30 - 2014 Синдром Альпорта.ppt
- Количество слайдов: 59



СИНДРОМ АЛЬПОРТА - одна из болезней коллагена IV типа С. Боровой, 2014

Синдром Альпорта – гетерогенная группа наследственных неиммунных гломерулопатий, связанная с изменением коллагена базальных мембран, проявляющаяся гематурией и протеинурией, прогрессирующим снижением функции почек, нередко сочетающаяся с патологией слуха и зрения.

Arthur Cecil Alport – английский врач, уроженец Южно. Африканской республики - родился в Beaufort West, Karoo region, Western Cape province, умер в Лондоне. 25. 01. 1880 – 17. 04. 1959 В 1927 г. описал три поколения в семье, у которой прогрессирующий наследст-вен -ный нефрит сочетался с тугоухостью. Кроме того заметил, что самым частым симптомом была гематурия, а мужчины поражались более тяжело, чем женщины. Впоследствии много подобных семейств было описано, а в 1961 г. болезнь назвали синдромом Альпорта.

Исторические аспекты наследственного нефрита 1875 – W. Disckinson – сообщение о семейной гематурии W. H. Dickinson. Diseases of the kidney and urinary derangements. London, Langmans, 1875; part 2: 278. 1902 – L. Cuthrie – первое описание семьи со случаями гематурии в нескольких поколениях L. B. Guthrie. ”Idiopathic, ” or congenital, hereditary and familial haematuria. Lancet, London, 1902, 1: 1243 -1246. 1923 – A. Hurst обнаружил развитие уремии у некоторых членов этой семьи A. F. Hurst. Hereditary familial congenital haemorrhagic nephritis occurring in sixteen individuals in three generations. Guy’s Hosp Rec, 1923, 3: 368 -370. 1927 – А. Аlport описал эту же семью и отметил, что у некоторых родственников имелась тугоухость, а уремия у мужчин развивалась раньше, чем у женщин. A. C. Alport. Hereditary familial congenital haemorrhagic nephritis. British Medical Journal, London, 1927, I: 504 -506.

1985 – L. Menlove et al. выявили ген, ответственный за синдром Альпорта. Он был обнаружен на длинном плече Х-хромосомы в зоне 21– 22 q.
")
Генеалогическое дерево семьи, изученной Альпортом и далее Crawfurd and Toghill (1968)

Эпидемилогия синдрома Альпорта Распространенность в России 17 : 100 000 [Вельтищев Ю. В. , Игнатова М. С. 1996] Встречается у лиц в разных регионах мира, независимо от расы и национальной принадлежности. Встречается чаще, чем выявляется. 1: 5000 – 1: 10000 (США, 1973 -2004)

Генетика синдрома Альпорта Генетическая основа наследственного нефрита мутация гена COL 4 A 5, кодирующего альфа-5 -цепь коллагена IV типа. Продукт гена - цепи коллагена IV типа. COL 4 A 3 и COL 4 A 4 находятся на 2 -й хромосоме. Их мутация характерна для аутосомно-рецессивного типа передачи наследственного нефрита. Аутосомно-рецессивный тип передачи выявляется у 6% больных выявляется, а аутосомно-доминантный у 16% больных [Игнатова М. С. , 2000]. Доминантный сцепленный с полом тип наследования характерен для классического с-ма Альпорта (наследственный нефрит с тугоухостью и ранним развитием почечной недостаточности).

Женщины, страдающие синдромом Альпорта, в отличие от мужчин редко входят в терминальную ХПН Синдром Aльпорта вызывается мутацией X хромосомы. У женщин две X хромосомы, у мужчин одна X, а другая Y. Поэтому у женщин Х хромосома, полученная от здоровой матери, «защищает» от большинства эффектов «плохой» хромосомы, полученной от отца. У мужчин же только одна Х хромосома, поэтому если она «больна» , то в их теле нет никакой защиты от ее неприятностей. Дочь отца, страдающего синдромом Альпорта, всегда наследует от него «больную» Х хромосому. В то же время сын больного отца никогда не будет страдать синдромом, поскольку от матери он получит «здоровую» Х хромосому, а от отца – «здоровую» Y хромосому. женщина мать дочь мужчина отец сын

