e48434405bcf4bb65bdf120c0263b793.ppt
- Количество слайдов: 72



St. Jude Leukemia/Lymphoma Board 20 July 2010 Bone Marrow Failure and Success Introduced by Winfred C. Wang, MD Ross Perko, MD Asha B. Pillai, MD Winfred C. Wang, MD

St. Jude Leukemia/Lymphoma Board 20 July 2010 Bone Marrow Failure and Success Ross Perko, MD

Case Presentation • 8 y. o. AA female presented to the Le Bonheur ED on 7 -4 -10. • CC: multiple pre-syncopal episodes over 2 -3 weeks, one occurring immediately prior to presentation.

• HPI: – On the day of admission, this patient was riding her bicycle with her older brother when she started to feel very tired and stopped to get off the bike. She then started to feel dizzy and lightheaded and slowly fell to the ground and scraped her knee. She states that her legs were trembling. She did not lose consciousness and her brother came to her aid and helped her to her feet and she collapsed again. The brother then carried her to the house to their mother for help.

• In the ED: – Patient’s mother reported that the child had two other episodes similar to this in the last 3 weeks, but did not have any loss of consciousness with them. The patient has been more pale over this timeframe. Over the past week, the patient has had increased weakness and clumsiness. She has had no recent illnesses. No one else at home has been sick.
, 8 lbs 13 oz. Clavicle broken during")
• PMH: Full term (38 weeks), 8 lbs 13 oz. Clavicle broken during delivery. Hospitalized at 2 yrs for bronchiolitis. Seasonal allergies. • PSH: PE tubes, age 5 yrs • Allergies: NKDA • Transfusion Hx: none • Immunizations: UTD per mother

• ROS: • Constitutional: weakness, fatigue and decreased activity of sudden onset. Decreased appetite with 5 pound weight loss over the past few months. • HEENT: : no scleral icterus. No bleeding gums, no epistaxis, no sore throat or congestion. • RESP: shortness of breath with activity, no cough. • CV: tachycardia, decreased exercise activity, no edema • GI/GU: loss of appetite, no nausea, emesis, diarrhea or constipation. No blood seen in stools or with bathroom use. No abdominal pain, no change in bowel habits. • Heme/Lymph: no recent easy bruising, no bleeding tendencies, no petichiae, no swollen lymph glands. • MSK: no back or joint pain. No joint swelling.
: 37. 40 C –")
• Vital signs in the ED – T (oral): 37. 40 C – Pulse: 140 – BP: 115/57 – RR: 24 – O 2 saturation: 100% on room air – Weight: 33 kg

• PE: – Lying in bed, NAD, very polite, inquisitive and bright female who was playful during the exam. – HEENT: no dysmorphic facies, eyes w/o scleral icterus or discharge. Nares patent. Mouth no lesions or plaques with normal dentition. No petechiae or purpura noted. No blood in the nares. – NECK: supple, full ROM, no lymphadenopathy. – CHEST: CTAB, no wheezes or rhonchi. – HEART: Regular rhythm with tachycardia, sinus based on the room cardiac monitor. Cap refill less than 2 seconds in upper and lower extremities. – ABD: Soft, nontender, and nondistended. No hepatopsplenomegaly, bowel sounds nl. – SKIN: No rashes or other lesions, no ecchymosis or petichiae. One isolated hyperpigmented birthmark on the left upper thigh 1 cm squared in size. – MSK: Normal appearance of hands and thumbs and toes with no thenar hypoplasia and no syndactyly.

• LABS: – – Na 141, K 4, Cl 106, CO 2 23, BUN 12, Cr 0. 61, glu 85, Ca 9. 5 Total protein 7. 8, albumin 4. 4 Total bili 0. 3, alk phos 174, AST 24, ALT 7 LDH 490, uric acid 4. 7 – – CBC: WBC 3. 4, hb 2. 8, platelet 4 Diff: 5 N, 94 L, 1 M; ANC 170 MCV 135. 8, MCH 34. 6, MCHC 25. 5, RDW 14. 9 Reticulocytes < 0. 1%, ARC 0 – UA: normal – AB positive, DAT negative

• Peripheral Blood Smear: – Decrease in all cell lines – WBC: predominantly small lymphocytes—normal, mature, nonactivated – Red blood cells macrocytic and normochromic – Scant platelets, normal morphology

• Family Hx: – Maternal: Mother and maternal aunt had history of “anemia” associated with heavy menstrual flow. Maternal aunt had received multiple transfusions thought to be secondary to fibroids. Mother had GB removed after delivering the patient. – Paternal: Father paraplegic secondary to accident. Pancreatic ca in maternal uncle. – No history of leukemia, sickle cell disease or thalassemia.

