Phylogeny_L4.ppt
- Количество слайдов: 50



СТРУКТУРНАЯ БИОИНФОРМАТИКА Карпов Павел Андреевич Лекция № 4
 «…великое Дерево Жизни заполняет земную кору своими мертвыми и сломанными ветвями и")
Деревья (trees) «…великое Дерево Жизни заполняет земную кору своими мертвыми и сломанными ветвями и покрывает поверхность вечно ветвящимися и прекрасными побегами» Ч. Дарвин

Задача построения филогенетического дерева The time will come, I believe, though I shall not live to see it, when we shall have fairly true genealogical trees of each great kingdom of Nature. Charles Darwin ь Биологические составляющие – • сравнение 3 -х и более объектов (кто на кого более похож. . ) • реконструкция эволюции (кто от кого, как и когда произошел…) ь Математическая составляющая – задача кластеризации, использование теории графов и комбинаторной оптимизации для того, чтобы на основе исходных биологических данных получить разумное с точки зрения эксперта-биолога дерево.

Реальные события : эволюция в природе или лаборатории, компьютерная симуляция в Данные: например, а. к. последовательности или количество усиков >Seq 1 ASGCTAFKL . . . ACGCTAFKL I -> L ACGCTAFKI A -> G GCGCTAFKI Построенное дерево >Seq 3 GCGCTLFKI >Seq 4 GCGCTGFKI. . . древовидный граф, вычисленный на основе данных, может отражать или не отражать реальные события

Рутинная процедура Составление выборки последовательностей Множественное выравнивание Построение дерева фрагмент записи в виде правильной скобочной структуры: (((((con 101: 38. 51018, (f 53969: 28. 26973, ((f 67220: 8. 39851, max 4: 27. 50591): 4. 92893, con 92: 30. 19677): 13. 62315): 9. 53075): 25. 83145, Визуализация и анализ дерева

Кладистический анализ Лист Узел Ветвь Корень «Клада»

Основные термины

Кладистический анализ • Кла да (от греч. κλάδος — «ветвь» , «ответвление» ; англ. clade) — группа организмов, которые являются потомками единственного общего предка и всех потомков этого предка. Термин используется в филогенетике. • Основное предположение кладистики заключается в том, что члены группы имеют общую эволюционную историю. Потому они более близко относятся друг к другу, чем к другим группам организмов. • Кладистические группы не зависят от того сходны ли организмы по физическим чертам, а зависят от их эволюционных взаимоотношений.

Допущения кладистического анализа • Виды являются только раздвоением, или отделением, из наследственной группы; • В случае гибридизации или горизонтального переноса генетической информации виды считаются исчезнувшими, а такие явления — редкими или отсутствующими. • Все виды в группе должны разделять общего предка и все виды, полученные от общего предка, должны войти в таксон.
, у которой все виды разделяют общего предка")
Возможные группы: • Монофилетическая группа (или клада), у которой все виды разделяют общего предка и все виды что происходят от этого общего предка включаются в группу. • Парафилетическая группа, у которой все виды разделяют общего предка, но не все виды, которые происходят от этого общего предка, включаются в группу. • Полифилетическая группа, в которой виды, которые не разделяют непосредственного общего предка, складываются одну группу, исключая виды, которые бы связали их.

Филогене тика • Филогене тика, или филогенети ческая система тика — область биологической систематики, которая занимается идентификацией и прояснением эволюционных взаимоотношений среди разных видов жизни на Земле, как современных, так и вымерших.

Молекулярная филогенетика • Молекулярная филогенетика — способ установления родственных связей между живыми организмами на основании изучения структуры полимерных макромолекул — ДНК, РНК и белков. Результатом молекулярно-филогенитического анализа является построение филогенетического дерева живых организмов. • • Макромолекулярные данные: нуклеотидные последовательности и белки. Поскольку геномы эволюционируют через постепенное накопление мутаций, количество отличий последовательности нуклеотидов между парой геномов разных организмов должно указать, как давно эти два генома разделили общего предка. Два генома, которые разделились в недавнем прошлом, должны иметь меньшие отличий, чем два генома, чей общий предок очень давний. Главная задача молекулярной филогенетики: сравнение разных геномов друг с другом, для получения сведений об эволюционных взаимоотношениях.

