

d9a3fa29785182389d0a963f92e3969f.ppt
- Количество слайдов: 59
 DEFINITION SSCP is the electrophoretic separation of singlestranded nucleic acids") Single-Strand Conformation Polymorphism (SSCP) DEFINITION SSCP is the electrophoretic separation of singlestranded nucleic acids based on subtle differences in sequence (often a single base pair) which results in a different secondary structure and a measurable difference in mobility through a gel.
Single-Strand Conformation Polymorphism (SSCP) DEFINITION SSCP is the electrophoretic separation of singlestranded nucleic acids based on subtle differences in sequence (often a single base pair) which results in a different secondary structure and a measurable difference in mobility through a gel.
 BACKGROUND • The mobility of double-stranded DNA in gel electrophoresis is dependent on strand size and length but is relatively independent of the particular nucleotide sequence. The mobility of single strands, however, is noticeably affected by very small changes in sequence, possibly one changed nucleotide out of several hundred. Small changes are noticeable because of the relatively unstable nature of single-stranded DNA; in the absence of a complementary strand, the single strand may experience intrastrand base pairing, resulting in loops and folds that give the single strand a unique 3 D structure, regardless of its length. A single nucleotide change could dramatically affect the strand's mobility through a gel by altering the intrastrand base pairing and its resulting 3 D conformation
BACKGROUND • The mobility of double-stranded DNA in gel electrophoresis is dependent on strand size and length but is relatively independent of the particular nucleotide sequence. The mobility of single strands, however, is noticeably affected by very small changes in sequence, possibly one changed nucleotide out of several hundred. Small changes are noticeable because of the relatively unstable nature of single-stranded DNA; in the absence of a complementary strand, the single strand may experience intrastrand base pairing, resulting in loops and folds that give the single strand a unique 3 D structure, regardless of its length. A single nucleotide change could dramatically affect the strand's mobility through a gel by altering the intrastrand base pairing and its resulting 3 D conformation
, SSCPs are allelic variants of inherited,") • Like restriction fragment length polymorphisms (RFLPs), SSCPs are allelic variants of inherited, genetic traits that can be used as genetic markers. Unlike RFLP analysis, however, SSCP analysis can detect DNA polymorphisms and mutations at multiple places in DNA fragments. As a mutation scanning technique, though, SSCP is more often used to analyze the polymorphisms at single loci, especially when used for medical diagnoses. • Most experiments involving SSCP are designed to evaluate polymorphisms at single loci and compare the results from different individuals.
• Like restriction fragment length polymorphisms (RFLPs), SSCPs are allelic variants of inherited, genetic traits that can be used as genetic markers. Unlike RFLP analysis, however, SSCP analysis can detect DNA polymorphisms and mutations at multiple places in DNA fragments. As a mutation scanning technique, though, SSCP is more often used to analyze the polymorphisms at single loci, especially when used for medical diagnoses. • Most experiments involving SSCP are designed to evaluate polymorphisms at single loci and compare the results from different individuals.
 A B C
A B C
 Figure 1: SSCP Procedure. The three equal-length doublestranded DNA fragments are shown with the corresponding single-stranded structures, the red fragment folding into the smallest molecule and the green the largest (Panel A). The desired polymorphism is selected with PCR primers; primer A is in excess to amplify only a single strand (Panel B). Both the doublestranded and single-stranded fragments are run through gel electrophoresis (Panel C). If not for the color labels, there would be no distinction between the double-stranded fragments. The single-stranded fragments, however, show considerable variation in mobility; the small red molecule migrates more quickly through the gel than either the blue or the large green molecule. Using this SSCP result, it becomes clear that the different lanes (red, blue, or green) contain strands with different sequences; the more far apart the bands, the less similar the nucleotide sequences
Figure 1: SSCP Procedure. The three equal-length doublestranded DNA fragments are shown with the corresponding single-stranded structures, the red fragment folding into the smallest molecule and the green the largest (Panel A). The desired polymorphism is selected with PCR primers; primer A is in excess to amplify only a single strand (Panel B). Both the doublestranded and single-stranded fragments are run through gel electrophoresis (Panel C). If not for the color labels, there would be no distinction between the double-stranded fragments. The single-stranded fragments, however, show considerable variation in mobility; the small red molecule migrates more quickly through the gel than either the blue or the large green molecule. Using this SSCP result, it becomes clear that the different lanes (red, blue, or green) contain strands with different sequences; the more far apart the bands, the less similar the nucleotide sequences
 • Procedure as illustrated in Figure 1: 1. A specific pair of PCR primers (forward and reverse) is used to amplify the desired DNA fragments from individuals. 2. Single-stranded DNA is produced by asymmetric PCR: the primer on one side of the fragment is greatly in excess over the other primer. After the low-concentration primer supply is exhausted, continued PCR produces only the target single strand. 3. The mobilities of the single stranded fragments are compared by electrophoresis on a neutral polyacrylamide gel. 4. Bands are detected by radioactive labeling or (more often) silver staining, and the pattern is interpreted
• Procedure as illustrated in Figure 1: 1. A specific pair of PCR primers (forward and reverse) is used to amplify the desired DNA fragments from individuals. 2. Single-stranded DNA is produced by asymmetric PCR: the primer on one side of the fragment is greatly in excess over the other primer. After the low-concentration primer supply is exhausted, continued PCR produces only the target single strand. 3. The mobilities of the single stranded fragments are compared by electrophoresis on a neutral polyacrylamide gel. 4. Bands are detected by radioactive labeling or (more often) silver staining, and the pattern is interpreted
. Figure 2: Sample SSCP Gel Result and Interpretation. DNA was isolated and amplified") ). Figure 2: Sample SSCP Gel Result and Interpretation. DNA was isolated and amplified from sand flies (Lutzomyia longipalpis). SCCP analysis of the DNA shows multiple haplotypes, or sets of alleles usually inherited as a unit. Lanes 3 and 4 were identical haplotypes from two individuals. The difference in band migration in adjacent lanes is associated with the number of nucleotide differences (in parentheses): lanes 2 -3 (2), lanes 3 -4 (0), lanes 4 -5 (3), lanes 5 -6 (1), lanes 6 -7 (3), lanes 7 -8 (1), lanes 8 -9 (1), and lanes 9 -10 (4).
). Figure 2: Sample SSCP Gel Result and Interpretation. DNA was isolated and amplified from sand flies (Lutzomyia longipalpis). SCCP analysis of the DNA shows multiple haplotypes, or sets of alleles usually inherited as a unit. Lanes 3 and 4 were identical haplotypes from two individuals. The difference in band migration in adjacent lanes is associated with the number of nucleotide differences (in parentheses): lanes 2 -3 (2), lanes 3 -4 (0), lanes 4 -5 (3), lanes 5 -6 (1), lanes 6 -7 (3), lanes 7 -8 (1), lanes 8 -9 (1), and lanes 9 -10 (4).
 SSCP LIMITATIONS AND CONSIDERATIONS • Single-stranded DNA mobilities are dependent on temperature. For best results, gel electrophoresis must be run in a constant temperature. • Sensitivity of SSCP is affected by p. H. Doublestranded DNA fragments are usually denatured by exposure to basic conditions: a high p. H. Kukita et al. found that adding glycerol to the polyacrylamide gel lowers the p. H of the electrophoresis buffer-more specifically, the Tris-borate buffer--and the result is increased SSCP sensitivity and clearer data.
SSCP LIMITATIONS AND CONSIDERATIONS • Single-stranded DNA mobilities are dependent on temperature. For best results, gel electrophoresis must be run in a constant temperature. • Sensitivity of SSCP is affected by p. H. Doublestranded DNA fragments are usually denatured by exposure to basic conditions: a high p. H. Kukita et al. found that adding glycerol to the polyacrylamide gel lowers the p. H of the electrophoresis buffer-more specifically, the Tris-borate buffer--and the result is increased SSCP sensitivity and clearer data.