• Social Hx: – Patient lives at home with mother, father, and 2 full siblings (14 y. o. brother and 1 y. o. sister). – Completed 3 rd grade, no academic concerns. • Exposures: No exposure to ionizing radiation, chemicals, pesticides or heavy metals. No recent travel outside the state.

• Patient admitted to the PICU and given 1 unit of PRBC (approximately 250 m. L) over two infusion periods; transfusion tolerated well. • 7/5/10: Patient transferred to St. Jude for further workup and preparation for a bone marrow biopsy and aspirate the following day (after receiving another unit of PRBC and a platelet transfusion). • 7/6/10: Bone marrow exam (to be shown). • 7/13/10: Placement of double-lumen Hickman line.
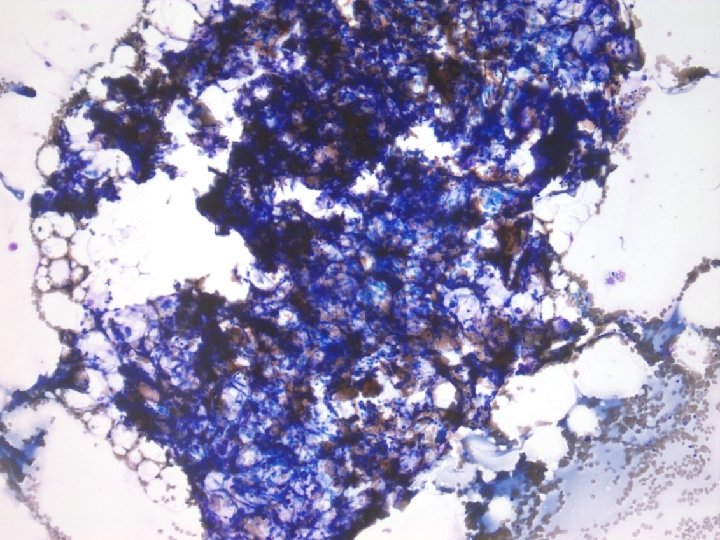
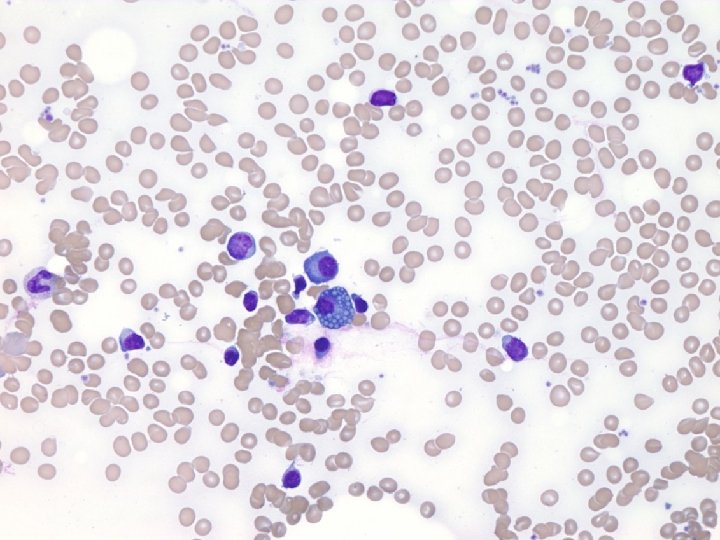

