Microsoft Office PowerPoint Presentation.pptx
- Количество слайдов: 17
 СРС НА ТЕМУ: Синдром Фелти. Синдром Стилла. ВЫПОЛНИЛА : НАЗАРОВА А. М ПРОВЕРИЛА : _______ ФАКУЛЬТЕТ: ОБЩАЯ МЕДИЦИНА ГРУППА: 004 -02 Р 2017
СРС НА ТЕМУ: Синдром Фелти. Синдром Стилла. ВЫПОЛНИЛА : НАЗАРОВА А. М ПРОВЕРИЛА : _______ ФАКУЛЬТЕТ: ОБЩАЯ МЕДИЦИНА ГРУППА: 004 -02 Р 2017
 Синдром Фелти – симптомокомплекс, включающий основную триаду признаков: ревматоидный артрит, спленомегалию и лейкопению. Кроме перечисленных симптомов, течение синдрома Фелти сопровождается лихорадкой, мышечной атрофией, пигментацией и язвами на коже голеней, полисерозитом, полиневропатией, эписклеритом, лимфаденопатией, гепатомегалией, склонностью к инфекционным заболеваниям. В диагностике синдрома Фелти решающее значение придается наличию в анамнезе ревматоидного артрита и лабораторным показателям (высоким титрам РФ, лейкопении, нейтропении, тромбоцитопении и др. ). Лечебный алгоритм при синдроме Фелти включает назначение кортикостероидов (солей лития или золота, D-пеницилламина), плазмафереза; проведение спленэктомии.
Синдром Фелти – симптомокомплекс, включающий основную триаду признаков: ревматоидный артрит, спленомегалию и лейкопению. Кроме перечисленных симптомов, течение синдрома Фелти сопровождается лихорадкой, мышечной атрофией, пигментацией и язвами на коже голеней, полисерозитом, полиневропатией, эписклеритом, лимфаденопатией, гепатомегалией, склонностью к инфекционным заболеваниям. В диагностике синдрома Фелти решающее значение придается наличию в анамнезе ревматоидного артрита и лабораторным показателям (высоким титрам РФ, лейкопении, нейтропении, тромбоцитопении и др. ). Лечебный алгоритм при синдроме Фелти включает назначение кортикостероидов (солей лития или золота, D-пеницилламина), плазмафереза; проведение спленэктомии.
 Синдром Фелти – самостоятельный вариант суставно-висцеральной формы ревматоидного артрита, сочетающий в себе полиартрит, лейкопению, увеличение селезенки, пигментацию кожи и другие клинико -лабораторные признаки. Синдром Фелти развивается примерно у 1 -5% больных ревматоидным артритом. В самостоятельную нозологию заболевание дифференцировано в 1929 году. Среди заболевших преобладают женщины (соотношение полов 3: 1) в возрасте старше 40 -50 лет. В ревматологии синдром Фелти встречается крайне редко, поэтому в вопросах этиологии, патогенеза, диагностической и лечебной тактики данной патологии остается много пробелов. Этиология синдром Фелти изучена еще в меньшей степени, чем ревматоидного артрита. Известно, что синдром является HLA-DRw 4 ассоциированным заболеванием. Механизм развития нейтропении объясняется тем, что образующиеся в организме больных с синдромом Фелти циркулирующие иммунные комплексы, нарушают функции нейтрофилов и активизируют их фагоцитоз, главным образом, в селезенке. Подтверждением тому служит обнаружение в цитоплазме нейтрофилов депозитов, содержащих иммуноглобулины (Ig. G, Ig. M) и комплемент, а также специфические антитела к лейкоцитам (в т. ч. антигранулоцитарные антитела). Кроме этого, в возникновении нейтропении предполагается участие гуморальных и клеточных факторов иммунитета, угнетающих лейкопоэз в костном мозге.