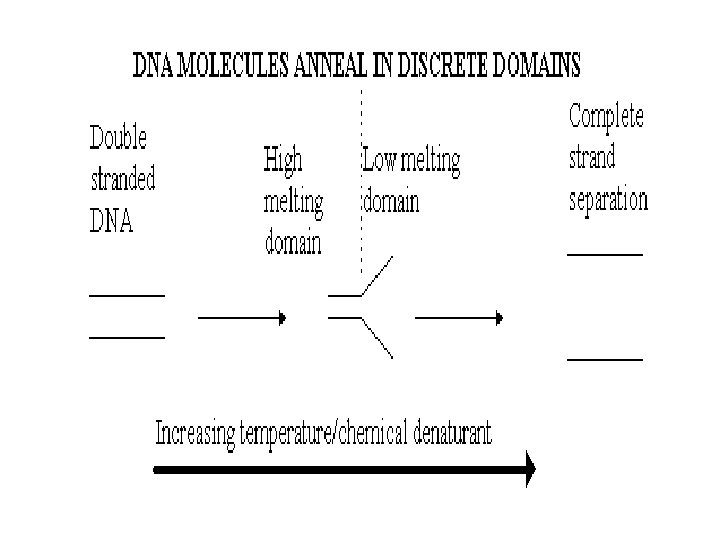
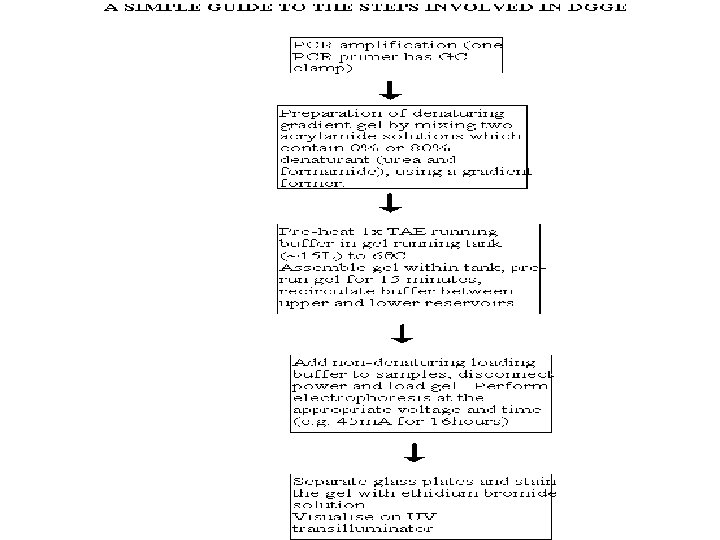
 THEORY The theory behind DGGE is very simple, the") DENATURING GRADIENT GEL ELECTROPHORESIS (DGGE) THEORY The theory behind DGGE is very simple, the two strands of a DNA molecule separate, or melt, when heat or a chemical denaturant is applied. The temperature at which a DNA duplex melts is influenced by two factors: 1. The hydrogen bonds formed between complimentary base pairs, GC rich regions melt at higher temperatures than regions that are AT rich. 2. The attraction between neighbouring bases of the same strand or "stacking" Consequently, a DNA molecule may have several melting domains with characteristic melting temperatures (Tm) determined by the nucleotide sequence.
DENATURING GRADIENT GEL ELECTROPHORESIS (DGGE) THEORY The theory behind DGGE is very simple, the two strands of a DNA molecule separate, or melt, when heat or a chemical denaturant is applied. The temperature at which a DNA duplex melts is influenced by two factors: 1. The hydrogen bonds formed between complimentary base pairs, GC rich regions melt at higher temperatures than regions that are AT rich. 2. The attraction between neighbouring bases of the same strand or "stacking" Consequently, a DNA molecule may have several melting domains with characteristic melting temperatures (Tm) determined by the nucleotide sequence.
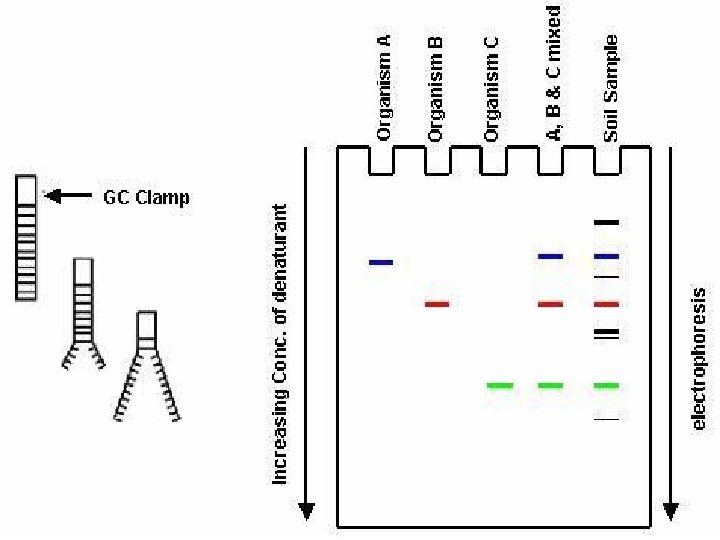
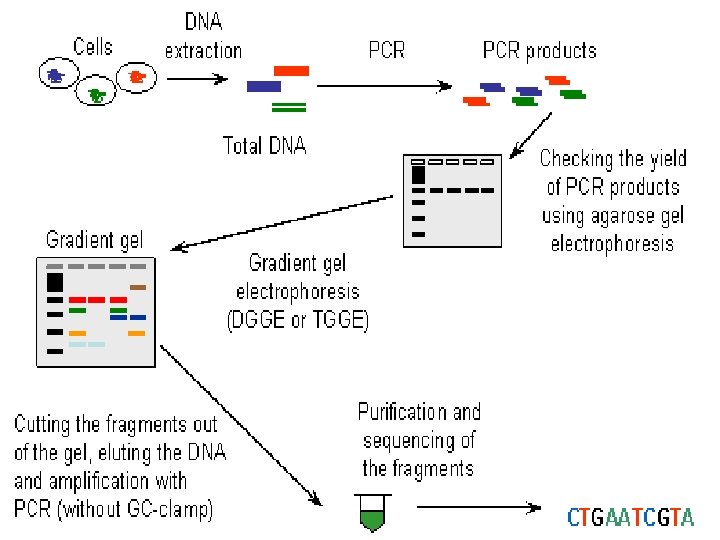
 DGGE exploits the fact that otherwise identical DNA molecules, which differ by only one nucleotide within a low melting domain will have different melting temperatures. When separated by electrophoresis through a gradient of increasing chemical denaturant (usually formamide and urea), the mobility of the molecule is retarded at the concentration at which the DNA strands of low melt domain dissociate. The branched structure of the single stranded moiety of the molecule becomes entangled in the gel matrix and no further movement occurs. Complete strand separation is prevented by the presence of a high melting domain, which is usually artificially created at one end of the molecule by incorporation of a GC clamp. This is accomplished during PCR amplification using a PCR primer with a 5' tail consisting of a sequence of 40 GC. PRELIMINARY PREPARATION: PRIMER DESIGN AND OPTIMISATION OF GEL RUNNING CONDITIONS. DGGE is usually performed on PCR products, primers must carefully be chosen so that the region to be screened for mutations has one or at the most two discrete melting domains (excluding the GC clamp).
DGGE exploits the fact that otherwise identical DNA molecules, which differ by only one nucleotide within a low melting domain will have different melting temperatures. When separated by electrophoresis through a gradient of increasing chemical denaturant (usually formamide and urea), the mobility of the molecule is retarded at the concentration at which the DNA strands of low melt domain dissociate. The branched structure of the single stranded moiety of the molecule becomes entangled in the gel matrix and no further movement occurs. Complete strand separation is prevented by the presence of a high melting domain, which is usually artificially created at one end of the molecule by incorporation of a GC clamp. This is accomplished during PCR amplification using a PCR primer with a 5' tail consisting of a sequence of 40 GC. PRELIMINARY PREPARATION: PRIMER DESIGN AND OPTIMISATION OF GEL RUNNING CONDITIONS. DGGE is usually performed on PCR products, primers must carefully be chosen so that the region to be screened for mutations has one or at the most two discrete melting domains (excluding the GC clamp).
 The GC clamp is usually positioned adjacent to the highest melting domain. Thus, full sequence data must be available so that a melt map of the molecule can be constructed, and primers can be designed to amplify a region of unit melting domain. The optimal gradient and gel running conditions must also be established. Computer Generation of Melt Maps and Primer Design The programs MELT 87, MELT 95 and MACMELT are used to generate a melt map from a known DNA sequence. The programs identify primer pairs that will amplify short segments of unit melting domain. Ideally the PCR products should be between 100 and 400 bp in size. The program predicts the effect on the melting temperature of the PCR product when a GC clamp is positioned at one of the four ends of the molecule.
The GC clamp is usually positioned adjacent to the highest melting domain. Thus, full sequence data must be available so that a melt map of the molecule can be constructed, and primers can be designed to amplify a region of unit melting domain. The optimal gradient and gel running conditions must also be established. Computer Generation of Melt Maps and Primer Design The programs MELT 87, MELT 95 and MACMELT are used to generate a melt map from a known DNA sequence. The programs identify primer pairs that will amplify short segments of unit melting domain. Ideally the PCR products should be between 100 and 400 bp in size. The program predicts the effect on the melting temperature of the PCR product when a GC clamp is positioned at one of the four ends of the molecule.