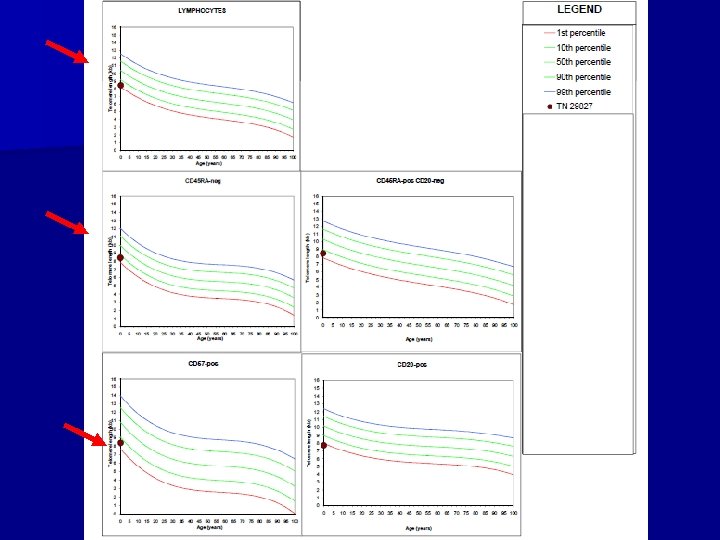
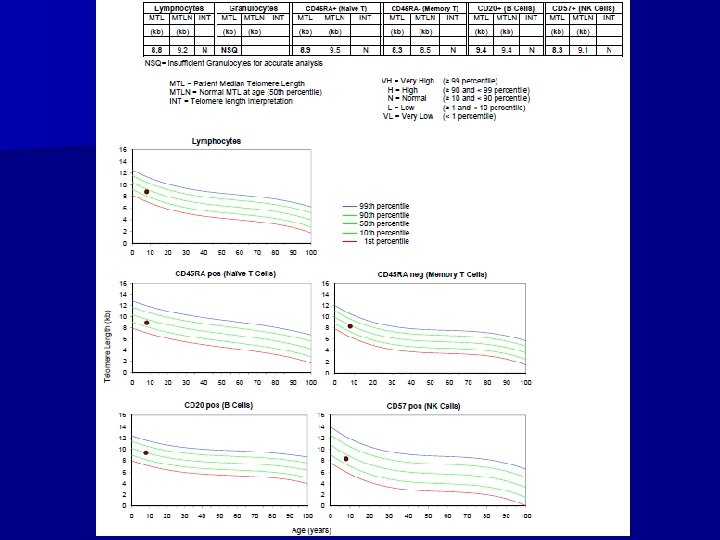
• Further work-up/lab studies. – Negative: • Active EBV, CMV, parvovirus, HIV and hepatitis infection • PNH screen – Normal: • Quantitative immunoglobulins • Serum folate and vitamin B 12 • Telomere length – Studies Pending: • Fanconi anemia testing (chromosome fragility) – HLA testing on family

St. Jude Leukemia/Lymphoma Board 20 July 2010 Bone Marrow Failure and Success Winfred C. Wang, MD

Aplastic Anemia Definition n Reduced or absent production of blood cells in the bone marrow – Acquired – Inherited (genetic, not present at birth) – Congenital (present at birth)

Acquired Aplastic Anemia Epidemiology n Incidence n Two peak ages: 15 -25 yrs, >60 yrs n More n No 2 cases/106 common in Asia (4 -7/106) sex or racial differences n Geographic variation is most likely due to environmental causes n Cause in an individual patient usually impossible to determine

Acquired Aplastic Anemia Etiology n Viruses – Non-A, non-B, non-C hepatitis § 0. 07% of hepatitis patients; 2 -5% of AA patients, 10% in Asia; male; <20 yrs old – Epstein-Barr virus – Cytomegalovirus – Human Immunodeficiency Virus – Human Parvovirus § Associated with immunocompromise § Differs from “aplastic crisis” in hemolytic anemia

Acquired Aplastic Anemia Etiology: Drugs and Toxins n Agents that regularly produce marrow depression – Benzene and chemicals containing benzene – Cytotoxic cancer chemotherapy, alkylating drugs: busulphan, melphalan, cyclophosphamide n Agents with low probability relative to use – Chloramaphenicol, chloroquine, sulfonamides – Insecticides: Chlorophenothane (DDT), γ-benzene hexachloride (lindane), parathion – Antioconvulsants: carbamazepine, hydantoins – Nonsteroidal anti-inflammatory agents: indomethacin, ibuprofen

Acquired Aplastic Anemia Etiology n Immune diseases – Hypogammaglobulinemia – Post-transfusion Gv. HD in immunodeficiencies n Paroxysmal nocturnal hemoglobinuria (PNH) n Myelodysplasia/myelofibrosis n Pregnancy

