

nucl_sec.ppt
- Количество слайдов: 45
 Современные технологии секвенирования
Современные технологии секвенирования
 История • • • 1975 – расшифрован геном бактериофага φХ 174 1977 – открытие метода дидезоксисеквенирования ДНК (Сэнгер) 1980 - Сэнгер получает нобелевскую премию по химии 1984 –расшифрован геном вируса Эпштейн-Барра (170 Kb) 1987 – появление первого автоматического секвенатора (Applied Biosystems) 1995 – расшифрован геном бактерии Haemophilius influenzae (1, 8 Mb) 1996 – геном эукариотической клетки (дрожжей) 1998 – геном многоклеточного организма (нематоды) 2000 – «черновой» вариант генома человека 2003 – полный вариант генома человека
История • • • 1975 – расшифрован геном бактериофага φХ 174 1977 – открытие метода дидезоксисеквенирования ДНК (Сэнгер) 1980 - Сэнгер получает нобелевскую премию по химии 1984 –расшифрован геном вируса Эпштейн-Барра (170 Kb) 1987 – появление первого автоматического секвенатора (Applied Biosystems) 1995 – расшифрован геном бактерии Haemophilius influenzae (1, 8 Mb) 1996 – геном эукариотической клетки (дрожжей) 1998 – геном многоклеточного организма (нематоды) 2000 – «черновой» вариант генома человека 2003 – полный вариант генома человека
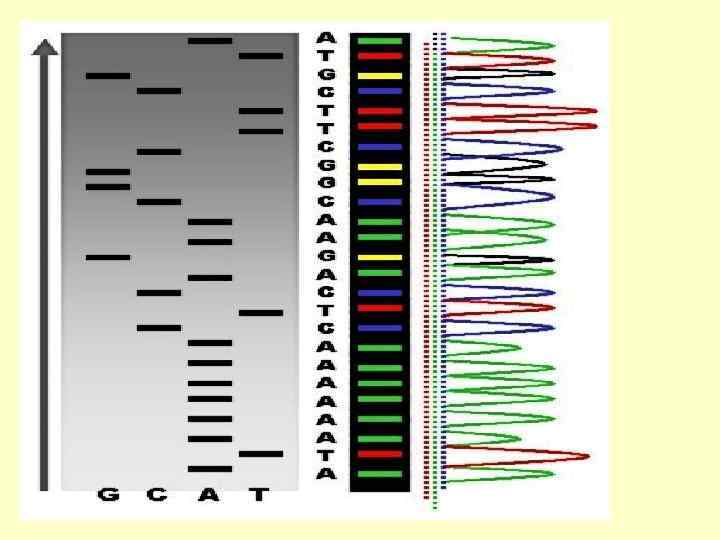
 Cеквенирование по Сэнгеру
Cеквенирование по Сэнгеру
") Секвенирование генома человека 1991 – 2000 • 10 лет • 300, 000 $ (Celera) • 3 млрд $ • Несколько сотен капиллярных секвенаторов 2007 – 10, 000 $
Секвенирование генома человека 1991 – 2000 • 10 лет • 300, 000 $ (Celera) • 3 млрд $ • Несколько сотен капиллярных секвенаторов 2007 – 10, 000 $
") 2005 - Genome Sequencer 20 System (454 Life Sciences Corp/Roche Applied Science)
2005 - Genome Sequencer 20 System (454 Life Sciences Corp/Roche Applied Science)
 • 1,") 2008 Стоимость ресеквенирования генома человека: • 2, 000 $ (454 Life Sciences) • 1, 000 $ (Illumina) • 500, 000 $ (Illumina) • 250, 000 $ (Illumina)
2008 Стоимость ресеквенирования генома человека: • 2, 000 $ (454 Life Sciences) • 1, 000 $ (Illumina) • 500, 000 $ (Illumina) • 250, 000 $ (Illumina)
 2009 • ABI – май 2009 – геном человека за 1 рабочий цикл прибора • Illumina – июнь 2009 – геном человека < 50 000$ • Helicos Biosciences – сентябрь 2009 – геном человека методом single molecule sequencing за 48 000$ • Complete Genomics – ноябрь 2009 - геном человека < 5 000$ • В России секвенирован геном человека (Курчатовский институт)
2009 • ABI – май 2009 – геном человека за 1 рабочий цикл прибора • Illumina – июнь 2009 – геном человека < 50 000$ • Helicos Biosciences – сентябрь 2009 – геном человека методом single molecule sequencing за 48 000$ • Complete Genomics – ноябрь 2009 - геном человека < 5 000$ • В России секвенирован геном человека (Курчатовский институт)
 200 Gb за прогон (8") 2010 • Январь 2010 – Hi. Seq 2000 (Illumina) 200 Gb за прогон (8 дней), ~ 25 Gb / день • ~10, 000 $ - геном человека
2010 • Январь 2010 – Hi. Seq 2000 (Illumina) 200 Gb за прогон (8 дней), ~ 25 Gb / день • ~10, 000 $ - геном человека
 2012 • Коммерческие услуги по секвенированию генома человека - ~ 2 недели, 4 -10 тыс. $ • Секвенирование экзома ( кодирующие последовательности всех известных генов) – 999$
