with following features: they are taller than normal people, has reduced intelligence, more aggressive than normal males, over produces male hormones like testosterone, has criminal and brutal tendencies.
47, XXX (triplo-X) About 1 in 1. 000 females born have three X-chromosomes instead of the normal two. These 47, XXX (triplo-X) females are mostly completely normal, although they are slightly less fertile and a small number have less than average intelligence. Barr bodies are two.
Biochemical method is a study of biological liquids such as blood, urine, amniotic fluid, liquor or spinal fluid, that allow to reveal the breakage of metabolism with help of chemical analysis.
Biochemical method There are 300 inborn metabolism or gene disease was discovered by biochemical methods. Biochemical method is useful for genetic counseling of couples at risk for having children with genetic disorders, gene diseases. Many human genetic diseases are caused by deficiencies in enzyme activities. Most of these diseases are inherited as recessive traits.
Beadle and Tatum’s “one gene — one enzyme” hypothesis Molecular genetics is a specific relationship between genes and enzymes, historically embodied in Beadle and Tatum’s “one gene — one enzyme” hypothesis , which states that each gene controls the synthesis or activity of a single enzyme. Since we know that enzymes may consist of more than one polypeptide and that genes code for individual polypeptide chains, a more modern title for this hypotheses is “ one gene-one polypeptide”.
Cause of gene diseases is breakage of metabolism. As known proteins may be structural; proteins, having specific functions like transfer of oxygen, protein-hormones and protein enzymes. Protein- enzymes are catalysators and participants of all biochemical reactions in the cell. Therefore the absence or inactivity of the enzyme is due to the absence of the normal form of the gene that controls the synthesis of the enzyme. Accumulation of derivatives or intermediate products of metabolism in blood, urine, amniotic fluid and other occurs, which damages the brain and other organs and causes the disease.
Enzymopathy Lack of the enzyme is due to the abnormal recessive gene. This mutant gene controls the synthesis of the “wrong” enzyme. It is also called enzymopathy.
Gene disease — a large group of diseases resulting from damage to DNA at the level of the gene . The term used in regard to monogenic diseases, in contrast to a broader group — hereditary disease.
Most genetic disorders caused by mutations in structural genes that carry out their function through the synthesis of polypeptides — proteins. Any gene mutation leads to a change in the structure or amount of protein. The beginning of any genetic disease associated with the primary effect of the mutant allele. The basic scheme of genetic diseases includes a number of links: mutant allele → product → change primary chain of biochemical processes in the cell bodies → organism
synthesis of abnormal protein development of excessive amounts of gene product; lack of development of the primary product; development of a reduced number of normal primary product.
General scheme of enzymopathy Mutant gene → changing nonfunctional enzyme → breakage of chemical reaction → absence of any substrates (impossible to synthesis) → accumulation of derivatives in the urine, blood, secret of bronches, amniotic fluid and other biological liquids ↓ non-function of organs, damage the brain, liver, kidney… ↓ disease (suffers whole organism)
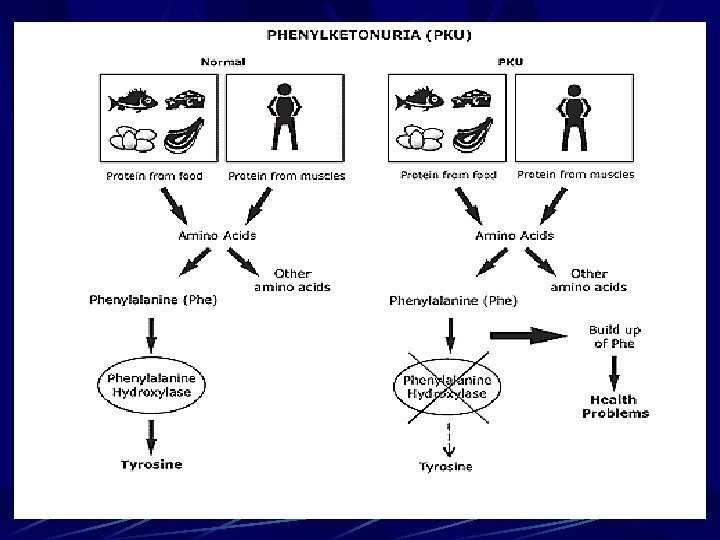
Phenylketonuria (PKU) For example, phenylketonuria (PKU) Cause: 1. Recessive mutation of gene on chromosome-1 which control the synthesis of enzyme phenylalanine hydroxylase. 2. The absence of that enzyme activity prevents the conversion of the amino acid phenylalanine to the amino acid tyrosine. Phenylalanine is one of the essential amino acid.