Aplastic Anemia Classification of Severity n Severe aplastic anemia – Bone marrow cellularity <25% – Two of three peripheral blood criteria: § ANC <500/cu. mm. § Platelets <20, 000/cu. mm. § Reticulocytes <20, 000/cu. mm. – No other hematologic disease n Moderate aplastic anemia – Bone marrow cellularity <50% – 2 or 3 cytopenias for >6 weeks: § ANC <1, 500/cu. mm. § Platelets <100, 000/cu. mm. § Reticulocytes <40, 000/cu. mm. Camitta BM et al. 1976, Khatib Z et al. 1994

Aplastic Anemia Presentation and Evaluation n CBC n Bone marrow aspirate and biopsy n Other blood tests – Morphology, reticulocyte count – Morphology, cellularity, cytogenetics – – – – LFTs, renal function tests Viral serologies PNH studies Chromosome breakage studies HLA typing Autoimmune disease evaluation Telomere length studies? SBDS gene mutation? C-mpl gene mutation?

Aplastic Anemia Clinical Course n Initial risk from infection, hemorrhage, transfusions n Response to immunosuppression: 75% at 6 months n Relapse after response to IS: 30% at 10 years n Clonal evolution and second malignancies – – – Overall risk: (10 -20% at 5 years) Paroxysmal nocturnal hemoglobinuria (10% at 2 -11 years) Myelodysplastic syndrome/AML (8% at 7 -10 years) Solid tumors (11%) Cytogenetic abnormalities (10%)
 Yes No HSCT")
Aplastic Anemia Treatment Algorithm Severe Moderate HLA-identical donor Observation (? ) Yes No HSCT ATG/Cs. A Non-responder Responder Allogeneic BMT Follow for relapse or late clonal disease
 (+Prednisone) 40 mg/kg/d x 4 days")
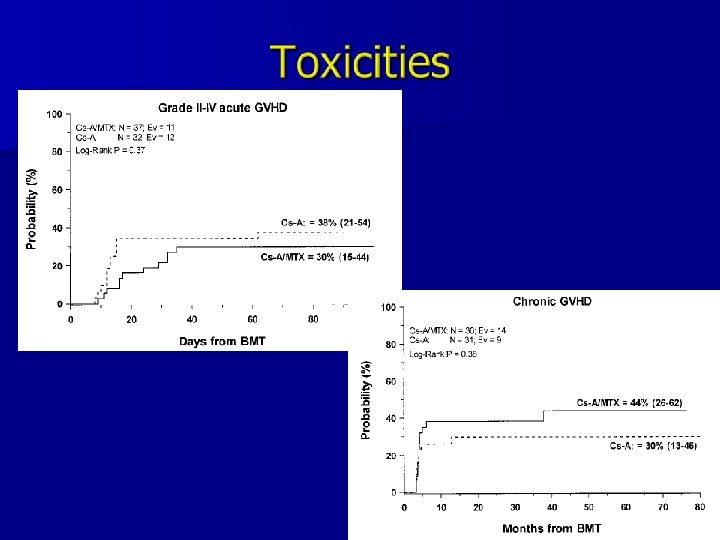
Severe Aplastic Anemia Immunosuppressive Therapy Anti-thymocyte globulin (ATG) (+Prednisone) 40 mg/kg/d x 4 days Cyclosporine (CSA) Therapeutic level until transfusion-independence for 2 months; then taper GM slowly; then CSA slowly GM-CSF (also possible: G-CSF or no growth factor) G-CSF => no difference in survival, but difference in time to neutrophil recovery Early identification of non-responders
 (Working Party Severe Aplastic Anemia) Bacigalupo A. Hematology 2007: 23")
Immunosuppressive Treatment (1991 -2002) (Working Party Severe Aplastic Anemia) Bacigalupo A. Hematology 2007: 23 -28
")
Aplastic Anemia Prognosis n Patients <20 years old: 10 yr. actuarial survival (with immunosuppression) = 73% n Age >16 y. o. worse outcome n Interval between dx and rx >23 d worse outcome n ANC at dx no effect on outcome

Clonal Evolution: Coefficients of selection in normal bone marrow and in aplastic anemia Bagby G. C. et al. Hematology 2007; 40 -46

