

9a5c20ba24b5c37d35f7bd6e085eceb1.ppt
- Количество слайдов: 57
 Spongiform Encephalopathies Dr. Cathal Collins 14/02/05
Spongiform Encephalopathies Dr. Cathal Collins 14/02/05
 Introduction n n Prion disease Neurodegenerative Long incubation period Progresses inexorably 5 human prion diseases: n n n Kuru Creutzfeldt-Jokob disease (CJD) New variant CJD Gerstmann-Straussler-Scheinker syndrome Fatal familial insomnia
Introduction n n Prion disease Neurodegenerative Long incubation period Progresses inexorably 5 human prion diseases: n n n Kuru Creutzfeldt-Jokob disease (CJD) New variant CJD Gerstmann-Straussler-Scheinker syndrome Fatal familial insomnia
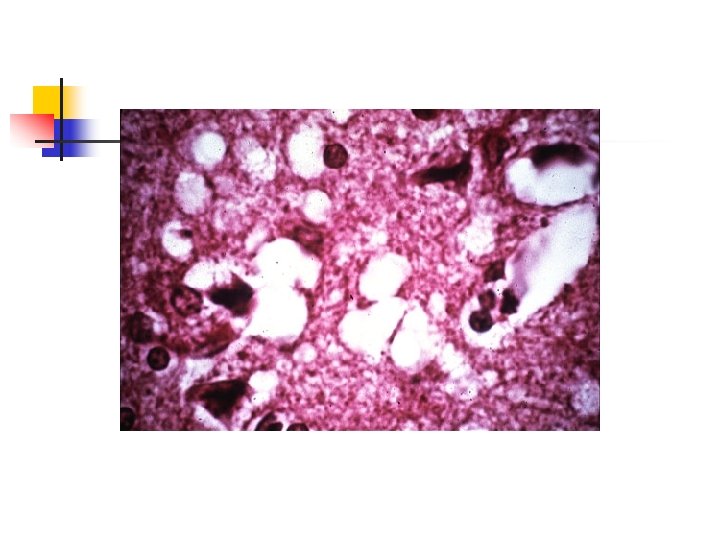
 Neuropathology n n Neuronal loss Proliferation of glial cells Absence of an inflammatory response Presence of small vacuoles which produces a spongiform appearance
Neuropathology n n Neuronal loss Proliferation of glial cells Absence of an inflammatory response Presence of small vacuoles which produces a spongiform appearance
 Animal prion diseases n n n Scrapie: sheep, goats; first described 1738 Transmissible Mink Encephalopathy: minks; 1964 Chronic Wasting Disease: mule, deer, elk; 1980 Bovine Spongiform Encephalopathy: cattle; 1986 Feline Spongiform Encephalopathy: cats; 1990
Animal prion diseases n n n Scrapie: sheep, goats; first described 1738 Transmissible Mink Encephalopathy: minks; 1964 Chronic Wasting Disease: mule, deer, elk; 1980 Bovine Spongiform Encephalopathy: cattle; 1986 Feline Spongiform Encephalopathy: cats; 1990
 BSE History in UK n n n 1985: first cases of BSE 1988: June BSE made notifiable July Ruminant feed banned August Compulsory slaughter and destruction of suspect cattle 1989 February Southwood report November Ban of sale of bovine offal for human consumption
BSE History in UK n n n 1985: first cases of BSE 1988: June BSE made notifiable July Ruminant feed banned August Compulsory slaughter and destruction of suspect cattle 1989 February Southwood report November Ban of sale of bovine offal for human consumption
 BSE History in UK n n n 1990 1992: 1996: November Bovine offal feed ban for all animals and birds Peak incidence of BSE First cases of nv. CJD Further restriction of bovine products from food and food chains
BSE History in UK n n n 1990 1992: 1996: November Bovine offal feed ban for all animals and birds Peak incidence of BSE First cases of nv. CJD Further restriction of bovine products from food and food chains
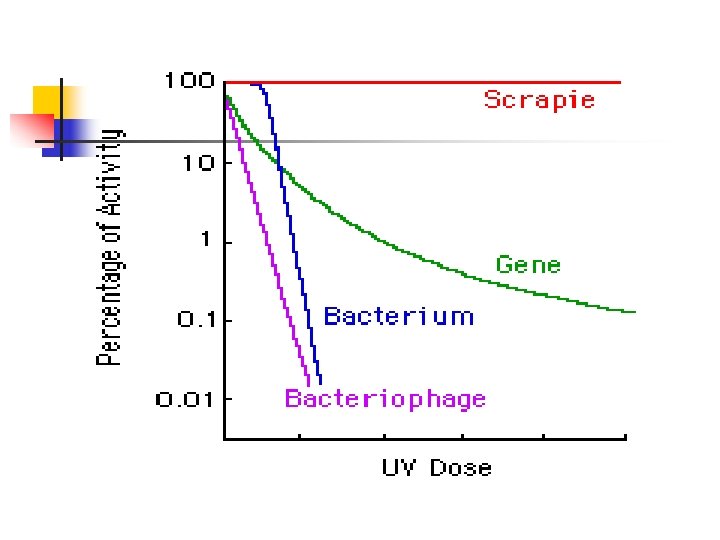
 Biology of Prions n n Dr. Stanley Prusiner: coined the term “prion” in 1982: proteinacious infectious particle Small infectious pathogen containing protein and lacking nucleic acid One characteristic feature is their resistance to a number of normal decontaminating procedures Resistant to: n n n Aldehydes e. g formaldehydes Nucleases Heat (80 C) UV and ionising radiation Non-ionic detergents
Biology of Prions n n Dr. Stanley Prusiner: coined the term “prion” in 1982: proteinacious infectious particle Small infectious pathogen containing protein and lacking nucleic acid One characteristic feature is their resistance to a number of normal decontaminating procedures Resistant to: n n n Aldehydes e. g formaldehydes Nucleases Heat (80 C) UV and ionising radiation Non-ionic detergents
 Biology of Prions n Inacticated by: n n n Prolonged autoclaving (at 121 C and 15 psi for 4. 5 h) Immersion in 1 M Na. OH (for 30 min, repeat 3 times) Immersion in strong organic solvents Inadequate autoclaving can establish heat resistant subpopulations Stainless steel instruments may also retain infectivity even after treatment with 10% formaldehyde
Biology of Prions n Inacticated by: n n n Prolonged autoclaving (at 121 C and 15 psi for 4. 5 h) Immersion in 1 M Na. OH (for 30 min, repeat 3 times) Immersion in strong organic solvents Inadequate autoclaving can establish heat resistant subpopulations Stainless steel instruments may also retain infectivity even after treatment with 10% formaldehyde
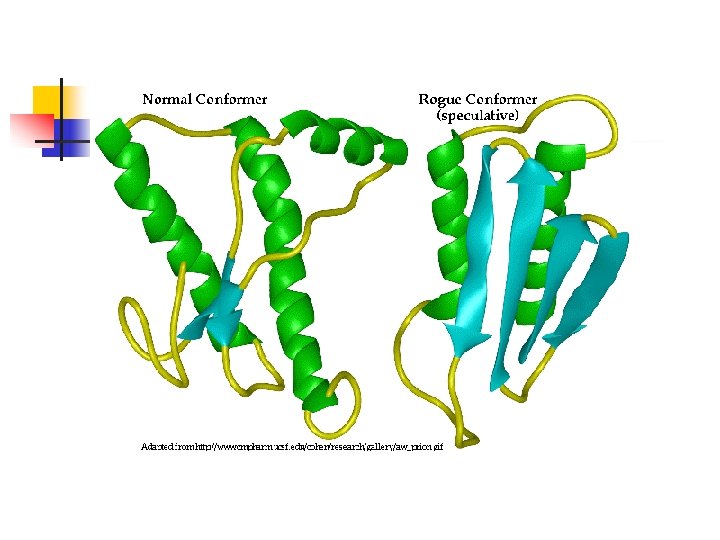
 n n The gene (PRPN) is located on the short") Prion Protein (Pr. P) n n The gene (PRPN) is located on the short arm of chromosome 20 Encodes a protein (Pr. Pc) found in normal brain (alpha helical); normal function unknown Pr. Psc (Pr. P in scrapie infected anmals) is a conformational isomer of Pr. Pc (B-pleated sheet) Pr. Psc is resistant to digestion with proteases and has a tendency to polymerise into scrapie -associated fibrils or prion rods