Phenylketonuria (PKU) 3. Phenylalanine, some of it changes into phenylpyruvic acid and metabolic block in the pathway occurs. 4. Phenylalanine accumulates in the tissues, accumulation of phenylketones and other derivatives in urine, blood and other fluids of an organism.
Phenylketonuria (PKU) 5. This substance drastically affects the cells of the central nervous system (CNS) and produces serious symptoms: severe mental retardation, a slow growth rate, and early death. 6. Disease is called phenylketonuria (PKU). PKU homozygotes develop normally on low phenylalanine diets.
By gene diseases in humans are numerous diseases of metabolism . They may be linked to the violation of the exchange of carbohydrates , lipids , steroids, purines and pyrimidines , bilirubin , metals, etc. So far, no single classification of hereditary diseases of metabolism.
Gene diseases (metabolism errors). Recessively inherited traits Phenylketonuria (PKU) Is an inborn error of metabolism. PKU occur in about 1 in 6. 000 -10. 000 births. PKU is most commonly caused by a recessive mutation of a gene on chromosome 1.
Phenylketonuria (PKU) The homozygous recessive individual lacks the enzyme phenylalanine hydroxylase needed to change one amino acid, phenylalanine, to another, tyrosine. The absence of that enzyme activity prevents the conversion of the amino acid phenylalanine to the amino acid tyrosine. Phenylalanine, phenylketones and other derivatives in urine, in the tissues accumulate, and some of it into phenylpyruvic acid which damages the brain and causes the disease.
Phenylketonuria (PKU) Lack of the enzyme is due to the abnormal recessive gene The affected baby is normal at the birth but within a few weeks, phenylalanine level in plasma starts rising, and by the age of 6 month he develops severe mental retardation If deficiency of phenylalanine hydroxylase is detected in the newborn, disorder can be usually prevented with a special diet, low in phenylalanine.
Albinism Is caused by an autosomal recessive mutation, and individuals must be homozygous for the mutation to exhibit the condition. Frequency is 1: 5. 000, 1: 25. 000. Albinism is characterized by lack of dark pigment melanin in the skin , hair and iris. It is caused by the absence of the enzyme tyrosinase which is necessary for the synthesis of the pigment melanin from dihydroxyphenylalanine. The gene for albinism (a) does not produce enzyme tyrosinase , but its normal allele (A) does. Thus, only homozygous individual (aa) is affected.
Alkaptonuria Is an inborn metabolic disorder produced by the action of a single gene. Frequency is 3 -5: 1. 000. Is caused by an autosomal recessive mutation leeds to lack of liver enzyme homogentisate oxidase is due to absence of the normal form of the gene that controls the synthesis of the enzyme. The symptoms include blackening of urine on exposure to air and darkening of certain cartilages. Urine of alkaptonuric person contains homogentisic acid, also called alkapton, which spontaneously combines with oxygen to form a black pigment. This pigment slowly deposits in the cartilages of ears and nose. It also causes mild arthritis.
Dominantly inherited traits These are caused by dominant genes. One parent carries the defective gene and half of the children inherit it. The defects produced by dominant genes include: polydactyly (extra digits), brachydactyly (abnormally short digits), some forms of dwarfism
Gene mutations in Sex chromosomes Haemophilia (Bleeder’s disease) was first reported by John Otto in 1803. Haemophilia is a defect of the blood which prevents its clotting. Haemophilics bleed excessively when injured. It is a sex-linked trait caused by a recessive gene, located in the X chromosome. This gene controls the synthesis of a protein needed for the clotting of blood.
Gene mutations in Sex chromosomes Red-green Colour Blindness is also a sex-linked recessive trait. Red-green Colour Blindness is the inability of certain human beings to distinguish red from green colour. Cause. Red-green Colour Blindness produced by an X-linked recessive gene by causing lack of one of the primary cone pigments of the retina. Normal eyesight is dominant to colour blindness.
