Obschee.ppt
- Количество слайдов: 177



СИБИРСКИЙ ГОСУДАРСТВЕННЫЙ ИНДУСТРИАЛЬНЫЙ УНИВЕРСИТЕТ Кафедра естественнонаучных дисциплин им. проф. В. М. Финкеля МЕТОДЫ КОНТРОЛЯ И АНАЛИЗА ВЕЩЕСТВ Составители студенты гр. ММ – 161 : Рябинин Александр Сергеевич Котов Дмитрий Алексеевич Шиманская Регина Владимировна Руководитель : Зенцова Светлана Витальевна СТРАНИЦА В КОНТАКТЕ: HTTP: //VK. COM/SIBSIUKOAX

СОДЕРЖАНИЕ Раздел 1. Введение. Знакомство с дисциплиной «Методы контроля и анализа веществ» Раздел 2. Аналитический контроль v 2. 1. Общие принципы аналитического контроля v 2. 2. Метрология и стандартизация аналитического контроля Раздел 3. Химические методы аналитического контроля. v Химические методы определения элементов v 3. 1. Титриметрический метод анализа v 3. 2. Гравиметрический метод анализа Раздел 4. Инструментальные методы контроля и анализа v 4. 1. Спектроскопические методы v 4. 1. 1. Атомно-эмиссионная спектроскопия v 4. 1. 2. Молекулярно абсорбционный метод анализа v 4. 2. Электрохимические методы анализа Заключение Список использованных источников
, промежуточных продуктов производства (электролитов, пульп, растворов")
ВВЕДЕНИЕ Изучение химического состава исходного сырья (руд, концентратов), промежуточных продуктов производства (электролитов, пульп, растворов различного состава), готовой продукции (металлов, сплавов и т. д. ), различных вспомогательных материалов осуществляется путем аналитического контроля производства.

Аналитический контроль дает возможность судить о ходе технологического процесса, соответствии его установленным режимам, о качестве используемого сырья и готовой продукции. Хорошо налаженный контроль способствует ритмичной работе предприятия, повышению качества продукции, уменьшению отходов производства, снижению брака.

1. Знакомство с дисциплиной «Методы контроля и анализа веществ» Определение химического состава продуктов металлургического производства является одним из специфических направлений производственной деятельности современного металлургического предприятия, позволяющее обеспечить стабильность технологических процессов и требуемое качество продукции. Стабилизация технологического процесса плавки достигается как стандартизацией состава исходных шихтовых материалов, так и прямым контролем и регулированием процесса плавки по составу используемых и выпускаемых продуктов. Для определения состава применяют различные методы анализа.

Важнейшей проблемой современного развития металлургической промышленности является повышение качества ее продукции. Требования к качеству, устанавливаемые стандартами, обязательны для всех предприятий, изготавливающих металлургическую продукцию. Критериями качества веществ являются стандарты. Государственный стандарт имеет силу закона и служит эталоном качества.

Первичными участками аналитического контроля на заводе являются цеховые лаборатории. Они непосредственно связаны с производством, ведут контроль исходных материалов, технологических процессов и готовой продукции. Типы анализов, выполняемых в цеховой лаборатории, определяются характером производства, которое она обслуживает.

2. Аналитический контроль - вид испытания, по 2. 1. Общие принципы аналитического контроля результатам которого оценивают содержание определяемых компонентов в контролируемом веществе. Определение химического состава продуктов металлургического производства осуществляется методами аналитической химии. Аналитическая химия — наука о методах определения состава веществ.

Основной принцип: качественный и количественный состав исследуемого объекта определяется по его аналитическим свойствам. В зависимости от способа измерения аналитического свойства, происходит классификация методов анализа. Виды аналитического контроля : Химический метод анализа Физико-химический метод анализа

Методы количественного анализа электролитический основанный на выделении отдельных металлов электролизом колориметрический производимый по сравнению интенсивности окраски данного раствора с окраской раствора определенной крепости органический анализ состоящий в сожжении органического вещества в углекислый газ С 02 и воду Н 20 и в определении по количеству их относительного содержания в веществе углерода и водорода газовый анализ состоящий в определении некоторыми специальными методами качественного и количественного состава газов или их смеси
 метод анализа Гравиметрический (весовой) метод анализа метод нейтрализации метод")
Химический метод анализа Титриметрический (объемный) метод анализа Гравиметрический (весовой) метод анализа метод нейтрализации метод оксидим етрии (окислен ия – восстан овления) (кислотноосновного титрования) Метод Комплексообразования Метод выделения МЕТОД ОТГОНКИ МЕТОД ОСАЖДЕНИЯ

Химические методы анализа 1. Титрометрический метод анализа В основе метода лежит химическая реакция. В зависимости от типа химической реакции различают: 1. 1. Метод нейтрализации или кислотно-основное титрование. Метод основан на передаче протона. В основе метода лежит реакция нейтрализации:

1. 2. Метод оксидиметрии или окислительновосстановительного титрования, основанный на передаче электрона.