Prion Protein (Pr. P) n n The gene (PRPN) is located on the short arm of chromosome 20 Encodes a protein (Pr. Pc) found in normal brain (alpha helical); normal function unknown Pr. Psc (Pr. P in scrapie infected anmals) is a conformational isomer of Pr. Pc (B-pleated sheet) Pr. Psc is resistant to digestion with proteases and has a tendency to polymerise into scrapie -associated fibrils or prion rods
 Biosynthesis of Pr. Pc n n Key step is modification of amino and carboxy terminals with the addition of a phosphatidylinositol glycolipid which serves to anchor the protein to the cell surface Pr. Pc is found attached to plasma membranes of neurons and may be concentrated at synaptic membranes Pr. Pc has transmembranous domains Degraded after endocytosis in acidic vesicles
Biosynthesis of Pr. Pc n n Key step is modification of amino and carboxy terminals with the addition of a phosphatidylinositol glycolipid which serves to anchor the protein to the cell surface Pr. Pc is found attached to plasma membranes of neurons and may be concentrated at synaptic membranes Pr. Pc has transmembranous domains Degraded after endocytosis in acidic vesicles
 Pr. Psc n n Accumulates within cells; does not normally appear on the cell surface Found predominantly in cytoplasmic vacuoles and secondary lysosomes Studies with mice either devoid of Pr. Pc or with abnormal isoforms indicate that host Pr. Pc must be present for the development of prion disease Prion diseases result from accumulation of abnormal isoforms of the Pr. P which is dependent upon conversion of Pr. Pc to Pr. Psc
Pr. Psc n n Accumulates within cells; does not normally appear on the cell surface Found predominantly in cytoplasmic vacuoles and secondary lysosomes Studies with mice either devoid of Pr. Pc or with abnormal isoforms indicate that host Pr. Pc must be present for the development of prion disease Prion diseases result from accumulation of abnormal isoforms of the Pr. P which is dependent upon conversion of Pr. Pc to Pr. Psc
 Pr. Psc n n How the first molecule of Pr. Psc appears in the host remains a mystery Exogenous source in sporadic and iatrogenic CJD Mutation in PRNP gene in familial forms The initial appearance (? de novo) probably triggers the replication of Pr. Psc; Pr. Pc acts as a nidus for the formation of Pr. Psc on challenge
Pr. Psc n n How the first molecule of Pr. Psc appears in the host remains a mystery Exogenous source in sporadic and iatrogenic CJD Mutation in PRNP gene in familial forms The initial appearance (? de novo) probably triggers the replication of Pr. Psc; Pr. Pc acts as a nidus for the formation of Pr. Psc on challenge
 Pr. Psc n n n Prior to transport to the nervous system, follicular dendritic cells within germinal centres of lymhoid tissue appear to act as a reservoir for the protein Two reports suggest that complement plays a role in early pathogenesis ( C 3, C 1 q, Bf/C 2, or complement receptors) Transport of Pr. Psc to the nervous system occurs via axons
Pr. Psc n n n Prior to transport to the nervous system, follicular dendritic cells within germinal centres of lymhoid tissue appear to act as a reservoir for the protein Two reports suggest that complement plays a role in early pathogenesis ( C 3, C 1 q, Bf/C 2, or complement receptors) Transport of Pr. Psc to the nervous system occurs via axons
 Neurotoxicity of prion protein n Pr. Psc causes apoptosis and cell death Misfolded Pr. P is transported to the cytosol for degradation- even small amounts of this protein in the cytosol are highly neurotoxic Accumulation of this protein may be an important step in prion disease pathogenesis
Neurotoxicity of prion protein n Pr. Psc causes apoptosis and cell death Misfolded Pr. P is transported to the cytosol for degradation- even small amounts of this protein in the cytosol are highly neurotoxic Accumulation of this protein may be an important step in prion disease pathogenesis
 Genetics n n PRNP encodes Pr. P and is located on the short arm of chromosome 20 A strong link has been established between mutations in the PRNP gene and forms of prion disease with a familial predisposition (f. CJD, GSS, FFI) More than 50 different mutations have been identified A single mutation may produce different clinical phenotypes in different individuals or families
Genetics n n PRNP encodes Pr. P and is located on the short arm of chromosome 20 A strong link has been established between mutations in the PRNP gene and forms of prion disease with a familial predisposition (f. CJD, GSS, FFI) More than 50 different mutations have been identified A single mutation may produce different clinical phenotypes in different individuals or families