Gene mutations in Sex chromosomes Night blindness is of two types: acquired and congenital. The former is due to vitamin A deficiency. The congenital is due to visual purple deficiency that interferes with the functions of the retinal rods. This night blindness is a sex-linked trait. It is caused by a recessive gene carried by the X-chromosome. More common in males. Gene mutations in Sex chromosomes
Gene mutations in Sex chromosomes Muscular Dystrophy. This disorder causes deterioration of muscles at an early age. The mutated gene on X chromosome fails to produce a protein called dystrophin. This protein is thought to relay the nerve’s signal to the calcium storage sacs in the muscle cell. As a result, calcium is not released, and muscle contraction is halted at the very first step. The victim becomes invalid by the age of 10 and usually dies by the age of 20. Frequency is 1: 3500 male births.
Haemoglobinopathy — Sickle-cell anaemia is hereditary disease found widely in tropical Africa and also in American blacks whose ancestors came from that part of Africa. It is characterize by sickle-shaped (crescentic) red blood cells formed under low oxygen conditions. Sickle-cell anemia is an genetically based human disease that results from an animo acid substitution in the hemoglobin protein that transports oxygen in the blood.
Sickle-cell anaemia Sickle-cell anemia was first described in 1910 by J. Herrick. He found that red blood cells from individuals with the disease lose the characteristics disk shape in condition of low oxygen tension and assume the shape of a sickle. The sickled red blood cells are more fragile than normal red blood cells and tend to break easily.
Tay-Sachs disease or Infantile Amaurotic Idiocy The child having this disease is born normal, but a few months later, due to an error in lipid metabolism, his brain and spinal cord are damaged. This results in mental retardation and paralysis. The victim dies in 3 or 4 years. These is no treatment for this disease. Tay-Sachs disease is caused by homozygosity for a rare recessive mutation that maps to chromosome 15. Lysosomes are membrane-bound organelles that contain 40 or more different digestive enzymes. These enzymes catalyze the breakdown of nucleic acids, proteins, polysaccharides, and lipids. A number of human diseases are caused by mutations in gene that code for lysosomal enzymes. These diseases called lysosomal-storage diseases , are generally caused by recessive mutations.
Lesch-Nyhan syndrome Is fatal human trait caused by a recessive, single gene mutation on the X-chromosome. An estimate 1 in 300. 000 male exhibit Lesch-Nyhan syndrome. Because this genetic disease is often lethal before reproductive age. Lesch-Nyhan syndrome is the result of a deficiency in the enzyme hypoxanthine guanine phosphoribosyl transferase , an enzyme essential to the utilization of purines (HG PRT). In this case, excess purines accumulate, and are converted to uric acid. At birth, Lesch-Nyhan individuals are healthy and develop normally for several months. Between three and eight months, delays in motor development lead to weak muscles. Later, the muscle tone change, radically, producing uncontrollable movements and involuntary spasms, seriously affecting feeding activities.
Galactosemia Is defect of metabolism sugar — lactosae. In blood accumulation of galactosae occurs, mental and developmental retardation, liver damages, nervous system, eyes and other organs. It is due to recessive mutation of gene. Autosomal-recessive type of inheritance. It is about of 1 in 16. 000 births.
Glycogen disease Glycogen accumulated in liver and kidney, can not converted again to glycosae and hypoglycosemya is developed due to lack enzyme glucosae-6 -phosphatasae in liver. Type of inheritance is autosomal-recessive. Symptoms: large head, short neck, large abdomen, dolly face, no mental retarded, but low growth rate.
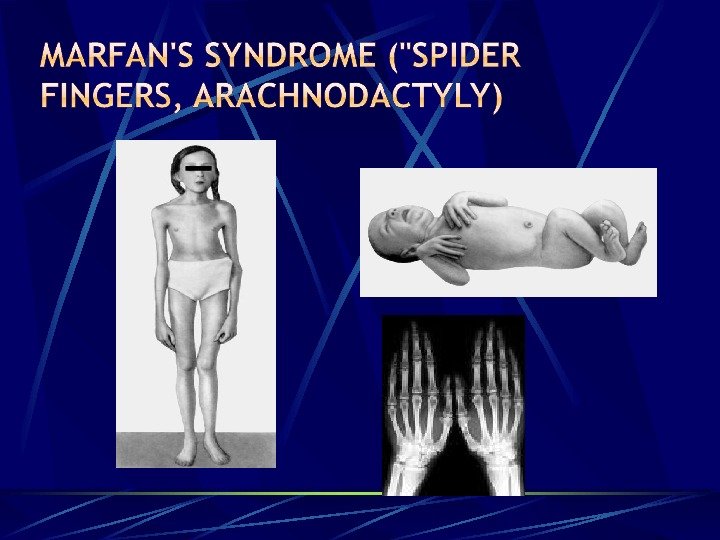
Marfan’s syndrome («spider fingers, arachnodactyly) — defeat of the connective tissue due to mutations in the gene responsible for the synthesis fibrillina; mukopolisaharidozy — a group of connective tissue diseases associated with metabolism of acid glycosaminoglycans. Fibrodisplations — a disease of connective tissue, associated with its progressive ossification of a mutation in the gene ACVR
Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) — контрактуры верхних и нижних конечностей. Синдром Моркио: типичные внешние проявления.