2012 • Коммерческие услуги по секвенированию генома человека - ~ 2 недели, 4 -10 тыс. $ • Секвенирование экзома ( кодирующие последовательности всех известных генов) – 999$
 Next Generation секвенирование • Скорость • Высокая производительность • Достаточно высокая точность (до 99, 995%) • Возможности дальнейшего развития технологий
Next Generation секвенирование • Скорость • Высокая производительность • Достаточно высокая точность (до 99, 995%) • Возможности дальнейшего развития технологий
 – единичный прочитанный фрагмент • Coverage (покрытие) – среднее") Термины • Read (рид, чтение) – единичный прочитанный фрагмент • Coverage (покрытие) – среднее количество ридов, которые содержат данный нуклеотид • Run (прогон) – время, за которое прибор совершает полный рабочий цикл • Output (производительность) – количество нуклеотидов, секвенируемых за один прогон прибора
Термины • Read (рид, чтение) – единичный прочитанный фрагмент • Coverage (покрытие) – среднее количество ридов, которые содержат данный нуклеотид • Run (прогон) – время, за которое прибор совершает полный рабочий цикл • Output (производительность) – количество нуклеотидов, секвенируемых за один прогон прибора
 Этапы • Получение одноцепочечных коротких фрагментов ДНК; создание библиотеки • Амплификация полученных фрагментов с помощью ПЦР • Секвенирование • Регистрация и преобразование сигнала • Обработка данных (сравнение с референсным геномом, сборка генома de novo и т. д. )
Этапы • Получение одноцепочечных коротких фрагментов ДНК; создание библиотеки • Амплификация полученных фрагментов с помощью ПЦР • Секвенирование • Регистрация и преобразование сигнала • Обработка данных (сравнение с референсным геномом, сборка генома de novo и т. д. )
; ПФ") Три основные технологии с флуоресцентной детекцией • Пиросеквенирование (присоединение нуклеотида высвобождает пирофосфат (ПФ); ПФ обращается в АТФ с помощью сульфурилазы; люцифераза + люциферин + АТФ = оксилюциферин (хемилюминисценция)) • Лигазное секвенирование (основано на способности лигазы сшивать фрагменты ДНК, если они комплементарны матричной цепи; при сшивке высвобождается флуоресцентная метка) • Секвенирование на молекулярных кластерах с использованием флуоресцентно-меченных нуклеотидов
Три основные технологии с флуоресцентной детекцией • Пиросеквенирование (присоединение нуклеотида высвобождает пирофосфат (ПФ); ПФ обращается в АТФ с помощью сульфурилазы; люцифераза + люциферин + АТФ = оксилюциферин (хемилюминисценция)) • Лигазное секвенирование (основано на способности лигазы сшивать фрагменты ДНК, если они комплементарны матричной цепи; при сшивке высвобождается флуоресцентная метка) • Секвенирование на молекулярных кластерах с использованием флуоресцентно-меченных нуклеотидов
 (пиросеквенирование) • ILLUMINA (секвенирование на") Основные фирмы - производители • 454 Life Sciences (Roche) (пиросеквенирование) • ILLUMINA (секвенирование на молекулярных кластерах) • Life Technologies (Applied Biosystems) (лигазное секвенирование)
Основные фирмы - производители • 454 Life Sciences (Roche) (пиросеквенирование) • ILLUMINA (секвенирование на молекулярных кластерах) • Life Technologies (Applied Biosystems) (лигазное секвенирование)