Какие генетические варианты синдрома Альпорта обсуждаются? Коллаген IV типа составляет семья из 4 -х белков (точнее – цепей), которые обозначаются как α-1, α-2, α-3, α-4, α-5 и α-6. Мутации, которые повреждают α-3, α-4 или α-5 типы цепей коллагена IV, могут вызывать синдром Альпорта. Имеется три генетических типа синдрома Альпорта: X-связанный – самая частая форма синдрома Альпорта – её имеют около 80% больных. При этом варианте мальчики всегда тяжелобольны и всегда входят в терминальную ХПН. Девочки с таким вариантом обычно имеют более легкие проявления болезни, но у них может развиваться почечная недостаточность. У остальных больных с синдромом Альпорта регистрируется либо аутосомно-рецессивный тип (15%), либо аутосомно -доминантный (5%).

ОРГАНЫ-МИШЕНИ ПРИ НАСЛЕДСТВЕННОМ НЕФРИТЕ При наследственном нефрите страдают: ç ç ç всегда – почки, а именно базальные мембраны клубочковых капилляров (микрогематурия, протеинурия, ХПН); во многих случаях: ç внутреннее ухо – базальные мембраны кортиева органа (снижение слуха вплоть до полной его потери); ç глаза (аномалия хрусталика – лентиконус, катаракта) базальные мембраны капсулы хрусталика.

Симптомы поражения почек при синдроме Альпорта n n Основной признак – гематурия, обычно микроскопическая. У мальчиков с Х-переносимым синдромом Альпорта она всегда регистрируется уже в периоде новорожденности. У большинства девочек тоже наблюдается гематурия, но она может быть непостоянной. Изредка у детей с синдромом Альпорта могут быть эпизоды – до нескольких дней – макрогематурии (как при болезни Берже). По мере взросления (чаще всего на втором десятилетии) у мальчиков появляются и другие симптомы – протеинурия и артериальная гипертензия. У большинства девочек с синдромом Альпорта протеинурия не регистрируется, а артериальное давление повышается уже в зрелые годы, но изредка бывают и исключения – все симптомы развиваются на 2 -3 десятилетии жизни.

Симптомы поражения кортиева органа при синдрома Альпорта n n n Не менее чем у 80% мальчиков с синдромом Альпорта в тот или иной период жизни развивается тугоухость, чаще на втором десятилетии (в возрасте 8 – 15 лет, иногда бывает первым симптомом болезни). Диагностика: аудиография (снижение слуха на высоких частотах по звуковоспринимающему сигналу) Глухота поражает оба уха! Fortunately, hearing aids are usually very effective in these people. Некоторые девочки с синдромом Альпорта также теряют слух, но в более поздние периоды жизни. Трансплантация почек не исправляет глухоту у больных синдромом Альпорта.

Симптомы поражения органа зрения при синдроме Альпорта Около 15% мужчин с синдромом Альпорта имеют аномалии the shape хрусталика, называемые лентиконусом. n n n Больные с передним лентиконусом могут иметь различные проблемы зрения, кроме того, у них может развиться катаракта. Нередко наблюдаются и другие изменения – астигматизм, сферофакия, миопия и др. The perifoveal dot and fleck retinopathy in a patient with X linked Alport syndrome (NH).

Хроническая почечная недостаточность – закономерный исход синдрома Альпорта p p p Не только клубочки, но и все остальные структуры почки замещаются соединительной тканью, что приводит к почечной недостаточности. Все мальчики с синдромом Альпорта вне зависимости от генетического типа постепенно развивают почечную недостаточность. Нередко уже на втором-третьем десятилетиях жизни они уже нуждаются в заместительной почечной терапии. В любом случае, терминальная ХПН возникает не позже 40 -50 -летнего возраста. У большинства девочек с X-связанным синдромом Альпорта снижения функции почек не регистрируют, но мере старения вероятность снижения функции у них почек увеличивается. Все мальчики и девочки с аутосомно-рецессивным синдромом Альпорта входят в почечную недостаточность около совершеннолетия. Больные с аутосомно-доминантным синдромом Альпорта входят в почечную недостаточность обычно в средние годы.