 Genetics n n The phenotype of a particular mutation may be influenced by the nature of the amino acids present at codon 129 Normal individuals have either valine or methionine at this site PRNP is an autosomal dominant gene; can be homozygous or heterozygous D 178 N mutation (asparagine for aspartic acid in codon 178): Homozygous for valine at codon 129 appear to develop CJD; those homozygous for methionine tend to have FFI
Genetics n n The phenotype of a particular mutation may be influenced by the nature of the amino acids present at codon 129 Normal individuals have either valine or methionine at this site PRNP is an autosomal dominant gene; can be homozygous or heterozygous D 178 N mutation (asparagine for aspartic acid in codon 178): Homozygous for valine at codon 129 appear to develop CJD; those homozygous for methionine tend to have FFI
 Genetics: Codon 129 n n n Molecular classification scheme for sporadic CJD based upon codon 129 polymorphism and characterisation of the properties of Pr. Psc A pattern of type 1 Pr. Psc plus at least 1 methionine at codon 129 was demonstrated in 70% Type 2 Pr. Psc plus codon 129 homozygous or heterozygous for valine was present in 25% and associated with ataxia
Genetics: Codon 129 n n n Molecular classification scheme for sporadic CJD based upon codon 129 polymorphism and characterisation of the properties of Pr. Psc A pattern of type 1 Pr. Psc plus at least 1 methionine at codon 129 was demonstrated in 70% Type 2 Pr. Psc plus codon 129 homozygous or heterozygous for valine was present in 25% and associated with ataxia
 Genetics: CJD n n Familial CJD: most common mutation is a substitution of lysine for glutamine in codon 200; phenotype may depend on codon 129 Sporadic CJD and iatrogenic CJD are not associated with PRNP gene mutations; however in these forms and nv. CJD, phenotyping at codon 129 appears to affect susceptibility and perhaps expression of the clinical illness
Genetics: CJD n n Familial CJD: most common mutation is a substitution of lysine for glutamine in codon 200; phenotype may depend on codon 129 Sporadic CJD and iatrogenic CJD are not associated with PRNP gene mutations; however in these forms and nv. CJD, phenotyping at codon 129 appears to affect susceptibility and perhaps expression of the clinical illness
 Genetics: GSS and FFI n n GSS - P 102 L mutation is the most common PRNP - large degree of phenotypic heterogeneity - polymorphism at codon 129 may play a modulating role FFI -D 178 N mutation predominant in those homozygous for methionine at codon 129
Genetics: GSS and FFI n n GSS - P 102 L mutation is the most common PRNP - large degree of phenotypic heterogeneity - polymorphism at codon 129 may play a modulating role FFI -D 178 N mutation predominant in those homozygous for methionine at codon 129
 Kuru n n First transmissible neurodegenerative disease to be identified and well studied Has served as the prototype of human prion diseases
Kuru n n First transmissible neurodegenerative disease to be identified and well studied Has served as the prototype of human prion diseases
 Kuru- Epidemiology n n n Was endemic in Papua New Guinea among the Fore tribes Felt to be transmitted from person to person by ritual cannibalism Still remains uncertain as to whether the brain was actually eaten, but it was handled after death, particularly by females and children No cases observed since these practices discontinued Primary cause of death in the tribes in 1960 s
Kuru- Epidemiology n n n Was endemic in Papua New Guinea among the Fore tribes Felt to be transmitted from person to person by ritual cannibalism Still remains uncertain as to whether the brain was actually eaten, but it was handled after death, particularly by females and children No cases observed since these practices discontinued Primary cause of death in the tribes in 1960 s
, ataxia and postural instability") Kuru- Clinical Features n n n Ambulatory phase: tremors (kuru=shivering), ataxia and postural instability Sedentary stage: loss of ambulation resulting from increased tremors and ataxia; involuntary movements Late stage: dementia, indifference Terminal stage: frontal release signs, cerebellar type dysarthria and inability to get out of bed Death typically due to pneumonia occurring within 9 -24 months form onset of disease
Kuru- Clinical Features n n n Ambulatory phase: tremors (kuru=shivering), ataxia and postural instability Sedentary stage: loss of ambulation resulting from increased tremors and ataxia; involuntary movements Late stage: dementia, indifference Terminal stage: frontal release signs, cerebellar type dysarthria and inability to get out of bed Death typically due to pneumonia occurring within 9 -24 months form onset of disease