 Alternatively, optimal gradient concentrations can be determined empirically by performing perpendicular gel electrophoresis. In such an experiment the denaturing gradient is perpendicular to the direction of electrophoresis. Optimisation of Gel Running Conditions The computer programs described above reduce the number of preliminary experiments required for optimisation of the gel running conditions. However, it is still necessary to run some preliminary gels to determine the optimal voltages and running times and to confirm the optimal denaturing gradient that has been chosen. The aim is to have well separated bands (normal and mutation positive control are simultaneously loaded on the gels) which are "focused" by the gradient. PCR products with two low melting domains require different gel conditions for the analysis of each domain. When optimised gel running conditions have been established the method can be used for mutation screening.
Alternatively, optimal gradient concentrations can be determined empirically by performing perpendicular gel electrophoresis. In such an experiment the denaturing gradient is perpendicular to the direction of electrophoresis. Optimisation of Gel Running Conditions The computer programs described above reduce the number of preliminary experiments required for optimisation of the gel running conditions. However, it is still necessary to run some preliminary gels to determine the optimal voltages and running times and to confirm the optimal denaturing gradient that has been chosen. The aim is to have well separated bands (normal and mutation positive control are simultaneously loaded on the gels) which are "focused" by the gradient. PCR products with two low melting domains require different gel conditions for the analysis of each domain. When optimised gel running conditions have been established the method can be used for mutation screening.
 Detection Rate and Sensitivity The detection rate is very high, in many cases it approaches 100%. It has been possible to identify a mutation present in a 100 bp sequence at the level of 0. 5%. Thus, DGGE is an ideal choice for mutation detection in the diagnostic laboratory ADVANTAGES of DGGE 1. High detection rate and sensitivity. 2. The methodology is simple and a non-radioactive detection method is used. 3. PCR fragments may be isolated from the gel and used in sequencing reactions. DISADVANTAGES of DGGE 1. Computer analysis and preliminary experiments are essential when setting up DGGE. 2. Purchase of DGGE equipment may be required. 3. Primers are more expensive because of the 40 bases of GC clamp.
Detection Rate and Sensitivity The detection rate is very high, in many cases it approaches 100%. It has been possible to identify a mutation present in a 100 bp sequence at the level of 0. 5%. Thus, DGGE is an ideal choice for mutation detection in the diagnostic laboratory ADVANTAGES of DGGE 1. High detection rate and sensitivity. 2. The methodology is simple and a non-radioactive detection method is used. 3. PCR fragments may be isolated from the gel and used in sequencing reactions. DISADVANTAGES of DGGE 1. Computer analysis and preliminary experiments are essential when setting up DGGE. 2. Purchase of DGGE equipment may be required. 3. Primers are more expensive because of the 40 bases of GC clamp.
 4. Additional primers may be required for sequencing? 5. Analysis of PCR fragments over 400 bp is less successful. 6. Genes which are exceptionally GC rich are not easily analysed by DGGE. VARIATIONS OF DGGE TEMPERATURE GRADIENT GEL ELECTROPHORESIS (TGGE) The chemical denaturant gradient is replaced by a gradient of increasing temperature down the gel. CONSTANT DENATURANT GEL ELECTROPHORESIS (CDGE) The chemical denaturant is at a constant concentration throughout the gel, equivalent to the melting temperature of the low melting domain. This approach requires different gel conditions for each PCR fragment to be analysed. The main application of CDGE is limited to the identification of known mutations.
4. Additional primers may be required for sequencing? 5. Analysis of PCR fragments over 400 bp is less successful. 6. Genes which are exceptionally GC rich are not easily analysed by DGGE. VARIATIONS OF DGGE TEMPERATURE GRADIENT GEL ELECTROPHORESIS (TGGE) The chemical denaturant gradient is replaced by a gradient of increasing temperature down the gel. CONSTANT DENATURANT GEL ELECTROPHORESIS (CDGE) The chemical denaturant is at a constant concentration throughout the gel, equivalent to the melting temperature of the low melting domain. This approach requires different gel conditions for each PCR fragment to be analysed. The main application of CDGE is limited to the identification of known mutations.

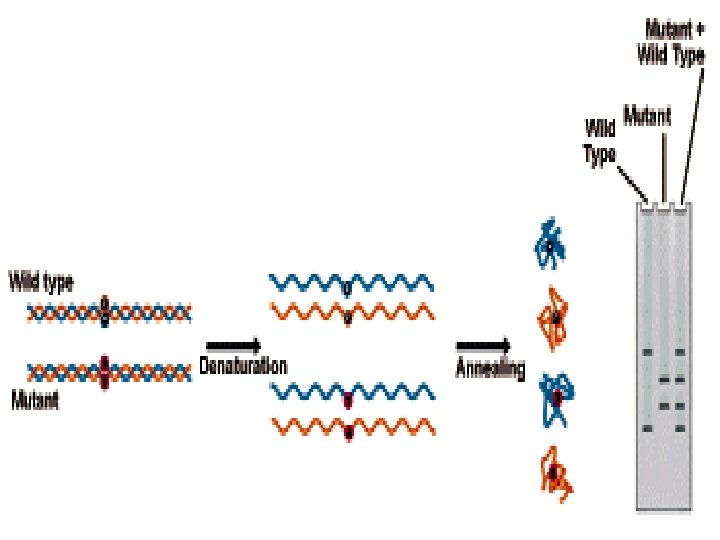
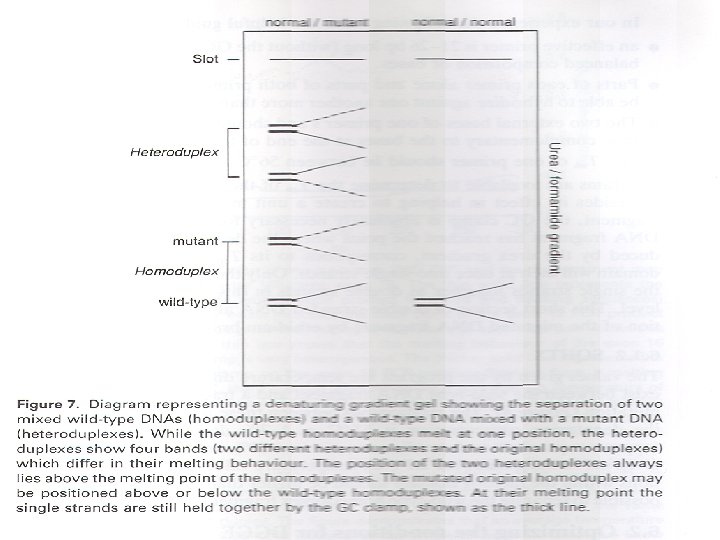
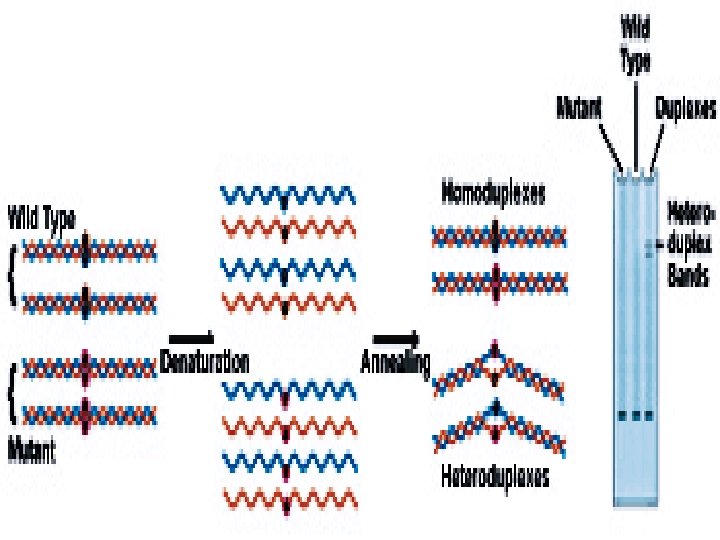
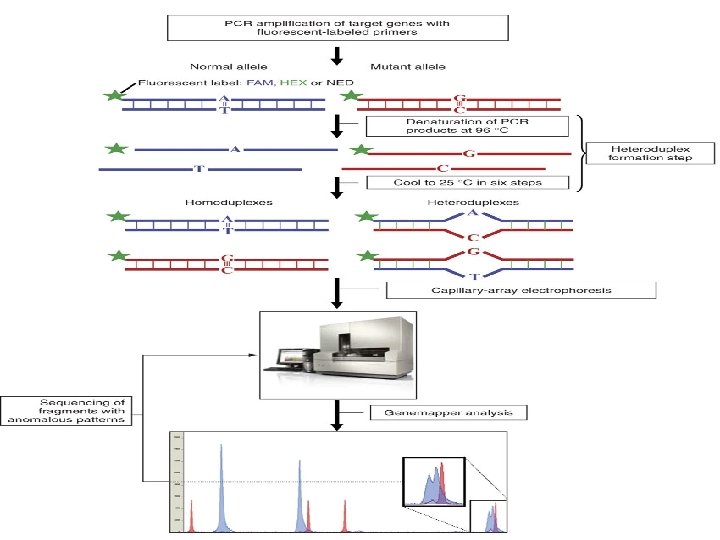
 Heteroduplex Analysis Mutations are detected by heteroduplex analysis based on the retardation of the heteroduplex compared with the corresponding homoduplex on a non-denaturing polyacrylamide gel. Heteroduplexes migrate more slowly than their corresponding homoduplexes due to a more open double-stranded configuration surrounding the mismatched bases. Basic Protocol Heteroduplexes are formed by mixing wild-type and mutant DNA amplified by PCR. The samples are denatured and 'reannealed' (usually by heating and cooling). Four distinct species are generated by this reassortment: wild-type homoduplex, mutant homoduplex, and two heteroduplexes. The formation of the heteroduplexes and their stability depend primarily on the type of mutation in the fragment. Single-base changes are more sensitive to temperature,
Heteroduplex Analysis Mutations are detected by heteroduplex analysis based on the retardation of the heteroduplex compared with the corresponding homoduplex on a non-denaturing polyacrylamide gel. Heteroduplexes migrate more slowly than their corresponding homoduplexes due to a more open double-stranded configuration surrounding the mismatched bases. Basic Protocol Heteroduplexes are formed by mixing wild-type and mutant DNA amplified by PCR. The samples are denatured and 'reannealed' (usually by heating and cooling). Four distinct species are generated by this reassortment: wild-type homoduplex, mutant homoduplex, and two heteroduplexes. The formation of the heteroduplexes and their stability depend primarily on the type of mutation in the fragment. Single-base changes are more sensitive to temperature,
 solvents, and ionic strength of the buffer. There is no way to predict the influence of these parameters on the stability of the heteroduplex, and thus electrophoretic conditions must be optimized , ( solvents, and ionic strength of the buffer).
solvents, and ionic strength of the buffer. There is no way to predict the influence of these parameters on the stability of the heteroduplex, and thus electrophoretic conditions must be optimized , ( solvents, and ionic strength of the buffer).