Клиническая картина синдрома Альпорта Ø Ø У женщин с наследственным нефритом часты спонтанные аборты, мертворождения, нефропатия беременных; Первые признаки болезни могут быть выявлены в любом возрасте: Ú гематурия (при случайном обследовании, м. б. изолированной); Ú протеинурия (ее степень определяет прогноз болезни, при большой протеинурии, а также развитии нефротического синдрома – прогноз неблагоприятный); Ú абактериальная лейкоцитурия (проявление тубулоинтерстициальных изменений). Стигмы дизэмбриогенеза (характерна однотипность стигм у больных членов семьи: аномалия развития почек, аномалии хрусталика глаза и др. ) Симптомы интоксикации (бледность, вялость, мышечная гипотония, анорексия без явных причин)

КЛИНИЧЕСКАЯ КАРТИНА СИНДРОМА АЛЬПОРТА Ø Головные боли, связанные с гипотензией (появление гипертензии при развитии ХПН) Ø Аномалия зрения ( 1/4 больных СА) Интеллект соответствует возрастным нормам Раннее развитие ХПН Ø Ø Ø Лабораторные данные: Ú азотемия; дизэлектролитемия; Ú анемия; метаболический ацидоз.
 и одной α 2(IV)")
СТРУКТУРА КОЛЛАГЕНА IV ТИПА Молекула состоит из двух α 1(IV) и одной α 2(IV) цепей. Они сшиты неколлагеновыми участками и оканчиваются тремя NC 1 доменами. Сигнальные пептиды и короткие N-терминальные неколлагеновые домены (7 S) указаны стрелкой. При полимеризации моллекулы коллагена IV типа сшиваются дисульфидными мостиками на обоих концах молекулы.
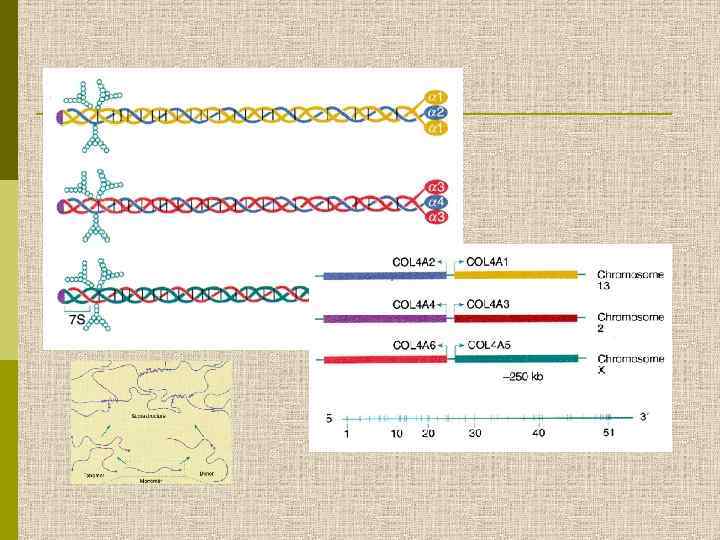

Цепи коллагена IV типа chrom chrom X

Геномная организация генов шести типов коллагена IV типа на хромосомах 13, 2 и Х

Состав коллагена IV типа у новорожденных и у взрослых

Молекула мономера коллагена IV типа Полимерная сеть димер мономер тетрамер
")
Полимерная сеть коллагена IV типа (Yourchenco and Schittney, 1990)
")
Состав коллагена в различных базальных мембранах Lamina densa Подоциты Эндотелий Lamina densa 3 (IV) 4 (IV) 5 (IV) Эпидермис 1 (IV), 2 (IV) 5 (IV), 6 (IV) Субэндотелий и мезангий 1 (IV) 2 (IV) коллаген V типа коллаген VI типа Дистальный каналец, капсула Боумена 1 (IV), 2 (IV) 3 (IV), 4 (IV) 5 (IV), 6 (IV)
 зрелый (с-м Альпорта) коллаген")
Динамика коллагена и изоформ ламинина в процессе развития ранний (норма) зрелый (с-м Альпорта) коллаген 1, 2 (IV) 3 -5 (IV) 1, 2 (IV) ламинин 1 2 2
 Wild-type ГБМ мышей без 3 (IV) Модель синдрома Альпорта J. H. Miner and")
a) Wild-type ГБМ мышей без 3 (IV) Модель синдрома Альпорта J. H. Miner and J. R. Sanes (1996)