Неукоренённые деревья A A B B C A C D B C D D E E Неукоренённое дерево следует понимать как множество возможных укоренений E C A B D E

Небинарные деревья A A B C B F D C E F D E Небинарное дерево следует понимать как множество возможных «разрешений» A B C E D F

Топология дерева =
 =")
Расстояния по дереву между листьями 16 47 8 22 10 6 D(MOUSE, CAEEL) = 6+31+92 = 129 52 31 6 8 92 Дерево с заданными длинами ветвей порождает метрическое пространство, элементами которого являются листья

Ультраметрические деревья Если на дереве можно найти точку такую, что расстояния от нее до всех листьев одинаковы, до дерево называется “ультраметрическим”. Ультраметрическое дерево можно однозначно укоренить (в эту самую точку). Содержательно ультраметрические деревья соответствуют случаю, когда длины ветвей суть время эволюции (а все последовательности современны) Время эволюции можно восстанавливать в предположении «молекулярных часов»
 - метод датирования филогенетических событий, основанный на аксиоматическом утверждении, что")
Молекулярные часы (molecular clock) - метод датирования филогенетических событий, основанный на аксиоматическом утверждении, что нуклеиновым кислотам присуща практически постоянная скорость эволюционно значимых замен молекулярных мономеров (нуклеотидов). Отсюда следует, что время, прошедшее с момента расхождения каждой пары ветвей родословного древа ("сестринских групп"), пропорционально числу молекулярных замен при условии отсутствия селективного давления. Разница между ортологичными молекулами двух разных видов используется для определения времени, прошедшего с тех пор, когда два вида в последний раз имели общего предка (для расчета времени различных эволюционных событий. Реально скорость мутаций неравномерна и различна для разных видов, поэтому расчеты на основе молекулярных часов весьма приблизительны. Гипотеза молекулярных часов была выдвинута при сравнении гемоглобина и цитохрома С из разных видов. Цукеркэндл и Полинг (Emile Zuckerkandl, a biologist, and Linus Pauling, a chemist: Zuckerkandl, Pauling, 1962 , 1965 ) заметили что темпы аминокислотных замен в этих белках приблизительно одинаковы при сравнении различных млекопитающих. Они предположили, что для любого белка во всех филогенетических линиях темпы эволюции постоянны. Эта гипотеза породила множество споров. Классические эволюционисты выступали против, так как на морфологическом уровне не наблюдается никакого постоянства скоростей. Возражения против этой гипотезы усилились после датировки молекулярными методами времени расхождения человека и африканских человекообразных обезьян. По молекулярным данным эта цифра равнялась 5 миллионам лет, а по общепринятому мнению палеонтологов - 15 миллионам лет. Проведенные исследования (например, Goodman, 1981 ), показали, что не во всех линиях наблюдается постоянный темп эволюции: разным таксонам присущи разные темпы эволюции. Скорее всего не существует глобальных молекулярных часов, но есть локальные, внутри таксонов. Действительно, внутри таксонов часто не наблюдается достоверных различий в темпах замен. Различия в темпах эволюции часто объясняют эффектом времени генерации или различной эффективностью систем репарации, скорости замен увеличиваются после генных дупликаций и во времена адаптивной радиации. Тем не менее, несмотря на спорность гипотезы молекулярных часов, она широко использутся для оценок времен дивергенции видов и построения филогенетических деревьев.

Алгоритмы реконструкции филогении по множественному выравниванию CAEEL HUMAN MOUSE PROWI MARPO BRANA VICFA ASWRQLRDVKRREQIQEVGADRMRLKAIKFNTILPQAIRDEAAEKMQKAR VDWRMWRDVKRRKMAYEYADERLRINSLRKNTILPKILQDVADEEIAALP VDWRMLRDLKRRKMAYEYADERLRINSLRKNTILPKDLQEMAGDEIAALP MFNSIKRDLKRRKLYKKYESKRLLYKALISDCNLNQDLRFILTQKLNKLP MSNQIIRDHKRRLLVAKYELKRMHYKAICQDRNLPNKIRYEYFFKLSKLP SEKQNSRDHKRRLLAAKFELRRKLYKAFCKDPDLPSDMRDKHRYKLSKLP SEKRNIRDHKRRLLAAKYELRRKLYKAFCKDSDLPSDMRDKLRYKLSKLP

Алгоритмов много! Прежде всего, алгоритм либо предполагает "молекулярные часы", и тогда реконструированное им дерево — укорененное и ультраметрическое, либо не предполагает, и тогда дерево всегда не ультраметрическое и как правило не укоренённое.