APLASTIC ANEMIA: S. J. EXPERIENCE [Jeng, et al. : GM-CSF + immunosuppression in the treatment of pediatric SAA. PBC (2005) 45: 170. ] n Retrospective review of rx of SAA (n = 18, 1996 -2001) n Median age = 7. 2 yr. (range, 1. 8 -17 yr. ) n Treatment: ATG (+short course of Pred), Cyclosporine, GM-CSF n Response to treatment: CR 10 (56%) PR 4 (22%) NR 4 (22%)
 n Median time to: ANC >500 41 days")
APLASTIC ANEMIA: S. J. EXPERIENCE (continued) n Median time to: ANC >500 41 days ANC >1500 51 PRBC tx independence 94 platelet tx independence 64 D/C treatment 287 n Infections: n = 17 (in 6 pts) over 36 pt. -yr. n Conclusions: – GM-CSF (+IS) may shorten time to ANC recovery – GM-CSF probably does not affect response rate
 Pancytopenias Single Cytopenias Fanconi anemia (FA)")
Inherited Bone Marrow Failure Syndromes (the Big 7) Pancytopenias Single Cytopenias Fanconi anemia (FA) Red blood cells Diamond-Blackfan anemia (DBA) Dyskeratosis congenita (DC) Shwachman Diamond syndrome (SDS) Congenital amegakaryocytic thrombocytopenia (CAMT) White blood cells Severe congenital neutropenia (Kostmann syndrome) Cyclic neutropenia Platelets Thrombocytopenia with absent radii (TAR)

Dyskeratosis Congenita n Underrecognized n Physical in pediatrics findings with increasing age – Lacey reticulated pigmentation – Dysplastic nails – Oral leukoplakia n Genetic patterns: Autosomal recessive, autosomal dominant, X-linked

Dyskeratosis Congenita n Leukoplakia n Dysplastic n Reticular nails Rash Viguié C. Dyskeratosis congenita (DKC). Atlas Genet Cytogenet Oncol Haematol. August 2001. http: //Atlas. Genetics. Oncology. org/Kprones/Dyskeratos. ID 10034. html

St. Jude Leukemia/Lymphoma Board 20 July 2010 Bone Marrow Failure and Success Asha B. Pillai, MD

Congenital/Constitutional Fanconi anemia Dyskeratosis congenita Shwachman-Diamond syndrome Amegakaryocytic thrombocytopenia Familial aplastic anemia n Acquired Idiopathic Secondary Drug/Toxin exposure Infection-associated Immune disorders including neoplasms Paroxysmal nocturnal hemoglobinuria (constitutional) n
")
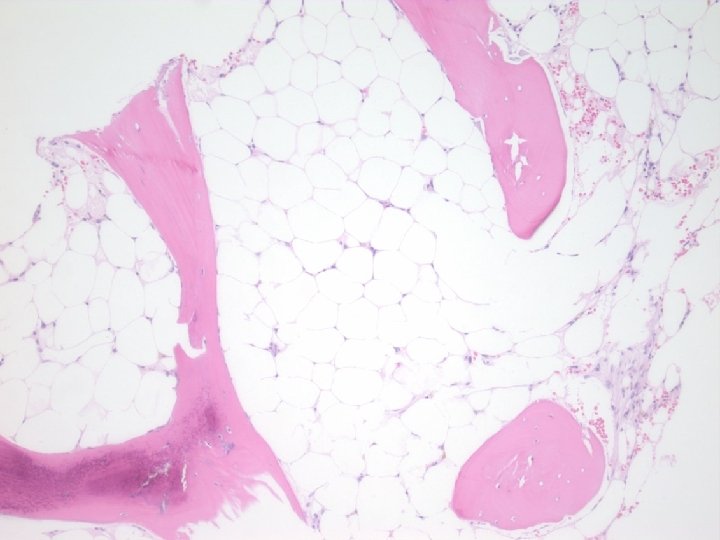
Aplastic Bone Marrow Biopsy (10 X)

n Constitutional AA are linked to DNA-repair defects or other types of chromosome instability n Stromal defects or decreased activity of multi-drug resistance P-glycoprotein has been suggested in drug induced AA n Acquired AA: the primary pathophysiologic mechanism is likely antigen driven, immune mediated, T-cell destruction of progenitor cells.
 provokes an aberrant immune response. n This")
n An inciting event (virus, drug, etc) provokes an aberrant immune response. n This immune response triggers oliogoclonal expansion of cytotoxic T cells that destroy hematopoietic stem cells. n Treatment of Acquired AA is based on attempts at eradicating or suppressing pathogenic T cell clones.