 (пиросеквенирование) • Первыми выпустили на рынок секвенатор нового поколения (GS") 454 Life Sciences (Roche) (пиросеквенирование) • Первыми выпустили на рынок секвенатор нового поколения (GS 20) • 2005 – расшифровка генома Mycoplasma genitalium и Streptococcus pneumoniae (первый геном с использованием Next gene технологии) • Конец 2005 г. – анализ генома мамонта • 2006 – расшифровка еще 4 -х бактериальных геномов • Помогали завершить секвенирование генома Дж. Уотсона (2008, 2, 000 $)
454 Life Sciences (Roche) (пиросеквенирование) • Первыми выпустили на рынок секвенатор нового поколения (GS 20) • 2005 – расшифровка генома Mycoplasma genitalium и Streptococcus pneumoniae (первый геном с использованием Next gene технологии) • Конец 2005 г. – анализ генома мамонта • 2006 – расшифровка еще 4 -х бактериальных геномов • Помогали завершить секвенирование генома Дж. Уотсона (2008, 2, 000 $)
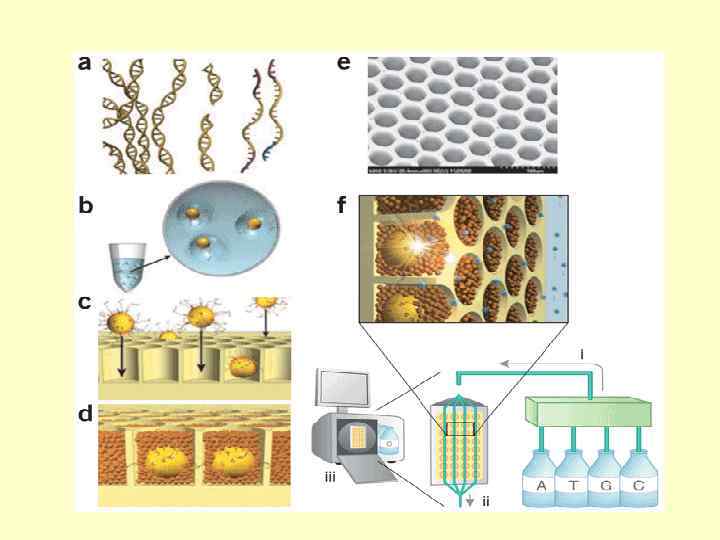

 Регистрация и преобразование сигнала
Регистрация и преобразование сигнала
 GS FLX Titanium XL+ • Длина рида – 1000 п. о. • 700 Mb за 23 часа
GS FLX Titanium XL+ • Длина рида – 1000 п. о. • 700 Mb за 23 часа
 1. Получение одноцепочечной ДНК 2. Генерация кластеров (гибридизация в проточной") Sequencing by synthesis (Illumina) 1. Получение одноцепочечной ДНК 2. Генерация кластеров (гибридизация в проточной ячейке с праймерами, амплификация с образованием мостиков) 3. Секвенирование
Sequencing by synthesis (Illumina) 1. Получение одноцепочечной ДНК 2. Генерация кластеров (гибридизация в проточной ячейке с праймерами, амплификация с образованием мостиков) 3. Секвенирование
 • 600 Gb за прогон • 55 Gb в день") Hi. Seq 2500/1500 (Illumina) • 600 Gb за прогон • 55 Gb в день
Hi. Seq 2500/1500 (Illumina) • 600 Gb за прогон • 55 Gb в день
 Лигазное секвенирование
Лигазное секвенирование
 • 20 Gb в день") 5500 x. L (Life Technologies) • 20 Gb в день
5500 x. L (Life Technologies) • 20 Gb в день
 • Полупроводниковое секвенирование") Другие технологии секвенирования • Cеквенирование на отдельных молекулах (Helicos, Pasific Biosciences) • Полупроводниковое секвенирование (Ion Torrent) • Секвенирование с использованием нанопоры (Oxford Nanopore) • Лигазное секвенирование с использованием “наношаров” (Complete Genomics)
Другие технологии секвенирования • Cеквенирование на отдельных молекулах (Helicos, Pasific Biosciences) • Полупроводниковое секвенирование (Ion Torrent) • Секвенирование с использованием нанопоры (Oxford Nanopore) • Лигазное секвенирование с использованием “наношаров” (Complete Genomics)
 Heliscope Single Molecule Sequencer
Heliscope Single Molecule Sequencer
™ 1 этап : пробоподготовка (5 часов) • •") True Single Molecule Sequencing (t. SMS)™ 1 этап : пробоподготовка (5 часов) • • • Обработка геномной ДНК ультразвуком для получения фрагментов длиной 100 -500 п. о. Получение коротких одноцепочечных фрагментов ДНК Синтез poly. A-хвоста и блок 3’конца – присоединение dd. T с меткой Нанесение на flow cell по 25 pg на линию (200 p. M) Гибридизация с олигоd. T (1 млн на см²) на flow cell