Что значит «реконструировать» ? • Выбрать топологию из множества вариантов • Ещё приписать длины всем ветвям (но не все алгоритмы это делают)

Два типа алгоритмов • a. Использующие вычисление расстояний между последовательностями • b. "Символьно-ориентированные" Выравнивание Матрица расстояний b a Дерево
")
Другая классификация алгоритмов • a. Переборные алгоритмы • b. Эвристические алгоритмы (UPGMA & Neighbor-Joining) • Эвристика — это не полностью математически обоснованный (или даже «не совсем корректный» ), но при этом практически полезный алгоритм.

• UPGMA основывается на применении UPGMA теории молекулярных часов ( реконструирует укоренённое ультраметрическое дерево) • Neighbor-Joining не предполагает Neighbor-Joining использования молекулярных часов и не укореняет деревья Оба алгоритма работают только с матрицей расстояний

Идея кластерного алгоритма Ультраметрическое дерево строится «от листьев к корню» • Находим два самых близких листа и объединяем их в кластер. • Кластеру сопоставляем узел, соединённый с этими листами. • Вычисляем (тем или иным способом) расстояние от кластера до остальных листьев. • Листья можно считать кластерами из одного элемента. Находим пару ближайших кластеров и объединяем их в новый кластер. • и т. д. , пока не останется один кластер. Разница между разными кластерными алгоритмами только в способе вычисления расстояний между кластерами.

Неукоренённые деревья A A B B C A C D B C D D E E Неукоренённое дерево следует понимать как множество возможных укоренений E C A B D E

Неукоренённые деревья
: Вычисляются эволюционные")
Основные алгоритмы построения филогенетических деревьев Методы, основанные на оценке расстояний (матричные методы): Вычисляются эволюционные расстояния между всеми вершинами (OTUs) и строится дерево, в котором расстояния между вершинами наилучшим образом соответствуют матрице попарных расстояний. • UPGMA (Unweighted Pair Group with Arithmetic Mean) • Ближайших соседей (Neighbor-joining, NJ) Символьно-ориентированные методы: Наибольшего правдоподобия, Maximum likelihood, ML Используется модель эволюции и строится дерево, которое наиболее правдоподобно при данной модели Максимальной экономии (бережливости), maximum parsimony, MP Выбирается дерево с минимальным количеством мутаций, необходимых для объяснения данных

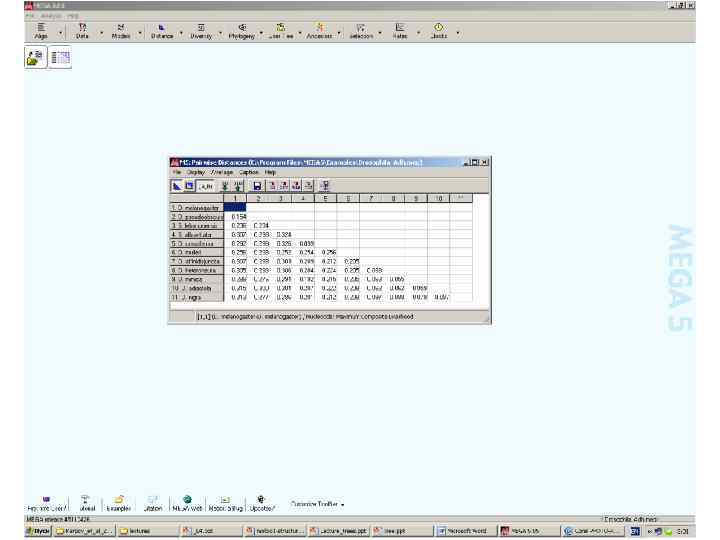
Методы, основанные на оценке расстояний • • Дано: М – матрица n x n, где Mij>=0 , Mij – эволюционное расстояние между листьями (OUT - Operational Units of Taxonomy). Задача: Построить реберно взвешенное (an edge-weighted) дерево, где каждая вершина (лист) соответствует объекту из M , а расстояние, измеренное по дереву между вершинами (листьями) i and j соответствует Mij.
 • Выбираем 2 наиболее похожие вершины a, c. • Строим")
UPGMA (алгоритм последовательной кластеризации) • Выбираем 2 наиболее похожие вершины a, c. • Строим новый узел k такой, что D(a, k)=D(b, k)=D(a, c)/2. • Пересчитываем матрицу попарных расстояний: D(b, a or c) = [ D(b, a) + D(b, c) ] /2 = (8+9)/2=8. 5 D(d, a or c) = [ D(d, a) + D(d, c) ] /2=(12+11)/2=11. 5 • Повторяем процедуру…. В конце концов получаем единственное ультраметрическое укорененное дерево • =11. 5