Синдром Фелти – самостоятельный вариант суставно-висцеральной формы ревматоидного артрита, сочетающий в себе полиартрит, лейкопению, увеличение селезенки, пигментацию кожи и другие клинико -лабораторные признаки. Синдром Фелти развивается примерно у 1 -5% больных ревматоидным артритом. В самостоятельную нозологию заболевание дифференцировано в 1929 году. Среди заболевших преобладают женщины (соотношение полов 3: 1) в возрасте старше 40 -50 лет. В ревматологии синдром Фелти встречается крайне редко, поэтому в вопросах этиологии, патогенеза, диагностической и лечебной тактики данной патологии остается много пробелов. Этиология синдром Фелти изучена еще в меньшей степени, чем ревматоидного артрита. Известно, что синдром является HLA-DRw 4 ассоциированным заболеванием. Механизм развития нейтропении объясняется тем, что образующиеся в организме больных с синдромом Фелти циркулирующие иммунные комплексы, нарушают функции нейтрофилов и активизируют их фагоцитоз, главным образом, в селезенке. Подтверждением тому служит обнаружение в цитоплазме нейтрофилов депозитов, содержащих иммуноглобулины (Ig. G, Ig. M) и комплемент, а также специфические антитела к лейкоцитам (в т. ч. антигранулоцитарные антитела). Кроме этого, в возникновении нейтропении предполагается участие гуморальных и клеточных факторов иммунитета, угнетающих лейкопоэз в костном мозге.
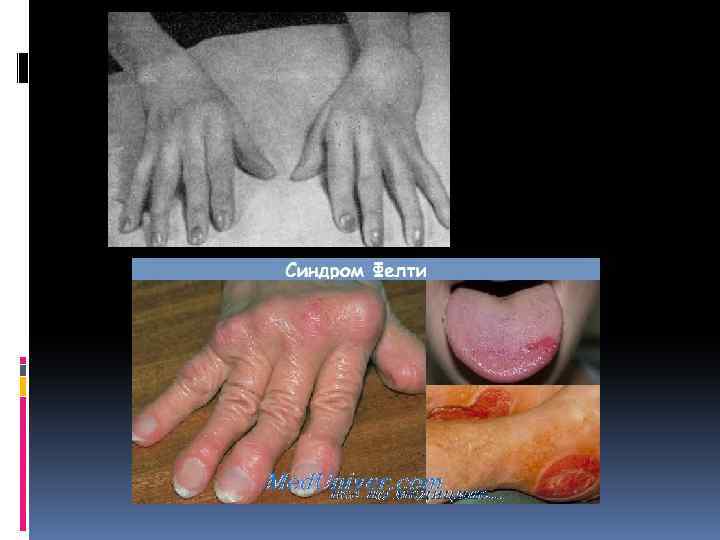
 Симптомы синдрома Фелти Синдром Фелти возникает у пациентов с серопозитивным ревматоидным артритом (т. е. с положительным ревматоидным фактором) в среднем спустя 10 лет после поражения суставов. Суставная симптоматика характеризуется развитием полиартрита с преимущественным вовлечением мелких суставов кистей и стоп. Характерно наличие и других проявлений ревматоидного артрита: подкожных узелков, лихорадки, полиневропатии, амиотрофии, генерализованного лимфаденита. Кроме этого, системные висцеральные поражения при синдроме Фелти включают миокардит, полисерозит, эписклерит, гепатомегалию. Селезенка на ощупь плотная, безболезненная; ее размеры обычно увеличиваются значительно, а вес в среднем в 4 раза превышает норму. В половине случаев развивается синдром Шегрена; возможно появление пигментации и язв на коже голеней. Вследствие лейкопении и угнетения иммунитета больные с синдромом Фелти склонны к инфекционной заболеваемости: повторным ОРВИ, пневмониям, рецидивирующим пиодермиям и пр. Течение синдрома Фелти может осложниться разрывом селезенки, портальной гипертензией, желудочно-кишечными кровотечениями, амилоидозом.
Симптомы синдрома Фелти Синдром Фелти возникает у пациентов с серопозитивным ревматоидным артритом (т. е. с положительным ревматоидным фактором) в среднем спустя 10 лет после поражения суставов. Суставная симптоматика характеризуется развитием полиартрита с преимущественным вовлечением мелких суставов кистей и стоп. Характерно наличие и других проявлений ревматоидного артрита: подкожных узелков, лихорадки, полиневропатии, амиотрофии, генерализованного лимфаденита. Кроме этого, системные висцеральные поражения при синдроме Фелти включают миокардит, полисерозит, эписклерит, гепатомегалию. Селезенка на ощупь плотная, безболезненная; ее размеры обычно увеличиваются значительно, а вес в среднем в 4 раза превышает норму. В половине случаев развивается синдром Шегрена; возможно появление пигментации и язв на коже голеней. Вследствие лейкопении и угнетения иммунитета больные с синдромом Фелти склонны к инфекционной заболеваемости: повторным ОРВИ, пневмониям, рецидивирующим пиодермиям и пр. Течение синдрома Фелти может осложниться разрывом селезенки, портальной гипертензией, желудочно-кишечными кровотечениями, амилоидозом.