 Microsatellite DNA Methodology Microsatellites (sometimes referred to as a variable number of tandem repeats or VNTRs) are short segments of DNA that have a repeated sequence such as CACA, and they tend to occur in non-coding DNA. In some microsatellites, the repeated unit (e. g. CA) may occur four times, in others it may be seven, or two, or thirty. In diploid organisms each individual animal will have two copies of any particular microsatellite segment. For example, a father might have a genotype of 12 repeats and 19 repeats, a mother might have 18 repeats and 15 repeats while their first born might have repeats of 12 and 15. On rare occasions, microsatellites can cause the DNA polymerase to make an extra copy of CA. Over time, as animals in a population breed, they will recombine their microsatellites during sexual reproduction and the population will maintain a variety of microsatellites that is characteristic for that population and distinct from other populations which do not interbreed.
Microsatellite DNA Methodology Microsatellites (sometimes referred to as a variable number of tandem repeats or VNTRs) are short segments of DNA that have a repeated sequence such as CACA, and they tend to occur in non-coding DNA. In some microsatellites, the repeated unit (e. g. CA) may occur four times, in others it may be seven, or two, or thirty. In diploid organisms each individual animal will have two copies of any particular microsatellite segment. For example, a father might have a genotype of 12 repeats and 19 repeats, a mother might have 18 repeats and 15 repeats while their first born might have repeats of 12 and 15. On rare occasions, microsatellites can cause the DNA polymerase to make an extra copy of CA. Over time, as animals in a population breed, they will recombine their microsatellites during sexual reproduction and the population will maintain a variety of microsatellites that is characteristic for that population and distinct from other populations which do not interbreed.
 The most common way to detect microsatellites is to design PCR primers that are unique to one locus in the genome and that base pair on either side of the repeated portion (figure 1). Therefore, a single pair of PCR primers will work for every individual in the species and produce different sized products for each of the different length microsatellites. Figure 1. Detecting microsatellites from genomic DNA. Two PCR primers (forward and reverse gray arrows) are designed to flank the microsatellite region. If there were zero repeats, the PCR product would be 100 bp in length. Therefore, by determining the size of each PCR product (in this case 116 bp), you can calculate how many CA repeats are present in each microsatellite (8 CA repeats in this example).
The most common way to detect microsatellites is to design PCR primers that are unique to one locus in the genome and that base pair on either side of the repeated portion (figure 1). Therefore, a single pair of PCR primers will work for every individual in the species and produce different sized products for each of the different length microsatellites. Figure 1. Detecting microsatellites from genomic DNA. Two PCR primers (forward and reverse gray arrows) are designed to flank the microsatellite region. If there were zero repeats, the PCR product would be 100 bp in length. Therefore, by determining the size of each PCR product (in this case 116 bp), you can calculate how many CA repeats are present in each microsatellite (8 CA repeats in this example).
 The PCR products are then separated by either gel electrophoresis or capillary electrophoresis. Either way, the investigator can determine the size of the PCR product and thus how many times the dinucleotide "CA" was repeated for each allele (figure 2). It would be nice if microsatellite data produced only two bands but often there are minor bands in addition to the major bands; they are called stutter bands and they usually differ from the major bands by two nucleotides.
The PCR products are then separated by either gel electrophoresis or capillary electrophoresis. Either way, the investigator can determine the size of the PCR product and thus how many times the dinucleotide "CA" was repeated for each allele (figure 2). It would be nice if microsatellite data produced only two bands but often there are minor bands in addition to the major bands; they are called stutter bands and they usually differ from the major bands by two nucleotides.
 Figure 2. Stylized examples of microsatellite data. Left half: four sets of data were produced by gel electrophoresis and so you can see the major (black) and stutter (gray) bands. MW; molecular weight standards. Right half: These data were produced by analysis on an automated capillary electrophoresis-based DNA sequencer. The data are line graphs with the location of each peak on the X-axis representing a different sized PCR product and the height of each peak indicates the amount of PCR product. The major bands produce higher peaks than the stutter peaks.
Figure 2. Stylized examples of microsatellite data. Left half: four sets of data were produced by gel electrophoresis and so you can see the major (black) and stutter (gray) bands. MW; molecular weight standards. Right half: These data were produced by analysis on an automated capillary electrophoresis-based DNA sequencer. The data are line graphs with the location of each peak on the X-axis representing a different sized PCR product and the height of each peak indicates the amount of PCR product. The major bands produce higher peaks than the stutter peaks.
 Capillary Electrophoresis Because the capillary tube has a high surface to volume ratio (25 -100 µm diameter), it radiates heat readily and thus samples do not over heat. Detection of the migrating molecules is accomplished by shining a light source through a portion of the tubing and detecting the light emitted from the other side (figure 1). Figure 1. Schematic of capillary electrophoresis system. Samples enter the tube from the right and travel to the left to the detection system which records the chromatogram output on a computer.
Capillary Electrophoresis Because the capillary tube has a high surface to volume ratio (25 -100 µm diameter), it radiates heat readily and thus samples do not over heat. Detection of the migrating molecules is accomplished by shining a light source through a portion of the tubing and detecting the light emitted from the other side (figure 1). Figure 1. Schematic of capillary electrophoresis system. Samples enter the tube from the right and travel to the left to the detection system which records the chromatogram output on a computer.
 Sample run times are very Short. Samples are applied to the capillary tubes when the cathode buffer is moved aside and sample chamber placed at the opening of the capillary tube. Either pressure is applied to the sample and 10 - 100 n. L is injected or an electrical current is applied through the sample and only the charged molecules enter the capillary. Once the electrophoretic separation is completed, the contents of the capillary are flushed out and fresh matrix fills the tube. Replacing the matrix within the capillary minimizes the possiblity of contaminating samples between runs.