True Single Molecule Sequencing (t. SMS)™ 1 этап : пробоподготовка (5 часов) • • • Обработка геномной ДНК ультразвуком для получения фрагментов длиной 100 -500 п. о. Получение коротких одноцепочечных фрагментов ДНК Синтез poly. A-хвоста и блок 3’конца – присоединение dd. T с меткой Нанесение на flow cell по 25 pg на линию (200 p. M) Гибридизация с олигоd. T (1 млн на см²) на flow cell
 2 этап: секвенирование • Камера фиксирует начальное положение фрагментов ДНК, связанных с олигоd. T. • Удаление метки, начало синтеза. Quad-цикл (поочередное добавление нуклеотидов с флуоресцентной меткой) • Добавление одного из 4 -х нуклеотидов с флуоресцентной меткой • Синтез (присоединение нуклеотида, комплементарного последовательности фрагмента) • Отмывка от свободных нуклеотидов и реагентов • Фиксирование наличия/отсутствия сигнала в определенных ранее позициях • Удаление флуоресцентной метки • Добавление следующего нуклеотида с меткой и т. д. 30 quad-циклов ~ 8 дней
2 этап: секвенирование • Камера фиксирует начальное положение фрагментов ДНК, связанных с олигоd. T. • Удаление метки, начало синтеза. Quad-цикл (поочередное добавление нуклеотидов с флуоресцентной меткой) • Добавление одного из 4 -х нуклеотидов с флуоресцентной меткой • Синтез (присоединение нуклеотида, комплементарного последовательности фрагмента) • Отмывка от свободных нуклеотидов и реагентов • Фиксирование наличия/отсутствия сигнала в определенных ранее позициях • Удаление флуоресцентной метки • Добавление следующего нуклеотида с меткой и т. д. 30 quad-циклов ~ 8 дней

") Секвенирование генома человека • 28 х покрытие генома • 4 рабочих цикла прибора (~месяц) • SNP – 99, 8% совпадений с SNP Arrays < 1% ложно-положительных результатов при сравнении с секвенированием по Сэнгеру • 48 000 $
Секвенирование генома человека • 28 х покрытие генома • 4 рабочих цикла прибора (~месяц) • SNP – 99, 8% совпадений с SNP Arrays < 1% ложно-положительных результатов при сравнении с секвенированием по Сэнгеру • 48 000 $
") Полупроводниковое секвенирование • Ion Torrent (Life Technologies)
Полупроводниковое секвенирование • Ion Torrent (Life Technologies)
 Секвенирование с использованием нанопоры
Секвенирование с использованием нанопоры
 Grid. ION (содержит 2000 нанопор, обещают 8000 нанопор и геном") Oxford Nanopore (февраль 2012) Grid. ION (содержит 2000 нанопор, обещают 8000 нанопор и геном человека за 15 минут) Mini. ION
Oxford Nanopore (февраль 2012) Grid. ION (содержит 2000 нанопор, обещают 8000 нанопор и геном человека за 15 минут) Mini. ION
 (Complete Genomics)") Секвенирование с использованием «наношаров» (наноболлов) (Complete Genomics)
Секвенирование с использованием «наношаров» (наноболлов) (Complete Genomics)
 • В проточной ячейке происходит секвенирование с использованием лигазной реакции и детекция флуоресцентного сигнала
• В проточной ячейке происходит секвенирование с использованием лигазной реакции и детекция флуоресцентного сигнала
 Что можно делать с помощью next gene технологий? • Геномное секвенирование de novo • Полное геномное ре-секвенирование • Частичное ре-секвенирование (расшифровка «экзома» , сиквенс отдельных генов) • Полный анализ транскриптома • Хроматиновую иммунопреципитацию • Определять экспрессию отдельных генов • Определять уровень метилирования • Секвенирование small RNA