 Kuru- Diagnosis&Pathology n n CSF unremarkable, EEG not characteristic Pathological hallmark is Pr. Psc-reactive plaques occurring with the greatest frequency in the cerebellum; neuronal loss and hypertrophy of astrocytes is also observed
Kuru- Diagnosis&Pathology n n CSF unremarkable, EEG not characteristic Pathological hallmark is Pr. Psc-reactive plaques occurring with the greatest frequency in the cerebellum; neuronal loss and hypertrophy of astrocytes is also observed
 Creutzfeld-Jakob Disease n n n Most frequent of the human prion diseases Still very rare Sporadic (s. CJD), familial (f. CJD), iatrogenic (i. CJD) and new variant (nv. CJD)
Creutzfeld-Jakob Disease n n n Most frequent of the human prion diseases Still very rare Sporadic (s. CJD), familial (f. CJD), iatrogenic (i. CJD) and new variant (nv. CJD)
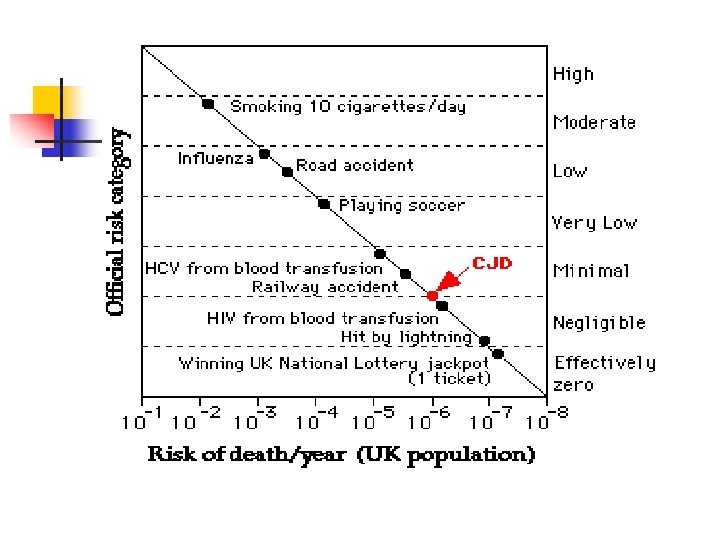
 CJD- Epidemiology n n n n Approx 1 case per 1 million population/year World-wide distribution Mean age of onset is 57 -62 Patients with nv. CJD and i. CJD tend to be much younger No gender predilection Incidence increased 30 -100 fold in certain areas of North Africa, Israel and Slovakia, due primarily to clusters of f. CJD Vast majoriy sporadic (85 -95%), 5 -15% f. CJD, <5% i. CJD
CJD- Epidemiology n n n n Approx 1 case per 1 million population/year World-wide distribution Mean age of onset is 57 -62 Patients with nv. CJD and i. CJD tend to be much younger No gender predilection Incidence increased 30 -100 fold in certain areas of North Africa, Israel and Slovakia, due primarily to clusters of f. CJD Vast majoriy sporadic (85 -95%), 5 -15% f. CJD, <5% i. CJD
 Iatrogenic CJD n n Following administration of cadaveric human pituitary hormones, dural graft transplants, use of dural mater in radiographic embolisation procedures, corneal transplants, liver transplants, and the use of contaminated neurosurgical instruments or stereotactic depth electrodes Hx of preceding infusion does not increase risk of developing CJD (epidemiology studies); however low levels of infectivity in in vitro studies
Iatrogenic CJD n n Following administration of cadaveric human pituitary hormones, dural graft transplants, use of dural mater in radiographic embolisation procedures, corneal transplants, liver transplants, and the use of contaminated neurosurgical instruments or stereotactic depth electrodes Hx of preceding infusion does not increase risk of developing CJD (epidemiology studies); however low levels of infectivity in in vitro studies
 CJD- Clinical Features n n Rapidly progressive mental deterioration and myoclonus are two cardinal manifestations of s. CJD Number of variants or subtypes of disease based upon area of involvement of the brain n n Visual Cerebellar Thalamic Striatal Variants of s. CJD also classified based on genotype of PRNP and the molecular properties of the pathological Pr. Psc
CJD- Clinical Features n n Rapidly progressive mental deterioration and myoclonus are two cardinal manifestations of s. CJD Number of variants or subtypes of disease based upon area of involvement of the brain n n Visual Cerebellar Thalamic Striatal Variants of s. CJD also classified based on genotype of PRNP and the molecular properties of the pathological Pr. Psc
 CJD- Clinical Features n n n Mental deterioration may manifest as dementia, behavioural abnormalities and deficits involving higher cortical function Concentration, memory and judgement difficulties are frequent early signs Mood changes such as apathy and depression are common Dementia becomes dominant and can advance rapidly Death usually occurs within one year
CJD- Clinical Features n n n Mental deterioration may manifest as dementia, behavioural abnormalities and deficits involving higher cortical function Concentration, memory and judgement difficulties are frequent early signs Mood changes such as apathy and depression are common Dementia becomes dominant and can advance rapidly Death usually occurs within one year
 CJD- Clinical Features n n n Myoclonus, especially provoked by startle, is present in more than 90% s. CJD should always be considered in a patient with a combination of a rapidly progressive dementia and myoclonus Extrapyramidal signs such as hypokinesia and cerebellar manifestations including nystagmus and ataxia occur in 2/3 Corticospinal tract involvement in 40 -80% Sensory signs and symptoms are common in nv. CJD; otherwise extremely atypical