Sample run times are very Short. Samples are applied to the capillary tubes when the cathode buffer is moved aside and sample chamber placed at the opening of the capillary tube. Either pressure is applied to the sample and 10 - 100 n. L is injected or an electrical current is applied through the sample and only the charged molecules enter the capillary. Once the electrophoretic separation is completed, the contents of the capillary are flushed out and fresh matrix fills the tube. Replacing the matrix within the capillary minimizes the possiblity of contaminating samples between runs.
 is commonly used to detect point mutations and other") Sequence-Specific PCR Sequence-specific PCR (SSP-PCR) is commonly used to detect point mutations and other single nucleotide polymorphisms. There are numerous modifications to the method, which involves careful design of primers such that the primer 3′ end falls on the nucleotide to be analyzed. Unlike the 5′ end, the 3′ end of a primer must match the template perfectly to be extended by Taq polymerase (Fig. 9 -12). By designing primers to end on a mutation, the presence or absence of product can be interpreted as the presence or absence of the mutation. Normal and mutant sequences can be analyzed simultaneously by making one primer longer than the other, resulting in differently sized products, depending on the sequence of the template. Multiplexed SSP-PCR was originally called Amplification Refractory Mutation System ( ARMS) PCR.
Sequence-Specific PCR Sequence-specific PCR (SSP-PCR) is commonly used to detect point mutations and other single nucleotide polymorphisms. There are numerous modifications to the method, which involves careful design of primers such that the primer 3′ end falls on the nucleotide to be analyzed. Unlike the 5′ end, the 3′ end of a primer must match the template perfectly to be extended by Taq polymerase (Fig. 9 -12). By designing primers to end on a mutation, the presence or absence of product can be interpreted as the presence or absence of the mutation. Normal and mutant sequences can be analyzed simultaneously by making one primer longer than the other, resulting in differently sized products, depending on the sequence of the template. Multiplexed SSP-PCR was originally called Amplification Refractory Mutation System ( ARMS) PCR.
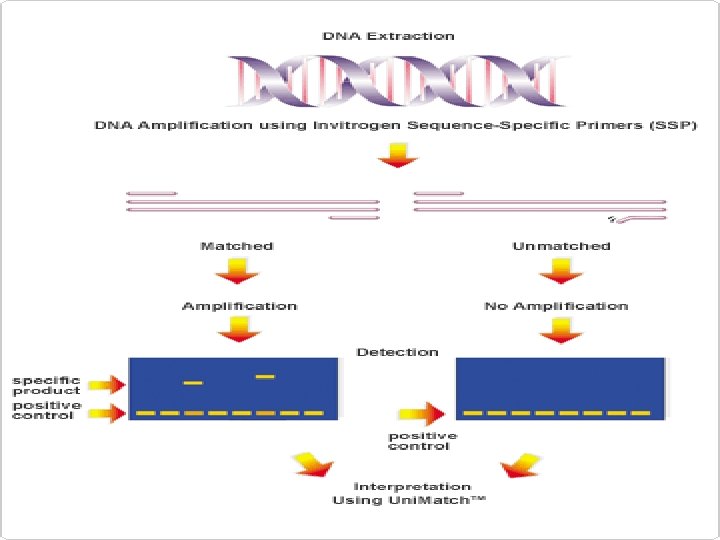
 Sequence – Specific PCR § multiplex SSP-PCR Figure 9 -14 Multiplex allele-specific PCR. The mutation (C→A) is detected by an allelespecific primer (3) that ends at the mutation. Primers 3 and 4 would then produce a midsized fragment (1– 3). If there is no mutation, a normal primer (2) binds and produces a smaller fragment (2– 4). Primers 1 and 4 always amplify the entire region (1– 4).
Sequence – Specific PCR § multiplex SSP-PCR Figure 9 -14 Multiplex allele-specific PCR. The mutation (C→A) is detected by an allelespecific primer (3) that ends at the mutation. Primers 3 and 4 would then produce a midsized fragment (1– 3). If there is no mutation, a normal primer (2) binds and produces a smaller fragment (2– 4). Primers 1 and 4 always amplify the entire region (1– 4).
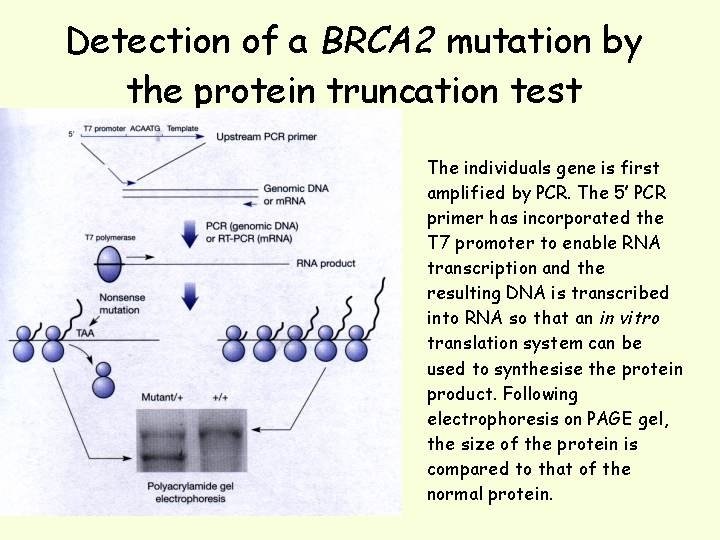
 Protein Truncation Test Nonsense or frameshift mutations cause premature truncation of proteins. The protein truncation test (also called in vitro synthesized protein or in vitro transcription/translation) is designed to detect truncated proteins as an indication of the presence of DNA mutations. This procedure uses a PCR product containing the area of the gene likely to have a truncating DNA mutation. The PCR product is transcribed and translated in vitro using commercially available coupled transcription/translation systems. When the peptide products of the reaction are resolved by polyacrylamide gel electrophoresis, bands below the normal control bands, representing truncated translation products, are indicative of the presence of DNA mutations
Protein Truncation Test Nonsense or frameshift mutations cause premature truncation of proteins. The protein truncation test (also called in vitro synthesized protein or in vitro transcription/translation) is designed to detect truncated proteins as an indication of the presence of DNA mutations. This procedure uses a PCR product containing the area of the gene likely to have a truncating DNA mutation. The PCR product is transcribed and translated in vitro using commercially available coupled transcription/translation systems. When the peptide products of the reaction are resolved by polyacrylamide gel electrophoresis, bands below the normal control bands, representing truncated translation products, are indicative of the presence of DNA mutations
 Most disease-causing mutations result in truncation of the protein product, so the protein truncation test is used most often because it will detect changes that are biologically significant. Method of choice for screening mutations in tumour supressors where over 90 -95% of mutations are chain terminating (BRCA 1, BRCA 2, APC). The gene (best to start with m. RNA as template) is amplified by PCR and the product of the PCR reaction is used to transcribe/translate the protein encoded by the gene using an in vitro translation system. The synthesised protein is run by SDS PAGE and compared to the wildtype protein where any differences in size can be visualised by staining the gel with Coomassie blue or silver stain.
Most disease-causing mutations result in truncation of the protein product, so the protein truncation test is used most often because it will detect changes that are biologically significant. Method of choice for screening mutations in tumour supressors where over 90 -95% of mutations are chain terminating (BRCA 1, BRCA 2, APC). The gene (best to start with m. RNA as template) is amplified by PCR and the product of the PCR reaction is used to transcribe/translate the protein encoded by the gene using an in vitro translation system. The synthesised protein is run by SDS PAGE and compared to the wildtype protein where any differences in size can be visualised by staining the gel with Coomassie blue or silver stain.
 Testing for a gene with many possible mutations For genes where there is the possibility of being affected by one or more of several different mutations the attempt is to find differences between the test sample and a reference wild-type sequence. Even if there is only one nucleotide difference between two sequences (SNP), several different tests are available which can differentiate between them 1. Single stranded conformational polymorphisms 2. Denaturing gradient gel electrophoresis 3. Heteroduplex analysis 4. Protein truncation test 5. DNA chips 6. DNA sequencing There are several monogenic disorders for which the mutations have been well characterised and the carrier incidence determined. For example, the cystic fibrosis gene is known to have more than 500 different mutations that affect its function.
Testing for a gene with many possible mutations For genes where there is the possibility of being affected by one or more of several different mutations the attempt is to find differences between the test sample and a reference wild-type sequence. Even if there is only one nucleotide difference between two sequences (SNP), several different tests are available which can differentiate between them 1. Single stranded conformational polymorphisms 2. Denaturing gradient gel electrophoresis 3. Heteroduplex analysis 4. Protein truncation test 5. DNA chips 6. DNA sequencing There are several monogenic disorders for which the mutations have been well characterised and the carrier incidence determined. For example, the cystic fibrosis gene is known to have more than 500 different mutations that affect its function.