Синдром Альпорта. Неспецифические гломерулярные изменения, атрофичные канальцы, лимфоцитарная инфильтрация интерстиция, пенистые клетки

1 2 5 6 НЕСПЕЦИФИЧЕСКИЕ ИЗМЕНЕНИЯ ПРИ СИНДРОМЕ АЛЬПОРТА 3 4 1. Нормальный клубочек 2. Мезангиальная пролиферация, утолщение капиллярной стенки 3. Пенистые клетки в интерстиции 4. Сегментарный фибриноидный некроз 5. Сегментарный склероз 6. Эпителиальная пролиферация

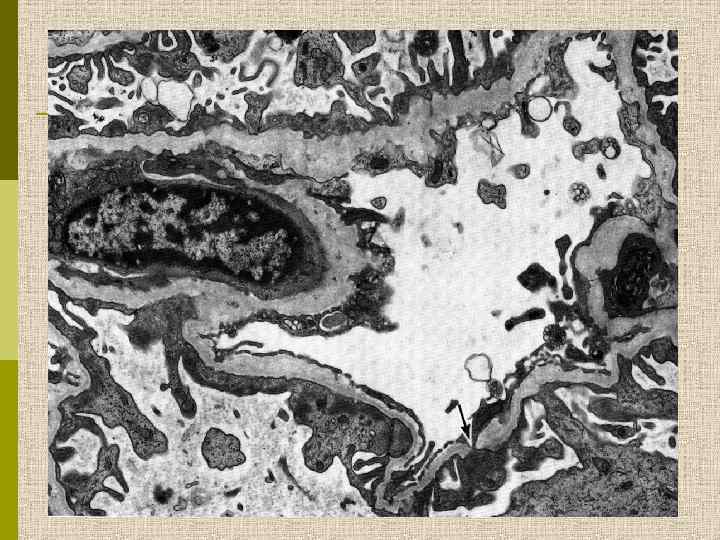
Ультраструктурные изменения ГБМ при синдроме Альпорта. ГБМ утолщена, слоиста, контур ее неровный. ( 9000. )


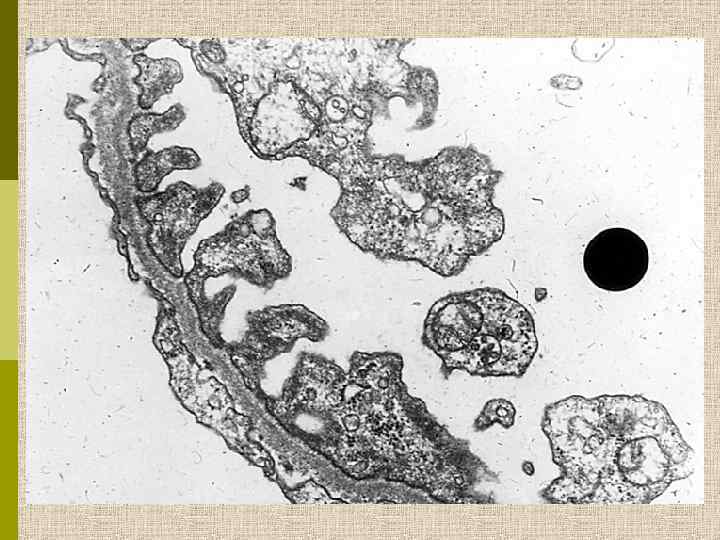
Непрямая иммунопероксидазная реакция при синдроме Альпорта Фиксация моноклональных анти-ГБМ антител МСА-Р 1 вдоль ГБМ, выявляемых кроличьей антисывороткой против мышиного иммуноглобулина, меченного пероксидазой. А. Здоровая почка Б. Сегментарное связывание МСА-Р 1 у больной с синдромом Альпорта В. Отсутствие связывания МСА-Р 1 у мужчины с синдромом Альпорта L. H. Noël, Hôpital Necker, Paris