CJD- Clinical Features n n n Myoclonus, especially provoked by startle, is present in more than 90% s. CJD should always be considered in a patient with a combination of a rapidly progressive dementia and myoclonus Extrapyramidal signs such as hypokinesia and cerebellar manifestations including nystagmus and ataxia occur in 2/3 Corticospinal tract involvement in 40 -80% Sensory signs and symptoms are common in nv. CJD; otherwise extremely atypical
 Subtypes of s. CJD n n Clinical phenotypes of s. CJD associated with molecular subtypes determined by the PRNP gene codon 129 genotype and the pathologic prion protein (Pr. Psc) type PRNP genotype homozygous or heterozygous for methionine (M) or valine (V) at codon 129 The Pr. Psc type is determined by Western blot analysis amd classified in the Parchi/Gambetti nomenclature as type 1 or type 2 Alternate Collinge nomenclature distinguishes 4 Pr. Psc subtypes: types 1 and 2 correspond with P&G Pr. Psc 1; 3 and 4 with P&G Pr. Psc 2
Subtypes of s. CJD n n Clinical phenotypes of s. CJD associated with molecular subtypes determined by the PRNP gene codon 129 genotype and the pathologic prion protein (Pr. Psc) type PRNP genotype homozygous or heterozygous for methionine (M) or valine (V) at codon 129 The Pr. Psc type is determined by Western blot analysis amd classified in the Parchi/Gambetti nomenclature as type 1 or type 2 Alternate Collinge nomenclature distinguishes 4 Pr. Psc subtypes: types 1 and 2 correspond with P&G Pr. Psc 1; 3 and 4 with P&G Pr. Psc 2
 Subtypes of s. CJD n n n 6 clinical phenotypes of s. CJD MM 1 and MV 1 (myoclonic, Heidenhain variant) account for about 70% of cases and correlate with the “classic CJD” phenotype VV 2 (ataxic variant) accounts for 16% MV 2 (Kuru plaque variant): 9% MM 2 thalamic, MM 2 cortical and VV 1 account for the rest
Subtypes of s. CJD n n n 6 clinical phenotypes of s. CJD MM 1 and MV 1 (myoclonic, Heidenhain variant) account for about 70% of cases and correlate with the “classic CJD” phenotype VV 2 (ataxic variant) accounts for 16% MV 2 (Kuru plaque variant): 9% MM 2 thalamic, MM 2 cortical and VV 1 account for the rest
 CJD- Diagnosis n n Clinical and laboratory features generally are sufficient for a ‘probable’ diagnosis of s. CJD WHO criteria for ‘probable’ diagnosis: n n Progressive dementia >/=2 of 4 of: myoclonus; visual or cerebellar disturbance; pyramidal/extrapyramidal dysfunction; akinetic mutism A typical EEG during an illness of any duration and/or positive 14 -3 -3 CSF assay with a clinical duration to death in less than 2 years Routine investigations should not suggest an alternative diagnosis
CJD- Diagnosis n n Clinical and laboratory features generally are sufficient for a ‘probable’ diagnosis of s. CJD WHO criteria for ‘probable’ diagnosis: n n Progressive dementia >/=2 of 4 of: myoclonus; visual or cerebellar disturbance; pyramidal/extrapyramidal dysfunction; akinetic mutism A typical EEG during an illness of any duration and/or positive 14 -3 -3 CSF assay with a clinical duration to death in less than 2 years Routine investigations should not suggest an alternative diagnosis
 CJD-Diagnosis n A definitive diagnosis requires these features in combination with one or more of the following: n n n Loss of neurons, gliosis, spongiform degeneration, or plaqes postive for Pr. Psc on histopathlogy of brain tissue Positive Pr. Psc staining following pretreatment of brain tissue to destroy Pr. Pc reactivity Positive histoblotting of brain tissue extracts for Pr. Psc after treatment to destroy Pr. Pc reactivity Transmission of characteristc neurodegenerative disease to experimental animals Demonstration of PRNP gene mutations
CJD-Diagnosis n A definitive diagnosis requires these features in combination with one or more of the following: n n n Loss of neurons, gliosis, spongiform degeneration, or plaqes postive for Pr. Psc on histopathlogy of brain tissue Positive Pr. Psc staining following pretreatment of brain tissue to destroy Pr. Pc reactivity Positive histoblotting of brain tissue extracts for Pr. Psc after treatment to destroy Pr. Pc reactivity Transmission of characteristc neurodegenerative disease to experimental animals Demonstration of PRNP gene mutations
 CJD- Diagnosis n n Neuroimaging: diffusion weighted MRI- can detect abnormalities as early as 3 weeks of symptom duration, CT generally normal EEG: aids diagnosis: characteristic pattern of periodic synchronous bi or triphasic sharp wave complexes (PSWCs) Protein markers: 14 -3 -3 protein in CSFespecially in those with classical subtypes of s. CJD Pathological studies of brain material to detect protease resistant Pr. Psc remains gold standard