2. Гравометрический метод анализа Гравиметрическим анализом называют метод количественного химического анализа, который базируется на точном измерении массы определяемого вещества или его составных частей, выделенных в химически чистом состоянии или в виде соответствующих соединений 2. 1. Метод отгонки определяемый компонент выделяют в виде летучего соединения действием кислоты или высокой температуры. • определяемый компонент выделяют в виде летучего соединения и поглощают поглотителем. Расчёт ведут по изменению массы поглотителя • Отгоняемое вещество отгоняют и отгон (дистиллят) взвешивают • Вещество взвешивают, совершают отгон и вновь взвешивают. • Расчёт производят по уменьшению массы навески. 2. 2. В методах осаждения определяемый компонент количественно осаждают химическими способами в виде малорастворимого химического соединения строго определенного состава.

3. Метод комплексообразования В методах осаждения определяемый компонент количественно осаждают химическими способами в виде малорастворимого химического соединения строго определенного состава. Методы 2 и 3 основаны на ассоциация ионов друг с другом. Эти же типы химических реакций часто используются в основе многих физико-химических методов.

Особенности химических методов анализа Преимущества : • Точность • Простота инструментального оформления Недостатки: • Низкая чувствительность и пределы обнаружения • Малая экспрессность методов
 люминесцентный рентгеноспектральный нейтронно-активизационный эмиссионный")
II. Физические методы анализа спектральный эмиссионный радиометрический (метод меченых атомов) люминесцентный рентгеноспектральный нейтронно-активизационный эмиссионный (пламенная фотометрия) атомно-абсорбционный ядерно-магнитный резонанс

Особенности физических методов анализа Преимущества : • возможность прогнозирования параметрических отказов изделий на основе ускоренных испытаний Недостатки: • необходимость испытаний в условиях многономенклатурного производства для новых изделий • непредсказуемость катастрофических отказов • отсутствие моделей связи показателей качества и результатов анализа отказов при производстве

III. Физико-химические, инструментальные методы анализа Делятся на: спектроскопические методы анализа электрохимические методы анализа хроматографические методы анализа термические методы анализа кинетические методы анализа структурные методы анализа

Особенности инструментальных методов анализа. Преимущества : • Высокая чувствительность, разрешающая способность • Экспрессность, возможность осуществления дистанционного контроля Недостатки: • Не всегда точные и неэкономичные (дорогая аппаратура и кадры).

Основные стадии аналитического контроля Любой метод включает этапы анализа: 1 • отбор пробы 2 • подготовка пробы к анализу 3 • выбор метода анализа 4 • отделение мешающих компонентов 5 • анализ – измерение аналитического свойства 6 • расчеты

2. 2. Метрология и стандартизация аналитического контроля Основные понятия. Стандартизация методик и стандартные образцы. Ошибки в количественном анализе. Математическая теория ошибок Предел обнаружения – это минимальное количество вещества, которое может быть найдено данным методом с допустимой погрешностью. Чувствительность – это изменение аналитического сигнала, приходящееся на единичное изменение концентрации. Воспринимаемый аналитический сигнал (Х) содержит информацию не только об исследуемом объекте (Х 1), но и может включать сигнал за счёт окружающего фона (Хф). X=X 1+Xф Для исключения Xф. необходимо: • пользоваться очищенными реактивами; • использовать различные операции разделения мешающих компонентов.

Между концентрацией и сигналом существует определённая зависимость. Характер которой находят опытным путём и выражают графически. Чувствительность может быть представлена в виде: tgα = ∆X/∆C, где α-угол наклона калибровочной зависимости, ∆X-изменение аналитического сигнала на единицу изменения концентрации ∆C. Чем меньше аналитический сигнал можно измерить, тем чувствительнее данная аналитическая методика. При определениях около предела обнаружения величина ошибки значительна. С достаточной точностью можно проводить определение, если измеряемая концентрация на порядок выше предела обнаружения.

Требования к величинам пределов обнаружения постоянно растут: 40 -е годы ХХ века – определение примеси в металле на уровне 0, 1 -0, 01 %; 50 -е годы ХХ века – требования к чистоте металлов выросли до 10 -4 -10 -5 %; 60 -е годы ХХ века – развитие радиоэлектроники, полупроводников потребовало оценивать добавки до 10 -8 % (германий, кремний); 70 -80 -е годы ХХ века появились методы с чувствительностью 10 -9 % и менее. Задачу снижения нижних пределов обнаружения можно решить двумя путями: 1. создание совершенно новых методов анализа; 2. разработка методов, включающих стадию предварительного концентрирования пробы.