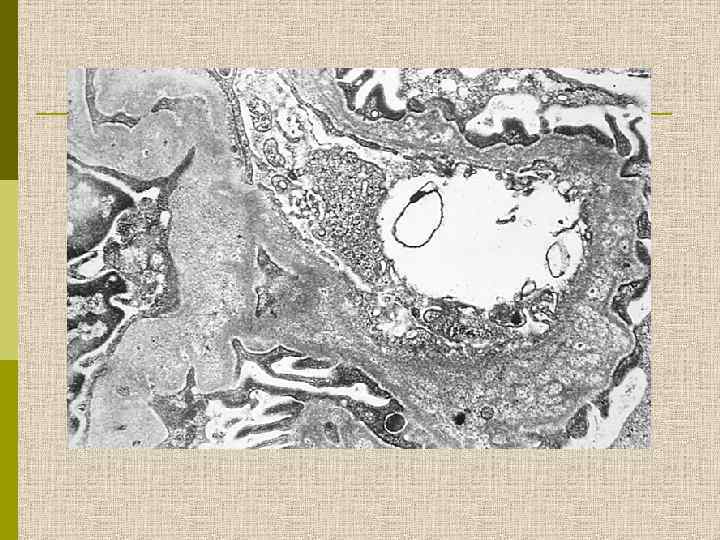
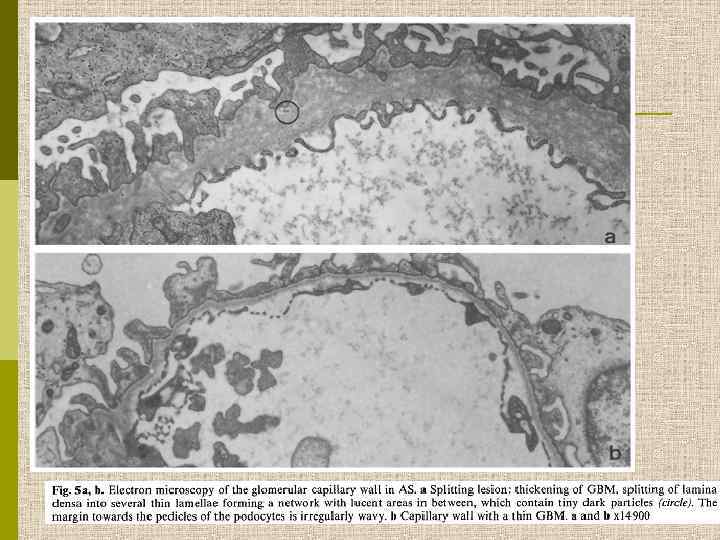
Электронограмма гломерулы больного с синдромом Альпорта. Утолщение ГБМ, splitting lamina densa и неровность субэпителиальной поверхности ГБМ. 7000

Диффузный лейомиоматоз пищевода. Pierre Cochat, Hôpital Edouard Herriot, Lyon, France

ВРОЖДЕННЫЕ БОЛЕЗНИ, ПРИ КОТОРЫХ НАБЛЮДАЕТСЯ СЕНСОРНО-НЕВРАЛЬНАЯ ТУГОУХОСТЬ В СОЧЕТАНИИ С ПОЧЕЧНЫМ ПОРАЖЕНИЕМ Синдром Альпорта и его варианты Синдром Muckle-Wells Болезнь Refsum Cockayne синдром (Sato et al. 1988) Почечный канальцевый ацидоз Ихтиоз, гиперпролинурия Синдром Charcot-Marie-Tooth Синдром Alstrцm Атаксия, гиперурикемия Псевдомиоклонус, сахарный диабет Семейная спастическая параплегия, снижение интеллекта (Fitzsimmons et al. 1988) Различные синдромы, включая почечные, генитальные и/или пальцевые нарушения Различные malformative синдромы, включая мочевой тракт и глаза, такие как брахио-ото-ренальная дисплазия (BOR синдром) (Fraser et al. 1980; Gimsing and Dyrmose 1986) и множественные lentigines синдромы Митохондриальные цитопатии Гипо/гиперпаратиреоидизм Адаптировано с работы Konigsmark and Gorlin (1976)