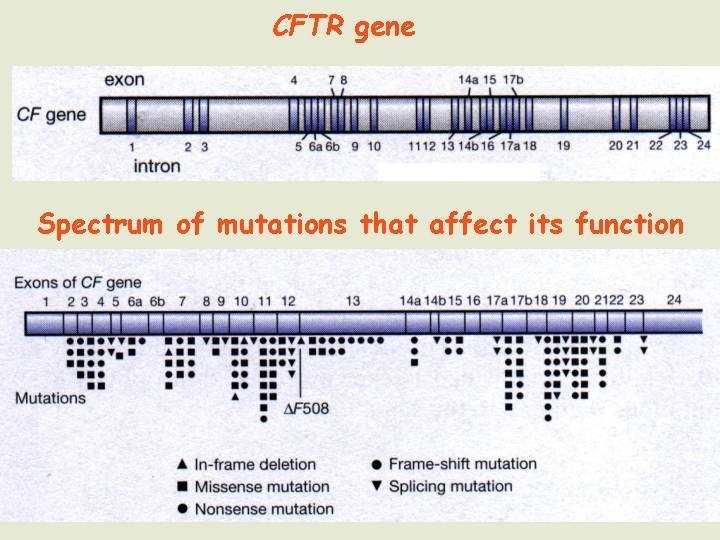
 The most common CFTR mutation is ∆F 508 (missing three nucleotides that encode the phenylalanine at position 508 in the protein's amino acid sequence). Oligonucleotide primers that flank the region are used to PCR amplify samples taken from each individual to determine if they carry this particular mutation. Two other mutations that affect specific restriction enzyme sites within the CFTR gene can be detected by a simple test using PCR followed by restriction digest analysis
The most common CFTR mutation is ∆F 508 (missing three nucleotides that encode the phenylalanine at position 508 in the protein's amino acid sequence). Oligonucleotide primers that flank the region are used to PCR amplify samples taken from each individual to determine if they carry this particular mutation. Two other mutations that affect specific restriction enzyme sites within the CFTR gene can be detected by a simple test using PCR followed by restriction digest analysis
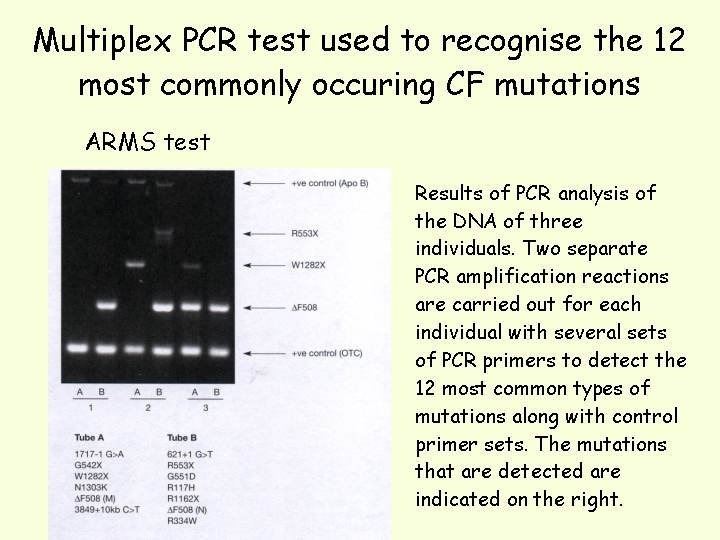
 A common method to detect the presence of more than one mutation is the amplification refractory mutation system (ARMS) which is a multiplex PCR technique, using allele Specific oligonucleotides (ASOs) to distinguish mutant and wild-type alleles. Two PCR reactions are carried out in parallel and the products run in adjacent lanes during electrophoresis. For each primer set, one primer is common to both reactions; the other primer is an ASO that anneals to the site of the mutation. In one reaction the ASO anneals to the wild-type allele and in the other, the ASO anneals to the mutant allele. Depending in which lane the band is detected will determine whether the mutant or wild-type allele is present. Multiple primer sets can be used to simultaneously screen for a number of mutations (just by making sure the length of product for each primer set is different so that each product can be resolved on a gel). It can be used to detect deletions and point mutations.
A common method to detect the presence of more than one mutation is the amplification refractory mutation system (ARMS) which is a multiplex PCR technique, using allele Specific oligonucleotides (ASOs) to distinguish mutant and wild-type alleles. Two PCR reactions are carried out in parallel and the products run in adjacent lanes during electrophoresis. For each primer set, one primer is common to both reactions; the other primer is an ASO that anneals to the site of the mutation. In one reaction the ASO anneals to the wild-type allele and in the other, the ASO anneals to the mutant allele. Depending in which lane the band is detected will determine whether the mutant or wild-type allele is present. Multiple primer sets can be used to simultaneously screen for a number of mutations (just by making sure the length of product for each primer set is different so that each product can be resolved on a gel). It can be used to detect deletions and point mutations.
 DNA microarrays - DNA chips- are a novel and still developing technology. These are the future of genetic diagnostic testing. They allow the identity of every nucleotide in a test DNA sequence to be examined in a single operation. Oligonucleotides are spotted on to specific positions on a glass chip or other solid matrix (array) and are hybridised with a fluorescently labeled test DNA. The sequence of each oligo is the same as the wild type except that a central base is systematically changed so that there is an oligo for every possible sequence variation. The test DNA or RNA preferentially hybridises to the oligo that matches its sequence. The chips are read by a computer which will also help analyse the data. DNA sequencing is the most direct way of determining whether a mutation is present
DNA microarrays - DNA chips- are a novel and still developing technology. These are the future of genetic diagnostic testing. They allow the identity of every nucleotide in a test DNA sequence to be examined in a single operation. Oligonucleotides are spotted on to specific positions on a glass chip or other solid matrix (array) and are hybridised with a fluorescently labeled test DNA. The sequence of each oligo is the same as the wild type except that a central base is systematically changed so that there is an oligo for every possible sequence variation. The test DNA or RNA preferentially hybridises to the oligo that matches its sequence. The chips are read by a computer which will also help analyse the data. DNA sequencing is the most direct way of determining whether a mutation is present
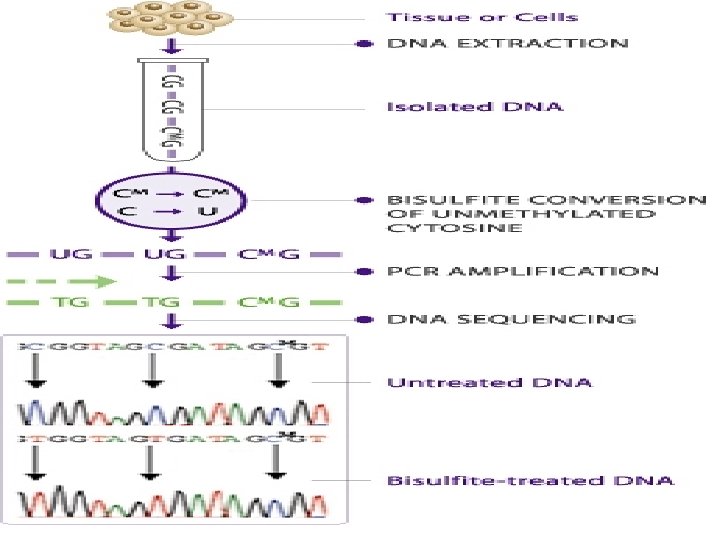
 Bisulfite sequencing is the use of bisulfite treatment of DNA to determine its pattern of methylation. Treatment of DNA with bisulfite converts cytosine residues to uracil but leaves 5 -methylcytosine residues unaffected. Thus, bisulfite treatment introduces specific changes in the DΝΑ . sequence that depend on the methylation status of individual cytosine residues, yielding single- nucleotide resolution information about the methylation status of a segment of DNA.
Bisulfite sequencing is the use of bisulfite treatment of DNA to determine its pattern of methylation. Treatment of DNA with bisulfite converts cytosine residues to uracil but leaves 5 -methylcytosine residues unaffected. Thus, bisulfite treatment introduces specific changes in the DΝΑ . sequence that depend on the methylation status of individual cytosine residues, yielding single- nucleotide resolution information about the methylation status of a segment of DNA.