Young, Neal. Rodrigo Calado and Philip Scheinberg. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006 108: 2509 -2519.

n Cellular and molecular pathways have been mapped in detail for T lymphocytes in patients with severe aplastic anemia. • Constitutive T-bet expression n Activation of T cells and oligoclonal expansion facilitate apoptosis of hematopoietic cells via a gammainterferon activated Fas-dependent pathway.

Neal Young , Rodrigo Calado and Philip Scheinberg. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006 108: 2509 -2519.

n Treatment failures reported in as many as 40 -50% of patients. n Clonal complications may develop in 2545% of patients receiving IST in 7 -11 years of therapy. – Myelodysplasia – Paroxysmal nocturnal hemoglobinuria.

n Rosenfeld S et al. 2003 – Chromosomal abnormalities 12/122 pts – Risk of PNH 10% at 2 -7 yrs n Frickhofen N et al. 2003 – PNH 10% at 11 yrs (5 pts, 2 clinical sx) – MDS/Leukemia 8%, 6. 6 -9. 5 yrs post tx – Solid tumor 11%, 1 -11 yrs post tx

n The risks of treatment complications, especially treatment related mortality, must be weighed against the probability of long term cure. n Bone marrow transplant is curative, but carries a significant risk of transplantrelated mortality and long-term complications, particularly in the unrelated donor setting.
 age (<20 years) n Short interval from")
n Matched n Younger Sibling Donor (MSD) age (<20 years) n Short interval from diagnosis to transplant n Conditioning without TBI n Higher resolution typing/HLA matching

n High rate of graft failure – Heavy transfusion exposure allosensitization in patients who do not go directly to transplant n Risk of treatment related toxicity – More intense preparative regimens to overcome allosensitization (i. e. TBI) can lead to increased Gv. HD and long term toxicities • Early/late toxicities of conditioning regimen
 § (1 -3% after HLA-identical")
n Graft Failure – Rates after HLA-identical sibling (~10%) § (1 -3% after HLA-identical sibling transplant in leukemia) – May result from persistence of donor immunocompetent cells, capable of recognizing minor histocompatibibility differences on donor cells or of immune reactions against both host and donor hematopoietic precursors. – Rate increase with increasing age – Exposure to increased numbers of transfusions and long durations of disease § Sensitization to donor histocompatibility antigens. Champlin RE, Horowitz MM, van Bekkum DW, et al: Graft failure following bone marrow transplantation for severe aplastic anemia: Risk factors and treatment results. Blood 73: 606 -613, 1989

n In severe aplastic anemia, HLA-identical related donor bone marrow transplant is considered the gold standard. n Storb et al. (2001) reported an 88% event -free survival for 81 patients with SAA receiving MSD transplantation.
 Blood Rev. 2005 May; 19(3): 143 -51.")
<10 years; n=156, 89%; 83 -94%) Blood Rev. 2005 May; 19(3): 143 -51.

• Requires conditioning • Cyclophosphamide/ATG current standard n High-dose necessary immune suppression is – Eliminate/suppress active immune processes involved in the primary pathogenesis of SAA – Eliminate/suppress immune-competent recipient cells capable of rejecting the donor graft

n 1/4 of otherwise eligible transplant candidates will have an HLA-identical related donor n First successful matched unrelated donor (MUD) HCT in SAA described in 1982 by Gordon-Smith and colleagues.

n MUD transplants have a higher rate of graftfailure and graft-versus-host disease than do HLA-identical related donor transplants – Greater degree of disparity for both major and minor histocompatibilityantigens than MSD. n Due to this, historically mortality is increased and risk-benefit ratio has not favored upfront MUD HCT for SAA pts lacking MSD
 n Most MUD transplant are performed relatively late, and in")
Matched Unrelated Donor (MUD) n Most MUD transplant are performed relatively late, and in patients who have failed to respond to 1 or more courses of immune suppression. n Availability of more sensitive DNA-based HLA typing techniques have also allowed improved selection of donors and hence better outcome by reducing graft rejection and Gv. HD.