Что можно делать с помощью next gene технологий? • Геномное секвенирование de novo • Полное геномное ре-секвенирование • Частичное ре-секвенирование (расшифровка «экзома» , сиквенс отдельных генов) • Полный анализ транскриптома • Хроматиновую иммунопреципитацию • Определять экспрессию отдельных генов • Определять уровень метилирования • Секвенирование small RNA
 Краткие результаты секвенирования экзомов и геномов за 2011/2012 • Секвенированы почти 700 образцов опухолей, относящихся к 17 -ти типам рака • Создан генетический атлас карциномы яичников и полностью описаны более 100 образцов рака груди (геном, экзом, РНК, SNP -чипы) • Около 100 публикаций по секвенированию экзомов, 60 из них связаны с открытием редких генетических вариантов, ассоциированных с заболеваниями.
Краткие результаты секвенирования экзомов и геномов за 2011/2012 • Секвенированы почти 700 образцов опухолей, относящихся к 17 -ти типам рака • Создан генетический атлас карциномы яичников и полностью описаны более 100 образцов рака груди (геном, экзом, РНК, SNP -чипы) • Около 100 публикаций по секвенированию экзомов, 60 из них связаны с открытием редких генетических вариантов, ассоциированных с заболеваниями.
 Недостаточные причины для секвенирования экзомов и геномов 1. Генотипирование по распространенным в популяции вариантам 2. Генетическое профилирование по аллелям риска, которые были выявлены в ходе полногеномных исследований ассоциации (GWAS) для распространенных заболеваний 1 и 2 - секвенирование невыгодно, поскольку не дает значимой медицинской информации (даже суммарный риск по таким маркерам достаточно низок) и дороже, чем обычное генотипирование
Недостаточные причины для секвенирования экзомов и геномов 1. Генотипирование по распространенным в популяции вариантам 2. Генетическое профилирование по аллелям риска, которые были выявлены в ходе полногеномных исследований ассоциации (GWAS) для распространенных заболеваний 1 и 2 - секвенирование невыгодно, поскольку не дает значимой медицинской информации (даже суммарный риск по таким маркерам достаточно низок) и дороже, чем обычное генотипирование
 Веские причины для секвенирования экзомов и геномов • Выявление редких наследственных вариантов при моногенной и семейной патологии • Выявление редких наследственных вариантов (аллелей высокого риска) при распространенных заболеваниях • Выявление соматических мутаций при раке • Поиск генов, связанных с заболеваниями • Клиническая или молекулярная диагностика
Веские причины для секвенирования экзомов и геномов • Выявление редких наследственных вариантов при моногенной и семейной патологии • Выявление редких наследственных вариантов (аллелей высокого риска) при распространенных заболеваниях • Выявление соматических мутаций при раке • Поиск генов, связанных с заболеваниями • Клиническая или молекулярная диагностика
 Примеры редких кодирующих аллелей при распространенных заболеваниях • • BRCA 1/2 ( и 11 других генов) – рак груди APP, PS 1/2, UBQLN 1 – ранняя форма болезни Альцгеймерa NRXN 1, SHANK 3 - аутизм ANGPTL 3/4/5, NPC 1 L 1, PCSK 9 – повышенный уровень холестерина и триглицеридов Подходы для выявления таких аллелей: 1) Секвенирование экзомов у большой выборки людей с заболеванием или у людей с крайней степенью фенотипического проявления признака (очень высокое/очень низкое значение параметра) 2) Целенаправленный анализ моногенных вариантов при распространенных заболеваниях