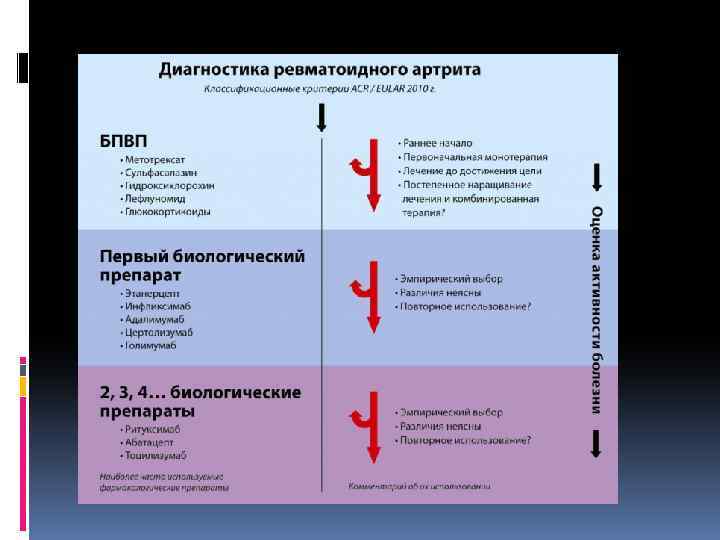
 Диагностика и лечение синдрома Фелти Основу для постановки клинического диагноза составляет сочетание ревматоидного полиартрита и спленомегалии. Для общего анализа крови характерны лейкопения, нейтропения, тромбоцитопения, анемия. Иммунологическое исследование крови при синдроме Фелти обнаруживает высокие титры РФ, наличие антинуклеарных антител (особенно антигранулоцитарных АТ), ЦИК, гипергаммаглобулинемию (повышение Ig. G и Ig. M). По данным миелограммы выявляется миелоидная гиперплазия костного мозга со сдвигом в сторону незрелых клеточных элементов. Данные инструментальных исследований (УЗИ суставов, рентгенографии, МРТ и КТ) малоинформативны. Синдром Фелти следует дифференцировать с лимфомами, саркоидозом, циррозом печени, гепатолиенальным синдромом. Поиск путей эффективного лечения синдрома Фелти продолжается. В настоящее время для стимуляции гранулоцитопоэза используются препараты лития, однако их применение может вызывать побочные эффекты: тремор, тубулоинтерстициальный нефрит, гипотиреоз. С целью уменьшения риска развития интеркуррентных инфекций целесообразно введение гранулоцитостимулирующего фактора. В рамках терапии, направленной на блокирование образования антигранулоцитарных AT, показано назначение глюкокортикоидов, базисных препаратов (D-пеницилламина, солей золота, метотрексата), проведение плазмафереза. При выраженной лейкопении и спленомегалии показана спленэктомия, однако у четверти больных даже после удаления селезенки нейтропения рецидивирует. Профилактика синдрома Фелти не разработана; прогноз весьма серьезный. Синдром Фелти увеличивает риск возникновения неходжкинских лимфом.
Диагностика и лечение синдрома Фелти Основу для постановки клинического диагноза составляет сочетание ревматоидного полиартрита и спленомегалии. Для общего анализа крови характерны лейкопения, нейтропения, тромбоцитопения, анемия. Иммунологическое исследование крови при синдроме Фелти обнаруживает высокие титры РФ, наличие антинуклеарных антител (особенно антигранулоцитарных АТ), ЦИК, гипергаммаглобулинемию (повышение Ig. G и Ig. M). По данным миелограммы выявляется миелоидная гиперплазия костного мозга со сдвигом в сторону незрелых клеточных элементов. Данные инструментальных исследований (УЗИ суставов, рентгенографии, МРТ и КТ) малоинформативны. Синдром Фелти следует дифференцировать с лимфомами, саркоидозом, циррозом печени, гепатолиенальным синдромом. Поиск путей эффективного лечения синдрома Фелти продолжается. В настоящее время для стимуляции гранулоцитопоэза используются препараты лития, однако их применение может вызывать побочные эффекты: тремор, тубулоинтерстициальный нефрит, гипотиреоз. С целью уменьшения риска развития интеркуррентных инфекций целесообразно введение гранулоцитостимулирующего фактора. В рамках терапии, направленной на блокирование образования антигранулоцитарных AT, показано назначение глюкокортикоидов, базисных препаратов (D-пеницилламина, солей золота, метотрексата), проведение плазмафереза. При выраженной лейкопении и спленомегалии показана спленэктомия, однако у четверти больных даже после удаления селезенки нейтропения рецидивирует. Профилактика синдрома Фелти не разработана; прогноз весьма серьезный. Синдром Фелти увеличивает риск возникновения неходжкинских лимфом.