n MUD – 218 patients – 3 year survival 53% n MSD – 732 patients – 3 year survival 82% Kennedy-Nasser, Alana, Kathryn Leung, Anita Mahajan, Heidi Weiss, James Arce, Stephen Gottschalk, George Carrum, Shakila Khan, Helen Heslop, Malcolm Brenner, Catherine Bollard, Robert Krance. Comparable outcomes of matched-related and alternative donor stem cell transplantation for pediatric severe aplastic anemia. Biology of Blood and Marrow Transplantation. 2006; 12: 1277 -1284

Horowitz, Mary. Current status of Allogeneic Bone Marrow Transplantation in Acquired Aplastic Anemia. Seminars in Hematology, vol 37, No 1, 2000: pg 30 -42
 Non-responder MUD HSCT")
Severe Aplastic Anemia HLA-identical donor Yes No MSD HSCTATG/Cs. A (IST) Non-responder MUD HSCT Responder Follow for relapse and/or late clonal disease
 retrospectively reviewed pediatric cohort who received allogeneic BMT (15")
n Kennedy-Nasser and colleagues (2006) retrospectively reviewed pediatric cohort who received allogeneic BMT (15 MSD, 23 alternative/non-MSD donors including MUD). – April 1997 to October 2005 – 36 consecutive pediatric patients
: 1277 -84")
Kennedy-Nassar Biol Blood Marrow Transplant. 2006 Dec; 12(12): 1277 -84
 – CD 34 selected peripheral blood")
n Stem cell products – bone marrow (n=34) – CD 34 selected peripheral blood (n=4) n Gv. HD prophylaxis – Cyclosporine – Tacrolimus – Methotrexate
 – Cyclophosphamide 50 mg/kg, 4 days (-5")
n Matched Sibling donor (n = 15) – Cyclophosphamide 50 mg/kg, 4 days (-5 to -2) – Equine ATG 30 mg/kg, 3 days (-5 to -3) n Alternative Donors (n = 23) – Reduced intensity regimen § Cyclophosphamide 50 mg/kg for 4 days (-6 to -3) § Low dose TBI – 200 c. Gy for MUD (day -2) § Prior to 2000 (n=9) – Equine ATG 30 mg/kg for 3 days (-5 to -3) § After 2000 (n=14) – Humanized anti-CD 52 monoclonal antibodies (CAMPATH) for 4 days (-4 to -1)
of patients survived – Median follow up was 52 months n")
n 86. 1% (31/36)of patients survived – Median follow up was 52 months n 4 year OS was 93% (MSD) versus 89% (all AD including MUD). – No statistical difference seen between the two populations.
 – Neutrophil count >0. 5 x 109 –")
n Defined as sustained (3 -day) – Neutrophil count >0. 5 x 109 – Unsupported platelet count >20 x 109 n 37/38 patients were evaluable for engraftment. n 36/37 patients engrafted. – Median time to neutrophil engraftment =17 days (11 -44 days) – Median time to platelet engraftment 32 days (11 -282 days)

n None of the 15 MSD patients developed severe acute Gv. HD (grade III or IV) n 4 of 23 AD patients developed severe acute Gv. HD – 2/9 with ATG – 2/14 with Campath n Chronic Gv. HD – 0/15 MSD – 3/23 AD (all 3 received ATG in preparative regimen)

n n BMT from a HLA-identical sibling is an established initial treatment for patients with SAA. For patients without a HLA-matched related donor immunosuppressive treatment with antithymocyte globulin and cyclosporine is an alternative. Younger patients with severe neutropenia have better outcomes usinganupfront transplant strategy. Patients failing immunosuppressive therapy with a suitable alternative donor referral for transplantation. Viollier, R. G Socie, A Tichelli, A Bacigalupo, ET Korthof, J Marsh, J Cornish, et al. Recent improvement in outcome of unrelated donor transplantation for aplastic anemia. Bone Marrow Transplantation. 2008: 41, 45 -50

End Ross Perko, MD Winfred C. Wang, MD Asha B. Pillai, MD More medical education materials are available at: Cure 4 Kids is an initiative of St. Jude Children’s Research Hospital You may print and download content for personal educational use only. All material is copyrighted by the author of the content or St. Jude Children’s Research Hospital. See legal terms and conditions at http: //www. Cure 4 Kids. org
e48434405bcf4bb65bdf120c0263b793.ppt