Не пользуйтесь UPGMA! Алгоритм строит ультраметрическое дерево, а это означает, что скорость эволюции одинакова для всех ветвей дерева. Использовать этот алгоритм имеет смысл только в случае ультраметрических данных (объектов эволюционирующих с одинаковой скоростью). реальное c точки зрения эксперта дерево UPGMA
 1. Рисуем «звездное» дерево и будем \"отщипывать\" от него")
Метод ближайших соседей (Neighbor-joining, NJ) 1. Рисуем «звездное» дерево и будем "отщипывать" от него по паре вершин, рассмотрим все возможные пары вершины. пусть - «среднее» расстояние до других вершин. 2. Выберем 2 вершины i и j с минимальным значением Mij – ui –uj т. е. выбираем 2 узла, которые близки друг к другу, но далеки ото всех остальных.
 3. Кластер (i, j) – новый узел дерева Расстояние")
Метод ближайших соседей (Neighbor-joining, NJ) 3. Кластер (i, j) – новый узел дерева Расстояние от i или от j до узла (i, j): di, (i, j) = 0. 5(Mij + ui-uj) dj, (i, j) = 0. 5(Mij + uj-ui) т. е. длина ветви зависит от среднего расстояния до других вершин. 4. Вычисляем расстояние от нового кластера до всех других M(ij)k = Mik+Mjk – Mij 2 5. В матрице М убираем i и j и добавляем (i, j). Повторяем, пока не останутся 2 узла. . .

Идея алгоритма Neighbor-Joining A B Находим пару листьев A, B, для которых сумма длин веток такого дерева минимальна. Длины получаются из матрицы расстояний.

Идея алгоритма Neighbor-Joining A B В наиболее распространённом варианте A и B — такая пара последовательностей, для которых минимальна величина (A, B) – M(A, B), где — расстояние из матрицы, а M — среднее расстояние от A и B до всех остальных последовательностей.

Идея алгоритма Neighbor-Joining A B Такие «соседи» дальше рассматриваются как один лист. «Объединение соседей» продолжается, пока не останутся только три «листа» . В отличие от кластерных алгоритмов, NJ не находит корня!
 • Строит бескорневое аддитивное дерево • • • Может")
Метод ближайших соседей (Neighbor-joining, NJ) • Строит бескорневое аддитивное дерево • • • Может работать с большим количеством данных Достаточно быстрый алгоритм Хорошо зарекомендовал себя на практике: если есть недвусмысленное с точки зрения эксперта дерево, то оно будет построено. Используется при множественном выравнивании с помощью программы Clustal. W Могут появиться ветви с длиной <0 • •

Достоверность топологии. Bootstraps. Есть множественное выравнивание и построенное по нему дерево. Верим ли мы в топологию дерева? • Создадим псевдоданные: N множественных выравниваний той же длины, что и исходное, каждое из псевдовыравниваний - случайный набор столбцов из исходного. • Построим N деревьев: на каждом внутреннем узле отметим долю случаев из N, в которых появлялся этот узел. Обычно верят в топологию, если метки узлов на бутстрепном дереве больше 70 -80%. Если меньше 30%, то не верим. В иных случаях – думаем… Бутстреп-анализ позволит оценить статистическую надёжность каждого из узлов построенного древа.

Traditional Human Chimp Gorilla Molecular Human Chimp Gorilla Orangutan Gibbon

Trees plagiarized by Chuck Staben, 1998 Sergeant Joyce Kilmer, 1914

Phylogeny_L4.ppt