 был впервые описан George Frederick Still в 1897 г. как") Ювенильный артрит (ЮА) был впервые описан George Frederick Still в 1897 г. как «особая форма болезни суставов, встречающаяся у детей» . Работа была основана на клиническом опыте автора как медицинского регистратора и патолога. G. F. Still был первым, кто подробно описал течение хронического артрита у 22 детей, 19 из которых он лечил. G. F. Still выделил у детей: ревматоидный артрит, артропатию Жакку и системное начало артрита, которое до сих пор носит его имя. Он также описал особенности поражения суставов у детей, отличающиеся от ревматоидного артрита взрослых, и подчеркнул, что заболевание начинается до потери молочных зубов, с половым диморфизмом 50: 50, лихорадкой, лимфаденопатией, спленомегалией, полисерозитом, анемией, отсутствием деформаций суставов и задержкой роста. Такая хроническая артропатия с острым началом ревматоидного артрита, сопровождавшимся лихорадкой, лимфаденопатией и/или спленомегалией, была описана у взрослых G. A. Bannatyne и A. Chauffard. Следует отметить, что описания единичных наблюдений с такой же клинической картиной встречались в литературе и до 1897 г. Характерная сыпь, названная «ревматоидной сыпью» или «сыпью Стилла» , в действительности была описана не самим G. F. Still, a M. E. Boldero, который в 1933 г. указал на преходящую эритематозную сыпь на разгибательных поверхностях тела. Комментарий профессора F. Langmead указывал на то, что сыпь связана с лихорадочными атаками. Более подробная характеристика экзантемы впоследствии была проведена I. S. Isdale и E. G. Bywaters, которые продемонстрировали строгую связь между «ревматоидной сыпью» и другими симптомами болезни: интермиттирующей лихорадкой, лимфаденопатией, спленомегалией, лейкоцитозом и увеличением СОЭ.
Ювенильный артрит (ЮА) был впервые описан George Frederick Still в 1897 г. как «особая форма болезни суставов, встречающаяся у детей» . Работа была основана на клиническом опыте автора как медицинского регистратора и патолога. G. F. Still был первым, кто подробно описал течение хронического артрита у 22 детей, 19 из которых он лечил. G. F. Still выделил у детей: ревматоидный артрит, артропатию Жакку и системное начало артрита, которое до сих пор носит его имя. Он также описал особенности поражения суставов у детей, отличающиеся от ревматоидного артрита взрослых, и подчеркнул, что заболевание начинается до потери молочных зубов, с половым диморфизмом 50: 50, лихорадкой, лимфаденопатией, спленомегалией, полисерозитом, анемией, отсутствием деформаций суставов и задержкой роста. Такая хроническая артропатия с острым началом ревматоидного артрита, сопровождавшимся лихорадкой, лимфаденопатией и/или спленомегалией, была описана у взрослых G. A. Bannatyne и A. Chauffard. Следует отметить, что описания единичных наблюдений с такой же клинической картиной встречались в литературе и до 1897 г. Характерная сыпь, названная «ревматоидной сыпью» или «сыпью Стилла» , в действительности была описана не самим G. F. Still, a M. E. Boldero, который в 1933 г. указал на преходящую эритематозную сыпь на разгибательных поверхностях тела. Комментарий профессора F. Langmead указывал на то, что сыпь связана с лихорадочными атаками. Более подробная характеристика экзантемы впоследствии была проведена I. S. Isdale и E. G. Bywaters, которые продемонстрировали строгую связь между «ревматоидной сыпью» и другими симптомами болезни: интермиттирующей лихорадкой, лимфаденопатией, спленомегалией, лейкоцитозом и увеличением СОЭ.