CJD- Diagnosis n n Neuroimaging: diffusion weighted MRI- can detect abnormalities as early as 3 weeks of symptom duration, CT generally normal EEG: aids diagnosis: characteristic pattern of periodic synchronous bi or triphasic sharp wave complexes (PSWCs) Protein markers: 14 -3 -3 protein in CSFespecially in those with classical subtypes of s. CJD Pathological studies of brain material to detect protease resistant Pr. Psc remains gold standard
 New Variant CJD n n n Initial reports of nv. CJD in 1996 quickly focused intense interest on the human prion diseases Linked with bovine spongiform encephalopathy Unique epidemiological features of this illness led to early recognition that this was indeed a ”new variant”
New Variant CJD n n n Initial reports of nv. CJD in 1996 quickly focused intense interest on the human prion diseases Linked with bovine spongiform encephalopathy Unique epidemiological features of this illness led to early recognition that this was indeed a ”new variant”
 nv. CJD- Epidemiology n n n First report of a case of nv. CJD in a 16 year old from the UK appeared in 1995 was quickly followed in 1996 by 22 other cases All bar one of the initial cases were from the UK January 2004 - 155 cases world-wide 145 from UK, 6 from France, 1 each from Ireland, Italy, Canada and the US All except the italian had resided in countries with known BSE
nv. CJD- Epidemiology n n n First report of a case of nv. CJD in a 16 year old from the UK appeared in 1995 was quickly followed in 1996 by 22 other cases All bar one of the initial cases were from the UK January 2004 - 155 cases world-wide 145 from UK, 6 from France, 1 each from Ireland, Italy, Canada and the US All except the italian had resided in countries with known BSE
 nv. CJD V s. CJD n nv. CJD is distinguished from s. CJD by n n n A considerably younger age of onset (mean age of onset 29 versus 65) Less rapid progression of disease (duration 14 months versus 4 -5) Differences in clinical presentation (sensory and psychiatric symptoms prominent in nv. CJD) Type 2 (P&G)/ type 4 (Collinge) Pr. Psc in nv. CJD Differences in neuropathology
nv. CJD V s. CJD n nv. CJD is distinguished from s. CJD by n n n A considerably younger age of onset (mean age of onset 29 versus 65) Less rapid progression of disease (duration 14 months versus 4 -5) Differences in clinical presentation (sensory and psychiatric symptoms prominent in nv. CJD) Type 2 (P&G)/ type 4 (Collinge) Pr. Psc in nv. CJD Differences in neuropathology
 nv. CJD- Clinical features n n n 63/100 of the 1 st cases presented initially with psychiatric symptoms Neurological symptoms preceded psychiatric in 15 Both were present in 22 Psychiatric symptoms include depression, apathy, anxiety, psychosis and intermittent delusions Sensory abnoramalities include dysaesthesias and paraesthesia of the face, hands, feet, legs or even hemibody
nv. CJD- Clinical features n n n 63/100 of the 1 st cases presented initially with psychiatric symptoms Neurological symptoms preceded psychiatric in 15 Both were present in 22 Psychiatric symptoms include depression, apathy, anxiety, psychosis and intermittent delusions Sensory abnoramalities include dysaesthesias and paraesthesia of the face, hands, feet, legs or even hemibody
 nv. CJD- Clinical features n n n Once neurological symptoms, typically ataxia, becomes evident, progression is more rapid Cognitive impairment, involuntary movements, immobility, unresponsiveness, and mutism are common signs as the disease progresses Paresis of upward gaze may be present (uncommon in other forms of CJD)
nv. CJD- Clinical features n n n Once neurological symptoms, typically ataxia, becomes evident, progression is more rapid Cognitive impairment, involuntary movements, immobility, unresponsiveness, and mutism are common signs as the disease progresses Paresis of upward gaze may be present (uncommon in other forms of CJD)
 nv. CJD- Diagnosis n n CSF studies rarely helpful: 14 -3 -3 is not a sensitive marker Combination of 14 -3 -3 and tau protein in CSF may be useful MRI better than CT: MRI may show signal hyperintensity in the pulvinar (pulvinar sign) or in both pulvinar and dorsomedial thalamus (hockey stick sign) EEG: abnormal in 70% but only slow wave pattern
nv. CJD- Diagnosis n n CSF studies rarely helpful: 14 -3 -3 is not a sensitive marker Combination of 14 -3 -3 and tau protein in CSF may be useful MRI better than CT: MRI may show signal hyperintensity in the pulvinar (pulvinar sign) or in both pulvinar and dorsomedial thalamus (hockey stick sign) EEG: abnormal in 70% but only slow wave pattern