Болезнь Шейе: типичные внешние проявления Рентгенологические признаки синдрома Моркио — деформация костей таза, вертлужных впадин, гипоплазия головок бедренных костей.
Синдром Марото — Лами: (типичные внешние проявления у девочки 9 лет) — грубые черты лица, бочкообразная грудная клетка. Синдром Гурлер: типичные внешние проявления.
Синдром Гунтера у мальчика 2 лет — изменения скелета слабо выражены, нет кифоза, контрактур.
Chondroplasia Due to a G to A transition mutation of the fibroplast growth factor receptor 3 gene on chromosome 4 p Short limbs and fingers Normal length of the trunk Bowed legs Relatively large head Depressed nasal bridge Prominent forehead
Methods of prenatal diagnosis (amniocentesis, chorionic-villus sampling)
Genetic counseling is the analysis of the probability that individuals have a genetic defect, or of the risk that prospective parents may produce a child with a genetic defect. In the latter case, genetic counseling involves presenting the available options for avoiding or minimizing those possible risks. Thus, genetic counseling provides people with an understanding of the genetic problems that are or may be inherent in their families or prospective families.
Genetic counseling generally starts with pedigree analysis of both families to determine the likelihood that a genetic disease is present in either group. Once evidence is found of a genetic disease in a family or families, prospective parents need to be informed of the probability that they will produce a child with that disease. Early detection of a genetic disease occurs at one or both of two levels. One is the detection of heterozygotes (carriers) of recessive mutations, and the other is the determination of whether or not the developing fetus shows the biochemical defect.
Genetic counseling In sum, genetic counseling is a lot more than the simple presentation of risk facts and figures to patients; it is the prevention of disease, the relief of pain, and the maintenance of health, all of which are goals of the medical profession in general.
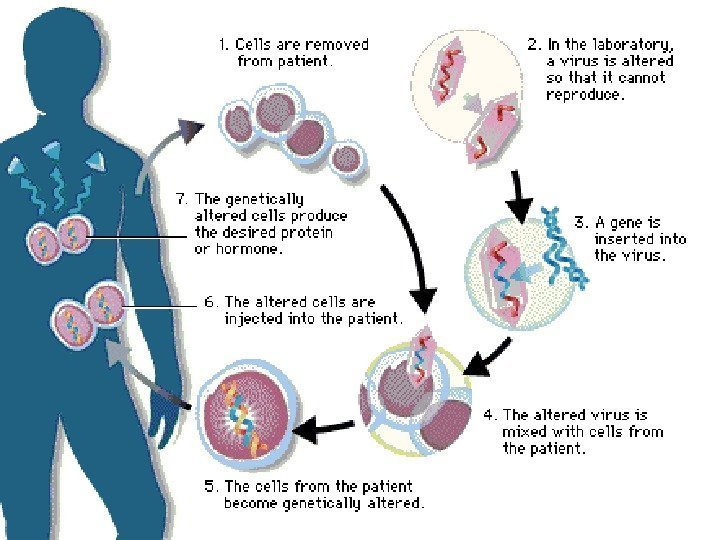
Gene therapy Diagnosis of Diseases Gene Therapy Gene therapy involves supplying a functional gene to cells lacking that function, with the aim of correcting a genetic disorder or acquired disease. It can be divided into two categories: 1)alteration of germ cells — a permanent genetic change for the whole organism and subsequent generations. 2)Somatic cell therapy, is analogous to an organ transplant.
Genetic therapy of Cancer 1. 1. Introduction in cell proteins – increasing immune system 2. 2. Introduction of virus genes – application of antivirus drugs 3. 3. Introduction of antioncogenes