 Эпидемиология Артрит поражает примерно одного из 1000 детей, рождающихся каждый год. Однако примерно у одного из 10 000 детей артрит будет прогрессировать. Многие дети страдают от острой воспалительной формы артрита, которому сопутствует вирусная или бактериальная инфекция. Эта форма артрита, часто очень тяжёлая и сложная, начинается в течение короткого отрезка времени и обычно проходит через несколько недель или месяцев. Классификация Существует несколько различных форм артрита, которые поражают преимущественно детей; некоторые врачи придерживаются следующего определения болезни Стилла: это детское заболевание, основными проявлениями которого являются артрит (который часто поражает несколько суставов), перемежающаяся лихорадка и быстропроходящая красная сыпь. Иногда при тяжёлом течении болезни она может распространиться на всё тело и осложниться увеличением селезёнки и лимфатических узлов, а также воспалением перикарда и радужки глаз. Впервые заболевание описано патологом Джорджем Стиллом в 1897 г. Типичные признаки данной болезни наблюдались и у взрослых; Эрик Байуотерс в 1971 г. представил подробный разбор этих случаев.
Эпидемиология Артрит поражает примерно одного из 1000 детей, рождающихся каждый год. Однако примерно у одного из 10 000 детей артрит будет прогрессировать. Многие дети страдают от острой воспалительной формы артрита, которому сопутствует вирусная или бактериальная инфекция. Эта форма артрита, часто очень тяжёлая и сложная, начинается в течение короткого отрезка времени и обычно проходит через несколько недель или месяцев. Классификация Существует несколько различных форм артрита, которые поражают преимущественно детей; некоторые врачи придерживаются следующего определения болезни Стилла: это детское заболевание, основными проявлениями которого являются артрит (который часто поражает несколько суставов), перемежающаяся лихорадка и быстропроходящая красная сыпь. Иногда при тяжёлом течении болезни она может распространиться на всё тело и осложниться увеличением селезёнки и лимфатических узлов, а также воспалением перикарда и радужки глаз. Впервые заболевание описано патологом Джорджем Стиллом в 1897 г. Типичные признаки данной болезни наблюдались и у взрослых; Эрик Байуотерс в 1971 г. представил подробный разбор этих случаев.
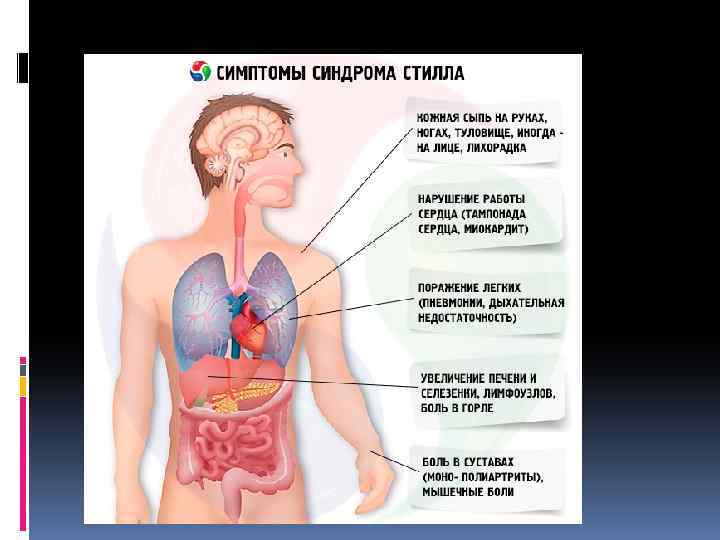