 nv. CJD- Diagnosis n n PRNP gene mutations are not present in nv. CJD, but all patients with clinically expressed nv. CJD have been homozygous for methionine at codon 129 Type 2 Pr. Psc (P&G nomenclature) or type 4 Pr. Psc in the Collinge nomenclature has been found in patients with nv. CJD (not characteristic of other human prion diseases)
nv. CJD- Diagnosis n n PRNP gene mutations are not present in nv. CJD, but all patients with clinically expressed nv. CJD have been homozygous for methionine at codon 129 Type 2 Pr. Psc (P&G nomenclature) or type 4 Pr. Psc in the Collinge nomenclature has been found in patients with nv. CJD (not characteristic of other human prion diseases)
 nv. CJD- Neuropathology n A number of neuropathological feature distinguish nv. CJD from s. CJD: n n n Presence of plaques, which stain intensely for Pr. Psc, distributed throughout the cerebrum and cerebellum and to a lesser extent the basal ganglia and thalamus The plaques have an eosinophilic centre and pale periphery with surrounding spongiform changes Cases of kuru and GSS have similar but not identical plaques The cerebellum is characteristically involved in nv. CJD has distinct type 4 Pr. Psc
nv. CJD- Neuropathology n A number of neuropathological feature distinguish nv. CJD from s. CJD: n n n Presence of plaques, which stain intensely for Pr. Psc, distributed throughout the cerebrum and cerebellum and to a lesser extent the basal ganglia and thalamus The plaques have an eosinophilic centre and pale periphery with surrounding spongiform changes Cases of kuru and GSS have similar but not identical plaques The cerebellum is characteristically involved in nv. CJD has distinct type 4 Pr. Psc
 nv. CJD- Links n n 2 cases of possible transfusion transmisssion of nv. CJD have been reported There is increasing evidence supporting the possibility that nv. CJD represents bovine-to-human transmission of BSE
nv. CJD- Links n n 2 cases of possible transfusion transmisssion of nv. CJD have been reported There is increasing evidence supporting the possibility that nv. CJD represents bovine-to-human transmission of BSE
 nv. CJD and BSE n n n The appearance of nv. CJD followed an epidemic of BSE in the UK The removal of organic solvents, which inactivate Pr. Psc, from the rendering process for bovine offal and the subsequent use of the offal as a component of feed for cattle has been suggested as a mechanism for amplifying the epidemic in animals Approx 50, 000 infected cattle are estimated to have entered the food chain
nv. CJD and BSE n n n The appearance of nv. CJD followed an epidemic of BSE in the UK The removal of organic solvents, which inactivate Pr. Psc, from the rendering process for bovine offal and the subsequent use of the offal as a component of feed for cattle has been suggested as a mechanism for amplifying the epidemic in animals Approx 50, 000 infected cattle are estimated to have entered the food chain
 nv. CJD and BSE n n The prohibition of ruminant-derived proteins in feeds for all animals and poultry in november 1990 and the banning of consumption of animals over the age of 30 months in March 1996 has led to a dramatic decline in cases of BSE Evidence in the favour of the association between nv. CJD and BSE includes the type 4 pattern of Pr. Psc which has not been seen in other prion diseases
nv. CJD and BSE n n The prohibition of ruminant-derived proteins in feeds for all animals and poultry in november 1990 and the banning of consumption of animals over the age of 30 months in March 1996 has led to a dramatic decline in cases of BSE Evidence in the favour of the association between nv. CJD and BSE includes the type 4 pattern of Pr. Psc which has not been seen in other prion diseases
 nv. CJD and BSE n Despite the apparent link between nv. CJD and BSE, the number of cases of nv. CJD has remained small. Possible reasons include: n n Low levels of Pr. Psc in milk and meat Inefficiency of oral route of infection Restriction of spread based upon a species barrier Low incidence of host genetic factors such as the frequency of homozygosity at codon 129
nv. CJD and BSE n Despite the apparent link between nv. CJD and BSE, the number of cases of nv. CJD has remained small. Possible reasons include: n n Low levels of Pr. Psc in milk and meat Inefficiency of oral route of infection Restriction of spread based upon a species barrier Low incidence of host genetic factors such as the frequency of homozygosity at codon 129