Таблица 2. 1. 1. Абсолютные пределы обнаружения методов Аналитической химии Методы Абсолютный предел обнаружения, г 10 -9 10 -11 10 -13 10 -15 10 -17 ///// Гравиметрия ХХ Титриметрия //// // Флуоресцентные ХХХХ ХХ/// ИВАМ ХХХХ ХХ/// Газовая хроматография ХХХХ ////// Радиоизотопные ХХХХ Масс-спектрометрия ХХХХ ////// ХХХ – достигается в большинстве случаев. /// - можно добиться.

Ошибки в количественном анализе. подразделяются на: • систематические ошибки это погрешности, одинаковые по знаку, происходящие от определенных причин, влияющие на результат либо в сторону увеличения, либо в сторону уменьшения его. • случайные ошибки это неопределенные по знаку и величине ошибки, в появлении каждой из которых не наблюдается какого-либо закономерности. • промахи это грубые ошибки, сильно искажающие результат анализа.

Виды систематических ошибок 1. Ошибки методические. Они зависят от особенностей метода анализа. Устранить их трудно. Например, частичная растворимость осадка, не вполне количественное протекание реакции и т. д. 2. Ошибки, зависящие от применяемых приборов и реактивов. 3. Ошибки оперативные. Неправильное или недостаточно тщательное выполнение операций. 4. Ошибки индивидуальные. Зависят от индивидуальных особенностей самого аналитика. Систематические ошибки влияют на правильность.

Правильность аналитического метода – это близость полученного результата к истинному значению. Каждый метод предполагает прямо пропорциональную зависимость между физико-химическим параметром (y) и составом вещества (x). У = f (x). Пусть xi – измеряемая величина, - истинное значение измеряемой величины. Тогда хi=/xi- / -абсолютная погрешность; хi / ∙ 100% - относительная погрешность.

Случайные ошибки происходят при всяком измерении, и в том числе при любом аналитическом определении, как бы тщательно оно ни проводилось. В отличие от систематических ошибок случайные ошибки не могут быть учтены или устранены введением каких-либо поправок. Однако они могут быть значительно уменьшены при увеличении числа параллельных определений. Случайные ошибки влияют на воспроизводимость.

Воспроизводимость результатов химического анализа отражает степень близости друг к другу результатов, полученных по данной методике. Анализ считают выполненным удовлетворительно лишь при условии хорошей воспроизводимости результатов отдельных определений. Из этих данных берут среднее арифметическое, которое и принимают за окончательный результат анализа. Количественно воспроизводимость характеризуют стандартным отклонением среднего результата. Если имеем n измерений, рассчитаем хi – среднее значение:

; . Найдем d – отклонение от среднего значения: d =/xi - xi/ и рассчитаем S – стандартное отклонение среднего результата или S 2 – дисперсию:

РАЗДЕЛ 3. ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИТИЧЕСКОГО КОНТРОЛЯ: ТИТРИМЕТРИЧЕСКИЙ МЕТОД АНАЛИЗА Сущность анализа. Титриметрический анализ основан на точном измерении количества реагента (объем, масса и др. ), израсходованного на реакцию с определяемым веществом. Это анализ, основанный на титровании. Титрование - медленное прибавление (небольшими порциями) титрованного (стандартного, титранта, рабочего) раствора к анализируемому раствору до достижения точки эквивалентности - конца реакции. Титрованный раствор - это раствор с точно известной концентрацией.
 - момент титрования, когда количество добавленного титранта химически эквивалентно количеству титруемого")
Точка эквивалентности (ТЭ) - момент титрования, когда количество добавленного титранта химически эквивалентно количеству титруемого вещества. ТЭ еще можно назвать стехиометрической точкой, теоретической конечной точкой. Конечная точка титрования (КТТ) - это точка (момент) титрования устанавливаемая экспериментально по изменению цвета индикатора или какого либо физико-химического свойства раствора. В общем случае она не совпадает с теоретически рассчитанной точкой эквивалентности. u

Индикатор вещество, которое изменяет свою окраску в ТЭ, характеризуется малой концентрацией и интервалом перехода. Интервал перехода индикатора - это область концентрации ионов водорода, ионов металла, потенциалов (Е) окисленной или восстановленной формы вещества, в пределах которой визуально можно обнаружить изменения в оттенках окраски индикатора.