 The gold standard in analyzing DNA methylation is by chemical treatment of DNA using bisulfite. Bisulfite treatment converts cytosine to uracil while 5 -methy cytosine is resistant to the conversion. After the treatment, genomic DNA is subject to PCR amplification, subcloning, and sequencing. The sequencing result is then compared to the original sequences, and any methylated/unmethylated cytosine is unambiguously determined. As a result, methylated Cs remains as Cs while unmethylated Cs become Ts (see figures below).
The gold standard in analyzing DNA methylation is by chemical treatment of DNA using bisulfite. Bisulfite treatment converts cytosine to uracil while 5 -methy cytosine is resistant to the conversion. After the treatment, genomic DNA is subject to PCR amplification, subcloning, and sequencing. The sequencing result is then compared to the original sequences, and any methylated/unmethylated cytosine is unambiguously determined. As a result, methylated Cs remains as Cs while unmethylated Cs become Ts (see figures below).

 Nucleotides in blue are unmethylated cytosines converted to uracils by bisulfite, while red nucleotides are 5 -methylcytosines resistant to conversion.
Nucleotides in blue are unmethylated cytosines converted to uracils by bisulfite, while red nucleotides are 5 -methylcytosines resistant to conversion.
 All strategies assume that bisulfite-induced conversion of unmethylated cytosines to uracil is complete, and this serves as the basis of all subsequent techniques. Ideally, the method used would determine the methylation status separately for each allele. The methodologies can be generally divided into strategies based on methylation-specific PCR (MSP) , and strategies employing polymerase chain reaction (PCR) performed under non-methylation-specific conditions. Non-methylation-specific PCR based methods The first reported method of methylation analysis using bisulfite-treated DNA utilized PCR and standard dideoxynucleotide DNA sequencing to directly determine the nucleotides resistant to bisulfite conversion. Primers are designed to be strand-specific as well as bisulfite-specific (i. e. , primers containing non-Cp. G cytosines such that they are not complementary to non-bisulfite-treated DNA), flanking (but not involving) the methylation site of interest. Therefore, it will amplify both methylated and unmethylated sequences, in contrast to methylation-specific PCR. All sites of unmethylated cytosines are displayed as thymines in the resulting amplified sequence of the sense strand, and as adenines in the amplified antisense strand. This technique required cloning of the PCR product prior to sequencing for adequate sensitivity, and therefore was a very labour-intensive method unsuitable for higher throughput. Alternatively, nested PCR methods can be used to enhance the product for sequencing.
All strategies assume that bisulfite-induced conversion of unmethylated cytosines to uracil is complete, and this serves as the basis of all subsequent techniques. Ideally, the method used would determine the methylation status separately for each allele. The methodologies can be generally divided into strategies based on methylation-specific PCR (MSP) , and strategies employing polymerase chain reaction (PCR) performed under non-methylation-specific conditions. Non-methylation-specific PCR based methods The first reported method of methylation analysis using bisulfite-treated DNA utilized PCR and standard dideoxynucleotide DNA sequencing to directly determine the nucleotides resistant to bisulfite conversion. Primers are designed to be strand-specific as well as bisulfite-specific (i. e. , primers containing non-Cp. G cytosines such that they are not complementary to non-bisulfite-treated DNA), flanking (but not involving) the methylation site of interest. Therefore, it will amplify both methylated and unmethylated sequences, in contrast to methylation-specific PCR. All sites of unmethylated cytosines are displayed as thymines in the resulting amplified sequence of the sense strand, and as adenines in the amplified antisense strand. This technique required cloning of the PCR product prior to sequencing for adequate sensitivity, and therefore was a very labour-intensive method unsuitable for higher throughput. Alternatively, nested PCR methods can be used to enhance the product for sequencing.
 DNA methylation analysis methods not based on methylation-specific PCR. Following bisulfite conversion, the genomic DNA is amplified with PCR that does not discriminate between methylated and non-methylated sequences. The numerous methods available are then used to make the discrimination based on the changes within the amplicon as a result of bisulfite conversion.
DNA methylation analysis methods not based on methylation-specific PCR. Following bisulfite conversion, the genomic DNA is amplified with PCR that does not discriminate between methylated and non-methylated sequences. The numerous methods available are then used to make the discrimination based on the changes within the amplicon as a result of bisulfite conversion.
 This alternative method of methylation analysis also uses bisulfitetreated DNA but") Methylation-specific PCR (MSP) This alternative method of methylation analysis also uses bisulfitetreated DNA but avoids the need to sequence the area of interest. Instead, primer pairs are designed themselves to be "methylated-specific" by including sequences complementing only unconverted 5 methylcytosines, or conversely "unmethylated-specific", complementing thymines converted from unmethylated cytosines. Methylation is determined by the ability of the specific primer to achieve amplification. This method is particularly useful to interrogate Cp. G islands with possibly high methylation density, as increased numbers of Cp. G pairs in the primer increase the specificity of the assay. Placing the Cp. G pair at the 3'-end of the primer also improves the sensitivity. The initial report using MSP described sufficient sensitivity to detect methylation of 0. 1% of alleles. In general, MSP and its related protocols are considered to be the most sensitive when interrogating the methylation status at a specific locus.
Methylation-specific PCR (MSP) This alternative method of methylation analysis also uses bisulfitetreated DNA but avoids the need to sequence the area of interest. Instead, primer pairs are designed themselves to be "methylated-specific" by including sequences complementing only unconverted 5 methylcytosines, or conversely "unmethylated-specific", complementing thymines converted from unmethylated cytosines. Methylation is determined by the ability of the specific primer to achieve amplification. This method is particularly useful to interrogate Cp. G islands with possibly high methylation density, as increased numbers of Cp. G pairs in the primer increase the specificity of the assay. Placing the Cp. G pair at the 3'-end of the primer also improves the sensitivity. The initial report using MSP described sufficient sensitivity to detect methylation of 0. 1% of alleles. In general, MSP and its related protocols are considered to be the most sensitive when interrogating the methylation status at a specific locus.
 Methylation-specific PCR is a sensitive method to discriminately amplify and detect a methylated region of interest using methylated-specific primers on bisulfite-converted genomic DNA. Such primers will only anneal to sequences that are methylated, and thus containing 5 -methylcytosines that are resistant to conversion by bisulfite. Alternatively, unmethylated-specific primers can be used.
Methylation-specific PCR is a sensitive method to discriminately amplify and detect a methylated region of interest using methylated-specific primers on bisulfite-converted genomic DNA. Such primers will only anneal to sequences that are methylated, and thus containing 5 -methylcytosines that are resistant to conversion by bisulfite. Alternatively, unmethylated-specific primers can be used.
 The Methy. Light method is based on MSP, but provides a quantitative analysis using real-time PCR. Methylated-specific primers are used, and a methylated-specific fluorescence reporter probe is also used that anneals to the amplified region. Alternatively, the primers or probe can be designed without methylation specificity if discrimination is needed between the Cp. G pairs within the involved sequences. Quantitation is made in reference to a methylated reference DNA. A modification to this protocol to increase the specificity of the PCR for successfully bisulfite-converted DNA (Con. Light-MSP) uses an additional probe to bisulfite-unconverted DNA to quantify this non-specific amplification. Further methodology using MSP-amplified DNA analyzes the products using (Mc-MSP). This method amplifies bisulfite-converted DNA with both methylated-specific and unmethylated-specific primers, and determines the quantitative ratio of the two products by comparing the differential peaks generated in a melting-curve analysis. A high resolution melting analysis method which using both real time quantification and melting analysis has been introduced particularly for sensitive detection of low level methylation.
The Methy. Light method is based on MSP, but provides a quantitative analysis using real-time PCR. Methylated-specific primers are used, and a methylated-specific fluorescence reporter probe is also used that anneals to the amplified region. Alternatively, the primers or probe can be designed without methylation specificity if discrimination is needed between the Cp. G pairs within the involved sequences. Quantitation is made in reference to a methylated reference DNA. A modification to this protocol to increase the specificity of the PCR for successfully bisulfite-converted DNA (Con. Light-MSP) uses an additional probe to bisulfite-unconverted DNA to quantify this non-specific amplification. Further methodology using MSP-amplified DNA analyzes the products using (Mc-MSP). This method amplifies bisulfite-converted DNA with both methylated-specific and unmethylated-specific primers, and determines the quantitative ratio of the two products by comparing the differential peaks generated in a melting-curve analysis. A high resolution melting analysis method which using both real time quantification and melting analysis has been introduced particularly for sensitive detection of low level methylation.