 Gerstmann-Straussler. Scheinker Syndrome n n n A rare human prion disease 1 -10 cases per 100 million population/year Autosomal dominant pattern with virtual complete penetrance Hallmark: progressive cerebellar degeneration accompanied by different degrees of dementia in patients entering mid-life (mean 43 -48) Course of illness advances for 5 years before culminating in death Myoclonus is typically absent
Gerstmann-Straussler. Scheinker Syndrome n n n A rare human prion disease 1 -10 cases per 100 million population/year Autosomal dominant pattern with virtual complete penetrance Hallmark: progressive cerebellar degeneration accompanied by different degrees of dementia in patients entering mid-life (mean 43 -48) Course of illness advances for 5 years before culminating in death Myoclonus is typically absent
 GSS n n Phenotypic variability due to differences in underlying PRNP mutation or in the polymorphism in codon 129 Lab or imaging studies not useful Demonstration of PRNP mutation useful means for diagnosis Neuropathological features consistent with other forms of prion disease: however kurulike plaques especially in the cerebellum are common findings; also neurofibrillary tangles and neuropil threads identical to those seen in Alzheimers
GSS n n Phenotypic variability due to differences in underlying PRNP mutation or in the polymorphism in codon 129 Lab or imaging studies not useful Demonstration of PRNP mutation useful means for diagnosis Neuropathological features consistent with other forms of prion disease: however kurulike plaques especially in the cerebellum are common findings; also neurofibrillary tangles and neuropil threads identical to those seen in Alzheimers
 Fatal Familial Insomnia n n Rapidly fatal with a mean duration of 13 months; midlife (35 -61 years) Develop progressive insomnia with loss of normal circadian sleep-activity pattern Impaired concentration and memory, confusion, inattention and behavioural changes occur but overt dementia is rare Myoclonus, ataxia and spasticity occur with disease progression
Fatal Familial Insomnia n n Rapidly fatal with a mean duration of 13 months; midlife (35 -61 years) Develop progressive insomnia with loss of normal circadian sleep-activity pattern Impaired concentration and memory, confusion, inattention and behavioural changes occur but overt dementia is rare Myoclonus, ataxia and spasticity occur with disease progression
 and") FFI n n Only prion disease to produce dysautonomia (increased T, BP, HR) and endocrine disturbances (ACTH, cortisol, GH, PRL) Genetic studies are diagnostic procedure of choice Most cases associated with D 178 N PRNP mutation Spongiform degeneration rarely detected; neuronal loss and gliosis is maximal within the thalamus
FFI n n Only prion disease to produce dysautonomia (increased T, BP, HR) and endocrine disturbances (ACTH, cortisol, GH, PRL) Genetic studies are diagnostic procedure of choice Most cases associated with D 178 N PRNP mutation Spongiform degeneration rarely detected; neuronal loss and gliosis is maximal within the thalamus
 Treatment of Prion Diseases n n No effective treatment has been identified for human prion diseases which are universally fatal; supportive treatment mainstay Flupirtine maleate is a centrally acting, nonopioid analgesic that has displayed cytoprotective activity in vitro in neurons treated with a prion protein fragment: better MMSe but survival not enhanced Chlorpromazine and quinacrine: inhibit Pr. Psc formation in vitro: studies needed Potential targets will include the steps in the conversion of Pr. Pc to Pr. Psc
Treatment of Prion Diseases n n No effective treatment has been identified for human prion diseases which are universally fatal; supportive treatment mainstay Flupirtine maleate is a centrally acting, nonopioid analgesic that has displayed cytoprotective activity in vitro in neurons treated with a prion protein fragment: better MMSe but survival not enhanced Chlorpromazine and quinacrine: inhibit Pr. Psc formation in vitro: studies needed Potential targets will include the steps in the conversion of Pr. Pc to Pr. Psc
 Summary n n n n Prions: do not have nucleic acid Conversion of Pr. Pc to Pr. Psc Codon 129 in PRNP gene Neurodegenerative diseases Long incubation period Progress inexorably; no treatment 5 types of human prion disease nv. CJD and links to BSE
Summary n n n n Prions: do not have nucleic acid Conversion of Pr. Pc to Pr. Psc Codon 129 in PRNP gene Neurodegenerative diseases Long incubation period Progress inexorably; no treatment 5 types of human prion disease nv. CJD and links to BSE