Примеры редких кодирующих аллелей при распространенных заболеваниях • • BRCA 1/2 ( и 11 других генов) – рак груди APP, PS 1/2, UBQLN 1 – ранняя форма болезни Альцгеймерa NRXN 1, SHANK 3 - аутизм ANGPTL 3/4/5, NPC 1 L 1, PCSK 9 – повышенный уровень холестерина и триглицеридов Подходы для выявления таких аллелей: 1) Секвенирование экзомов у большой выборки людей с заболеванием или у людей с крайней степенью фенотипического проявления признака (очень высокое/очень низкое значение параметра) 2) Целенаправленный анализ моногенных вариантов при распространенных заболеваниях
 и") Проблемы секвенирования больших геномов • Инсерции, делеции, повторы - Использование сцепленно-парного (Mate paired) и парноконцевого (Paired ends) секвенирования • Гомополимерные последовательности (например, GGGGG) - Использование модифицированных нуклеотидов и полимераз
Проблемы секвенирования больших геномов • Инсерции, делеции, повторы - Использование сцепленно-парного (Mate paired) и парноконцевого (Paired ends) секвенирования • Гомополимерные последовательности (например, GGGGG) - Использование модифицированных нуклеотидов и полимераз
 Mate pair ends 1. 2. 3. Получение одноцепочечных фрагментов ДНК определенного размера (например, 500 bp), создание библиотек Нас интересуют только короткие последовательности на концах фрагмента (35 -100 bp каждая). Последовательность в середине нам не нужна. После ряда процедур удаляется последовательность из середины, остаются только концы, которые секвенируем.
Mate pair ends 1. 2. 3. Получение одноцепочечных фрагментов ДНК определенного размера (например, 500 bp), создание библиотек Нас интересуют только короткие последовательности на концах фрагмента (35 -100 bp каждая). Последовательность в середине нам не нужна. После ряда процедур удаляется последовательность из середины, остаются только концы, которые секвенируем.
 Mate pair ends - 2 • - После сиквенса у нас есть: концевые последовательности 1 и 2 мы знаем, что в геноме они разделены ~500 bp у нас огромное количество таких фланкирующих последовательностей, полученных от разных фрагментов С помощью специальной программы картируем эти последовательности на референсный геном. По изменению расстояния между последовательностями можем судить о наличии инсерции/делеции. Если один конец не картируется из -за высокого содержания повторов в регионе, а второй – картируется, можем локализовать и первую последовательность, т. к. знаем растояние между ними.
Mate pair ends - 2 • - После сиквенса у нас есть: концевые последовательности 1 и 2 мы знаем, что в геноме они разделены ~500 bp у нас огромное количество таких фланкирующих последовательностей, полученных от разных фрагментов С помощью специальной программы картируем эти последовательности на референсный геном. По изменению расстояния между последовательностями можем судить о наличии инсерции/делеции. Если один конец не картируется из -за высокого содержания повторов в регионе, а второй – картируется, можем локализовать и первую последовательность, т. к. знаем растояние между ними.