 Taq. Man probes used in conjunction with methylation-specific primers in semiquantitative, real-time PCR Taq. Man probes are used in between methylation specific primer sites. Bisulfite converted DNA of unmethylated Cp. G sites require a different primer sequence to that of methylated Cp. G sites. Therefore, two different primer pairs are used, one specific for methylated and converted DNA, the other primer pair specific for unmethylated and converted DNA. If the methylation specific primer binds to the DNA, it will be elongated during the extension phase and the 5'→ 3' exonuclease activity of Taq DNA Polymerase will lead to the degradation of the primer and the release of the fluorophore. As the fluorophore is now separated from the quencher moiety, a fluorescence is detectable.
Taq. Man probes used in conjunction with methylation-specific primers in semiquantitative, real-time PCR Taq. Man probes are used in between methylation specific primer sites. Bisulfite converted DNA of unmethylated Cp. G sites require a different primer sequence to that of methylated Cp. G sites. Therefore, two different primer pairs are used, one specific for methylated and converted DNA, the other primer pair specific for unmethylated and converted DNA. If the methylation specific primer binds to the DNA, it will be elongated during the extension phase and the 5'→ 3' exonuclease activity of Taq DNA Polymerase will lead to the degradation of the primer and the release of the fluorophore. As the fluorophore is now separated from the quencher moiety, a fluorescence is detectable.
 Methylation-specific Taq. Man probes in quantitative Methy. Light real-time PCR Dual-labeled probes, including Taq. Man probes, are sequence-specific oligonucleotides with a fluorophore and a quencher moiety attached. The fluorophore is at the 5' end of the probe, and the quencher moiety is usually located at the 3' end or internally. During the extension phase of PCR, the probe is cleaved by the 5'→ 3' exonuclease activity of Taq DNA polymerase, separating the fluorophore and the quencher moiety. This results in detectable fluorescence that is proportional to the amount of accumulated PCR product.
Methylation-specific Taq. Man probes in quantitative Methy. Light real-time PCR Dual-labeled probes, including Taq. Man probes, are sequence-specific oligonucleotides with a fluorophore and a quencher moiety attached. The fluorophore is at the 5' end of the probe, and the quencher moiety is usually located at the 3' end or internally. During the extension phase of PCR, the probe is cleaved by the 5'→ 3' exonuclease activity of Taq DNA polymerase, separating the fluorophore and the quencher moiety. This results in detectable fluorescence that is proportional to the amount of accumulated PCR product.
 METHYL-SENSITIVE RESTRICTION ENZYMES The ability of methyl-sensitive restriction enzymes possessing the Cp. G dinucleotidecontaining recognition site to cut only unmethylated sites is used for various approaches. Parallel hydrolysis of DNA with such a restriction enzyme and its methyl-insensitive isoschizomer can present information about the number and distribution of the corresponding sites in the specimens containing 5 -methylcytosine. Most often, the pair Msp. I/Hpa. II is used which recognizes the CCGG sequence, with Hpa. II sensitive to methylation of the second C in the site. At present, more than 300 methyl-sensitive restriction endonucleases are known, and ~30 of them have methyl-insensitive isoschizomers. PCR-amplification of the genomic region under study becomes more sensitive after pretreatment of the sample with endonuclease. Only the initially methylated and thus non-hydrolyzed fragment of DNA will be amplified exponentially.
METHYL-SENSITIVE RESTRICTION ENZYMES The ability of methyl-sensitive restriction enzymes possessing the Cp. G dinucleotidecontaining recognition site to cut only unmethylated sites is used for various approaches. Parallel hydrolysis of DNA with such a restriction enzyme and its methyl-insensitive isoschizomer can present information about the number and distribution of the corresponding sites in the specimens containing 5 -methylcytosine. Most often, the pair Msp. I/Hpa. II is used which recognizes the CCGG sequence, with Hpa. II sensitive to methylation of the second C in the site. At present, more than 300 methyl-sensitive restriction endonucleases are known, and ~30 of them have methyl-insensitive isoschizomers. PCR-amplification of the genomic region under study becomes more sensitive after pretreatment of the sample with endonuclease. Only the initially methylated and thus non-hydrolyzed fragment of DNA will be amplified exponentially.
![]. Use of methyl-sensitive restriction endonucleases for analyzing methylation of specific Cp. G sites.](https://present5.com/presentation/d9a3fa29785182389d0a963f92e3969f/image-57.jpg "]. Use of methyl-sensitive restriction endonucleases for analyzing methylation of specific Cp. G sites.") ]. Use of methyl-sensitive restriction endonucleases for analyzing methylation of specific Cp. G sites. Genomic DNA containing unmethylated (white asterisks) and methylated (black asterisks) Cp. G sites is restricted analyzed by PCR with primers (shown by arrows) flanking the site under study. Nevertheless, it is obvious that these approaches require that the genomic region under study be presequenced. It is also essential that by these approaches methylation is studied not of the whole sequence but only of its regions that contain the recognition site of the endonuclease used.
]. Use of methyl-sensitive restriction endonucleases for analyzing methylation of specific Cp. G sites. Genomic DNA containing unmethylated (white asterisks) and methylated (black asterisks) Cp. G sites is restricted analyzed by PCR with primers (shown by arrows) flanking the site under study. Nevertheless, it is obvious that these approaches require that the genomic region under study be presequenced. It is also essential that by these approaches methylation is studied not of the whole sequence but only of its regions that contain the recognition site of the endonuclease used.
 Limitations Incomplete conversion Bisulphite sequencing relies on the conversion of every single unmethylated cytosine residue to uracil. If conversion is incomplete, the subsequent analysis will incorrectly interpret the unconverted unmethylated cytosines as methylated cytosines, resulting in false positive results for methylation. Only cytosines in single-stranded DNA are susceptible to attack by bisulphite, therefore denaturation of the DNA undergoing analysis is critical. It is important to ensure that reaction parameters such as temperature and salt concentration are suitable to maintain the DNA in a single-stranded conformation and allow for complete conversion.
Limitations Incomplete conversion Bisulphite sequencing relies on the conversion of every single unmethylated cytosine residue to uracil. If conversion is incomplete, the subsequent analysis will incorrectly interpret the unconverted unmethylated cytosines as methylated cytosines, resulting in false positive results for methylation. Only cytosines in single-stranded DNA are susceptible to attack by bisulphite, therefore denaturation of the DNA undergoing analysis is critical. It is important to ensure that reaction parameters such as temperature and salt concentration are suitable to maintain the DNA in a single-stranded conformation and allow for complete conversion.
 Degradation of DNA during bisulphite treatment A major challenge in bisulphite sequencing is the degradation of DNA that takes place concurrently with the conversion. The conditions necessary for complete conversion, such as long incubation times, elevated temperature, and high bisulphite concentration, can lead to the degradation of about 90% of the incubated DNA. Given that the starting amount of DNA is often limited, such extensive degradation can be problematic. The degradation occurs as depurinations resulting in random strand breaks. Therefore the longer the desired PCR amplicon , the more limited the number of intact template molecules will likely be. This could lead to the failure of the PCR amplification, or the loss of quantitatively accurate information on methylation levels resulting from the limited sampling of template molecules. It is thus important to assess the amount of DNA degradation resulting from the reaction conditions employed, and consider how this will affect the desired amplicon. A potentially significant problem following bisulphite treatment is incomplete desulfonation of pyrimidine residues due to inadequate alkalization of the solution. This may inhibit some DNA polymerases, rendering subsequent PCR difficult. However this situation can be avoided by monitoring the p. H of the solution to ensure that desulphonation will be complete
Degradation of DNA during bisulphite treatment A major challenge in bisulphite sequencing is the degradation of DNA that takes place concurrently with the conversion. The conditions necessary for complete conversion, such as long incubation times, elevated temperature, and high bisulphite concentration, can lead to the degradation of about 90% of the incubated DNA. Given that the starting amount of DNA is often limited, such extensive degradation can be problematic. The degradation occurs as depurinations resulting in random strand breaks. Therefore the longer the desired PCR amplicon , the more limited the number of intact template molecules will likely be. This could lead to the failure of the PCR amplification, or the loss of quantitatively accurate information on methylation levels resulting from the limited sampling of template molecules. It is thus important to assess the amount of DNA degradation resulting from the reaction conditions employed, and consider how this will affect the desired amplicon. A potentially significant problem following bisulphite treatment is incomplete desulfonation of pyrimidine residues due to inadequate alkalization of the solution. This may inhibit some DNA polymerases, rendering subsequent PCR difficult. However this situation can be avoided by monitoring the p. H of the solution to ensure that desulphonation will be complete