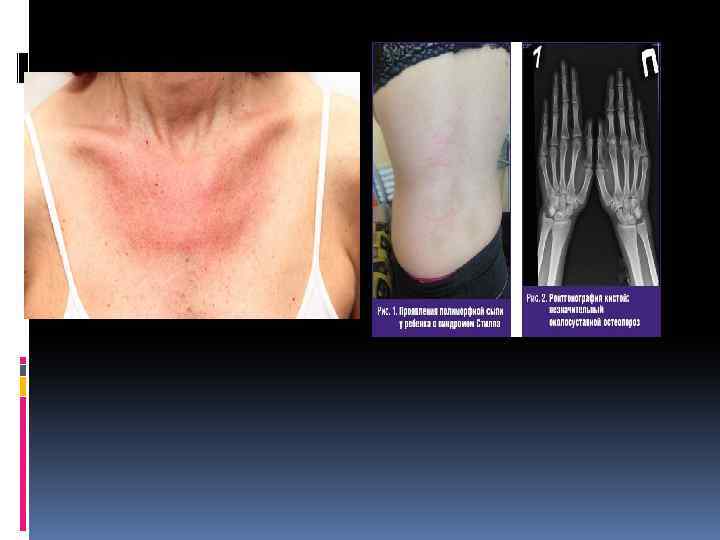
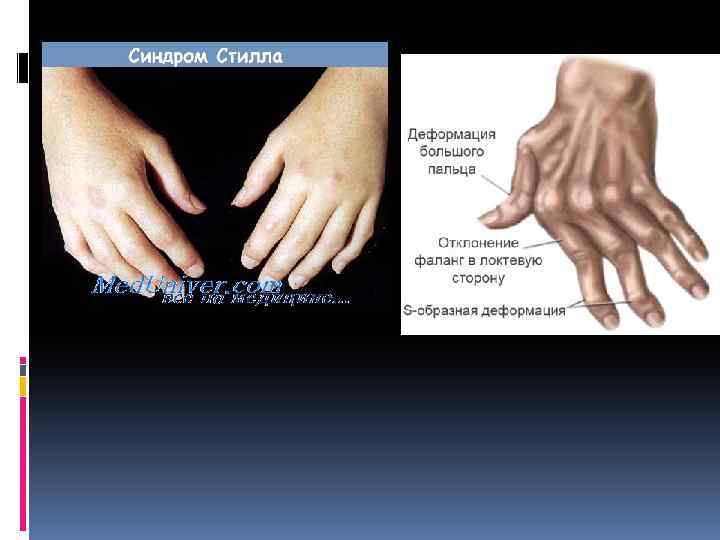
 Клиника Синдром Стилла характеризуется острым началом полиартрита с поражением крупных и мелких суставов и позвоночника, резкими болями и выраженными экссудативными явлениями с последующим развитием у трети больных деформации суставов. Наряду с этим отмечается высокая лихорадка, нередко предшествующая другим симптомам болезни, и полиморфные высыпания на коже лица, туловища и конечностей. Пятнистая или пятнистопапулезная экзантема появляется в часы максимального повышения температуры тела (чаще вечером) и исчезает при ее снижении. Отличительной особенностью кожной сыпи при синдроме Стилла считается возможность возникновения ее после трения или под воздействием теплой воды. Быстро развивается лимфаденопатия: лимфатические узлы плотные, безболезненные, подвижные, величиной не более мелкого ореха или вишни. Увеличиваются селезенка и печень, значительно нарушается общее состояние, наблюдается прогрессирующее похудание и выпадение волос. В дальнейшем температура тела снижается, кожные высыпания исчезают, но выявляются поражения внутренних органов: плеврит, пневмония, экссудативный перикардит (реже — миокардит, эндокардит), нефрит, полисерозит, полинейропатия и др. Редко наблюдается поражение глаз и амилоидоз почек. В периферической крови обнаруживаются стойкое повышение СОЭ, анемия, лейкоцитоз со сдвигом влево (иногда лейкопения), СРВ, повышение уровня серомукоида, трансаминаз и щелочной фосфатазы, гиперα 2 - и γглобулинемия. Характерным для синдрома Стилла считается отсутствие в крови ревматоидного (РФ) и антинуклеарного факторов (АНФ). На рентгенограммах суставов определяются остеопороз, некоторое сужение суставной щели, кисты. Выраженных эрозивных изменений костей и анкилозирования у большинства больных не происходит. Обычно болезнь протекает с обострениями и ремиссиями, причем высокая активность процесса может сохраняться до 5 лет. В отдельных случаях активность процесса и прогрессирование болезни заканчиваются в пубертатном возрасте, не оставляя при этом значительных деформаций. У других больных заболевание принимает непрерывнорецидивирующее течение с резким похуданием и задержкой развития ребенка и может закончиться через несколько летальным исходом от интеркуррентной инфекции или амилоидоза. У взрослых прогноз более благоприятный.