ФЕНОТИПЫ СИНДРОМА АЛЬПОРТА Тип Способ передачи Средний возраст Непочечные симптомы при т. ХПН у мужчин I недифференцированный доминантный < 31 потеря слуха ± глазные симптомы II доминантный (X-хромосома) < 31 потеря слуха ± глазные симптомы III доминантный (X-хромосома) аутосомнодоминантный > 31 потеря слуха IV V VI аутосомнодоминантный > 31 ? < 31 макротромбоцитопения ± включения в гранулоцитах потеря слуха ± глазные симптомы
 цепь Хромосомная локализация")
ХАРАКТЕРИСТИКА РАЗЛИЧНЫХ СОСТОЯНИЙ, ОПИСЫВАЕМЫХ КАК СИНДРОМ АЛЬПОРТА Клиническая картина Дефектная α(IV) цепь Хромосомная локализация COL 4 A 5 Х-сцепленная болезнь a-5 с сенсорно-невральной тугоухостью или без нее Мутантный ген Xq 22 X-сцепленная болезнь a-5 и a-6 Xq 22 с диффузным лейомиоматозом COL 4 A 5 и Аутосомно-рецессивная q 37 болезнь q 37 a-3 или a-4 Аутосомно-доминантная болезнь a-3 или a-4? COL 4 A 6 COL 4 A 3 2 q 35 - COL 4 A 4 2 q 35 - COL 4 A 3/ COL 4 A 4?
 и α 6(IV) гены находятся")
КОРРЕЛЯЦИЯ МЕЖДУ ДЕЛЕЦИЯМИ И ФЕНОТИПАМИ СИНДРОМА АЛЬПОРТА α 5(IV) и α 6(IV) гены находятся на X хромосоме и разделены зоной в 1, 4 kb. Стрелки указывают направление транскрипции. Мутации α 5(IV) вызывают только синдром Альпортв. Мутации обоих генов - α 5(IV) и α 6(IV) – приводят к синдрому Альпорта, сочетаемым с диффузным

РАСПРЕДЕЛЕНИЕ α-ЦЕПЕЙ КОЛЛАГЕНА IV ТИПА У ЗДОРОВЫХ И У БОЛЬНЫХ С Х-СЦЕПЛЕННЫМ ИЛИ АУТОСОМНО-РЕЦЕССИВНЫМ СИНДРОМОМ АЛЬПОРТА a 1 a 2 a 3 a 4 a 5 a 6 ГБМ + Капсула клубочка + Проксимальный каналец – Петля Генле + Дистальный каналец + Собирательные трубки + Сосуды – Кожа + Передняя капсула хруст. ? + + +/– – + + – – + + + – +/– – – + + + + – – – + + – – + – ГБМ + – – +
 a-3")
ГЕНЕТИЧЕСКИЕ ВАРИАНТЫ СИНДРОМА АЛЬПОРТА Х-сцепленный Аутосомно- Аутосомнодоминантный Мутированый ген Неизвестный рецессивный a-5 (IV) a-3 (IV) or a-4 (IV) Тип мутации Гемизиготный Предположительно (one copy mutated) гемизиготный Фенотип Раннее начало у мужчин Клинические лентиконус, Гомозиготный (one copy mutated) Раннее начало у мужчин и женщин Глухота, лентиконус, Глухота,
 контроль 1 (IV) синдром Альпорта")
1 (IV) контроль 1 (IV) синдром Альпорта
 контроль 3(IV) синдром Альпорта")
3 (IV) контроль 3(IV) синдром Альпорта
 контроль 5(IV) синдром Альпорта")
5 (IV) контроль 5(IV) синдром Альпорта

Диагностика синдрома Альпорта Клинические признаки Семейный анамнез Биопсионные данные: - электронномикроскопический анализ почечного биоптата - в почке – наличие или отсутствие α-3, α-4 и α-5 цепей коллагена IV типа - альтернативный вариант – биопсия кожи α-5 цепь коллагена IV типа в норме в коже присутствует. У большинства мужчин с X-связанной формой синдрома Альпорта в коже она отсутствует полностью.
![ДИАГНОСТИКА СИНДРОМА АЛЬПОРТА Критерии диагностики: [Kashtan C. et al. , 1993] гематурия или летальный](https://present5.com/presentation/26844889_437029672/image-47.jpg "ДИАГНОСТИКА СИНДРОМА АЛЬПОРТА Критерии диагностики: [Kashtan C. et al. , 1993] гематурия или летальный")
ДИАГНОСТИКА СИНДРОМА АЛЬПОРТА Критерии диагностики: [Kashtan C. et al. , 1993] гематурия или летальный исход от ХПН в семье; гематурия и/или протеинурия в семье; характерные изменения гломерулярной базальной мембраны, обнаруживаемые при электронной микроскопии; снижение слуха по данным аудиографии; врожденная патология зрения. Для установления диагноза необходимо наличие 3 из 5 критериев Для подтверждения диагноза: электронная микроскопия биоптата и ДНК-исследование (идентификация Х-хромосомы у родственников).
 гематурией 1. Ig. A-нефропатия (болезнь Берже) - спорадическая,")
Гломерулонефропатии, проявляющиеся изолированной ( «доброкачественной» ) гематурией 1. Ig. A-нефропатия (болезнь Берже) - спорадическая, с возможным прогрессированием к т. ХБП 2. Болезнь тонкой базальной мембраны - аутосомно-доминантная, без развития т. ХБП ( «доброкачественная семейная гематурия» ) 3. Синдром Альпорта (диффузное истончение/утолщение, расщепление гломерулярной базальной мембраны) - Х-связанная, с развитием т. ХБП

ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА СИНДРОМА АЛЬПОРТА Ø С гематурической формой гломерулонефрита: Ú Ú Ú Ø С метаболической нефропатией (оксалатно-кальциевой кристаллурией: Ú Ú Ú Ø При ГН нет семейных случаев одинаковых почечных заболеваний, тугоухости, ХПН; При морфологическом исследовании выявляется иммунный вариант ГН; При электронной микроскопии (ЭМ) отсутствуют специфические признаки с-ма Альпорта. В анамнезе указания на наличие МКБ; При морфологическом исследовании выявляется тубулоинтерстициальный нефрит и обнаруживаются кристаллы оксалатов в канальцах; При ЭМ нет специфических изменений. С болезнью тонких мембран: Ú Ú при ЭМ - равномерное истончение мембран гломерул; характерен благоприятный прогноз.
 присутствует a 5(IV) отсутствует STOP биопсия")
подозрение на синдром Альпорта биопсия кожи a 5(IV) присутствует a 5(IV) отсутствует STOP биопсия почки (с анализом коллагена IV типа) с-м Альпорта ? генетический анализ идентификация носителя пренатальный диагноз – + прогноз терапия STOP
 контроль 1 (IV) синдром Альпорта")
1 (IV) контроль 1 (IV) синдром Альпорта
 контроль 3 (IV) синдром Альпорта")
3 (IV) контроль 3 (IV) синдром Альпорта
 контроль 5 (IV) синдром Альпорта")
5 (IV) контроль 5 (IV) синдром Альпорта

ЛЕЧЕНИЕ БОЛЬНЫХ С СИНДРОМОМ АЛЬПОРТА p Сегодня специфического лечения больных с синдромом Альпорта нет p Лечение симптоматическое – такое же, как у больных с артериальной гипертензией или хроническими болезнями почек p Трансплантация почки является оптимальным способом терапии в стадии терминальной почечной недостаточности p Ведутся интенсивные исследования в части распознавания путей развития ХПН при синдроме Альпорта и поиску методов, которые бы замедлили предотвратили развитие т. ХПН. Некоторые исследования на экспериментальных животных являются обнадеживающими.

ЛЕЧЕНИЕ БОЛЬНЫХ С СИНДРОМОМ АЛЬПОРТА Специфического лечения не существует. Цель: замедлить прогрессирование снижения функции почек. Ø Исключение занятий спортом; Ø Сбалансированная диета; Ø Избегать контактов с инфекционными больными; Ø Вакцинация только по эпид. показаниям; Ø Санация очагов инфекции; Ø Витаминотерапия (vit гр. В), анаболические стероиды (ретаболил); Ø Гипербарическая оксигенация. Радикальный метод лечения – трансплантация почки

Acquired GBM Disease An interesting scenario of "acquired" anti-GBM disease can occur when a patient with Alport's Syndrome gets a renal transplant. Patients with Alport's Syndrome--a cause of hematuria with progressive renal failure and hearing loss--have mutations in genes encoding certain subunits of the collagen IV molecule which comprises a major structural component of the glomerular basement membrane. When they undergo a renal transplant, their immune system may well recognize the donor collagen IV as "foreign" since it has never been exposed to this antigen previously, and this can lead to the formation of circulating antiglomerular basement membrane antibodies and subsequent allograft dysfunction.
 1 : 422 -427")
H-J. Rumpelt. Pediatr Nephrol (1987) 1 : 422 -427
30 - 2014 Синдром Альпорта.ppt