Клиника Синдром Стилла характеризуется острым началом полиартрита с поражением крупных и мелких суставов и позвоночника, резкими болями и выраженными экссудативными явлениями с последующим развитием у трети больных деформации суставов. Наряду с этим отмечается высокая лихорадка, нередко предшествующая другим симптомам болезни, и полиморфные высыпания на коже лица, туловища и конечностей. Пятнистая или пятнистопапулезная экзантема появляется в часы максимального повышения температуры тела (чаще вечером) и исчезает при ее снижении. Отличительной особенностью кожной сыпи при синдроме Стилла считается возможность возникновения ее после трения или под воздействием теплой воды. Быстро развивается лимфаденопатия: лимфатические узлы плотные, безболезненные, подвижные, величиной не более мелкого ореха или вишни. Увеличиваются селезенка и печень, значительно нарушается общее состояние, наблюдается прогрессирующее похудание и выпадение волос. В дальнейшем температура тела снижается, кожные высыпания исчезают, но выявляются поражения внутренних органов: плеврит, пневмония, экссудативный перикардит (реже — миокардит, эндокардит), нефрит, полисерозит, полинейропатия и др. Редко наблюдается поражение глаз и амилоидоз почек. В периферической крови обнаруживаются стойкое повышение СОЭ, анемия, лейкоцитоз со сдвигом влево (иногда лейкопения), СРВ, повышение уровня серомукоида, трансаминаз и щелочной фосфатазы, гиперα 2 - и γглобулинемия. Характерным для синдрома Стилла считается отсутствие в крови ревматоидного (РФ) и антинуклеарного факторов (АНФ). На рентгенограммах суставов определяются остеопороз, некоторое сужение суставной щели, кисты. Выраженных эрозивных изменений костей и анкилозирования у большинства больных не происходит. Обычно болезнь протекает с обострениями и ремиссиями, причем высокая активность процесса может сохраняться до 5 лет. В отдельных случаях активность процесса и прогрессирование болезни заканчиваются в пубертатном возрасте, не оставляя при этом значительных деформаций. У других больных заболевание принимает непрерывнорецидивирующее течение с резким похуданием и задержкой развития ребенка и может закончиться через несколько летальным исходом от интеркуррентной инфекции или амилоидоза. У взрослых прогноз более благоприятный.
 Поражение глаз при болезни Стилла: иридоциклит лентовидная дегенерация роговицы осложненная катаракта
Поражение глаз при болезни Стилла: иридоциклит лентовидная дегенерация роговицы осложненная катаракта
 Диагностика Основана на учете клинических проявлений и лабораторных показателей, среди которых ведущими и обязательными являются лихорадка (до 39 °С), эритематозно-папулезная мультиформная сыпь, артралгии (миалгии), артрит, увеличение СОЭ, лейкоцитоз со сдвигом формулы влево, отсутствие РФ и АНФ.
Диагностика Основана на учете клинических проявлений и лабораторных показателей, среди которых ведущими и обязательными являются лихорадка (до 39 °С), эритематозно-папулезная мультиформная сыпь, артралгии (миалгии), артрит, увеличение СОЭ, лейкоцитоз со сдвигом формулы влево, отсутствие РФ и АНФ.
 Большие критерии Лихорадка 39 °С и выше продолжительностью не менее одной недели. Артралгия длительностью 2 недели и более. Типичная сыпь. Лейкоцитоз (> 10 х109/л), > 80% гранулоцитов. Малые критерии Боли в горле. Лимфаденопатия и/или спленомегалия. Печеночная дисфункция. Негативные ревматоидный и антинуклеарный факторы. Для классификации болезни как синдрома Стилла у взрослых необходимо наличие 5 и более критериев, включая 3 и более больших критерия, и исключение других заболеваний. Лечение Аналогично лечению ревматоидного артрита.
Большие критерии Лихорадка 39 °С и выше продолжительностью не менее одной недели. Артралгия длительностью 2 недели и более. Типичная сыпь. Лейкоцитоз (> 10 х109/л), > 80% гранулоцитов. Малые критерии Боли в горле. Лимфаденопатия и/или спленомегалия. Печеночная дисфункция. Негативные ревматоидный и антинуклеарный факторы. Для классификации болезни как синдрома Стилла у взрослых необходимо наличие 5 и более критериев, включая 3 и более больших критерия, и исключение других заболеваний. Лечение Аналогично лечению ревматоидного артрита.