ПРИЕМЫ ТИТРОВАНИЯ В методах прямого титрования определяемое вещество непосредственно реагирует с титрантом. Для проведения анализа этим методом достаточно одного рабочего раствора. В методах обратного титрования (или, как их еще называют, методах титрования по остатку) используются два тиосульфатом натрия. Основной и вспомогательный. Широко известно, например, обратное титрование хлорид-иона в кислых растворах. К анализируемому раствору хлорида сначала добавляют заведомый избыток титрованного раствора нитрата серебра (основного рабочего раствора). При этом происходит реакция образования малорастворимого хлорида серебра: Ag + + Cl- → Ag. Cl. Не вступившее в реакцию избыточное количество Ag. NO 3 оттитровывают раствором тиоцианата аммония (вспомогательного рабочего раствора): Ag+ + SCN- → Ag. SCN. Содержание хлорида можно рассчитать, т. к. известно общее количество вещества (моль), введенное в раствор, и количество вещества Ag. NO 3, не вступившее в реакцию с хлоридом.
. В")
Третьим основным видом титриметрических определений является титрование заместителя (титрование по замещению, косвенное титрование). В этом методе к определяемому веществу добавляют специальный реагент, вступающий с ним в реакцию. Один из продуктов взаимодействия затем оттитровывают рабочим раствором. Например, при иодометрическом определении тиосульфатом натрия. к анализируемому раствору добавляют заведомый избыток. KI. Происходит реакция 2 Cu 2+ + 4 I- → 2 Cu. I + I 2. Выделившийся йод оттитровывают тиосульфатом натрия. Известны и более сложные титриметрические методики анализа, являющиеся комбинацией этих трех основных.

Степень оттитрованности f - это отношение количества оттитрованного исследуемого вещества в данный момент титрования (оно эквивалентно количеству введенного титранта) к его исходному количеству. n. Т = CТVТ, а n 0 =C 0 V 0. f = n. Т / n 0 = CТVТ / C 0 V 0. ; При C 0 = CТ; f = VТ/V 0. ; где VТ - объема титранта, добавленный в данный момент титрования эквивалентно количеству исследуемого вещества. V 0 исходный объем титруемого вещества; C 0 и CТ молярные концентрации эквивалентов (нормальные концентрации) титруемого вещества и титранта. Степень оттитрованности выражают в долях и %. Если выделить 4 этапа титрования, то 1) до начала титрования f = 0; (0%) 2) до точки эквивалентности f < 1 (< 100%); 3) в ТЭ f =1 (100%); 4) за ТЭ f > 1 (> 100%)

УСТАНОВКА ДЛЯ ТИТРОВАНИЯ

РЕАКЦИИ, ПРИМЕНЯЕМЫЕ В ТИТРИМЕТРИЧЕСКОМ АНАЛИЗЕ ДОЛЖНЫ УДОВЛЕТВОРЯТЬ СЛЕДУЮЩИМ ОСНОВНЫМ ТРЕБОВАНИЯМ: Точка эквивалентности должна фиксироваться четко и точно. Реакция должна протекать количественно, т. е. константа равновесия реакции должна быть достаточно велика. Скорость аналитической реакции должна быть велика. Реакция не должна осложняться протеканием побочных реакций. Вещества, мешающие определению точки эквивалентности и протеканию основной реакции, должны отсутствовать. Раствор титранта должен быть стандартизирован.

МЕТОД НЕЙТРАЛИЗАЦИИ Метод нейтрализации – это один из видов титриметрического анализа, который широко используется в лабораториях различного медицинского и экологического профиля: клинических, диагностических, санитарногигиенических, судебноэкспертных, контроля состояния окружающей среды, стандартизации и контроля лекарственных форм.
 — взаимодействие кислоты и")
Нейтрализа ция (от лат. neuter — ни тот, ни другой) — взаимодействие кислоты и основания (щелочи) между собой с образованием соли и малодиссоциирующего вещества (воды). В большинстве своем, реакции нейтрализации экзотермичны. К примеру, реакция гидроксида натрия и соляной кислоты: НСl + Na. OH = Na. Cl + Н 2 О В ионном виде уравнение записывают так: Н+ + ОН− = Н 2 О
 в растворе гидроксида")
РАСЧЕТЫ В МЕТОДЕ НЕЙТРАЛИЗАЦИИ Требуется рассчитать массу щелочи. (m. Na. OH) в растворе гидроксида натрия. В расчетах будем использовать выражение СН 1∙V 1 = СН 2∙V 2 закона эквивалентов для растворов: СН(Na. OH)∙V(Na. OH) = СН(HCl)∙V(HCl), где Na. OH – анализируемое вещество, а HCl – рабочее вещество. Подставим в это уравнение выражение нормальности Na. OH: Отсюда выразим массу щелочи:

ИОННОЕ ПРОИЗВЕДЕНИЕ ВОДЫ Вода – слабый электролит. Запишем уравнение диссоциации воды и выражение константы равновесия К. H 2 O↔H++OH-; где [Н+] , [ОН-] , [Н 2 О]–равновесные концентрации, моль/л; 1, 8∙ 10 -16 – табличная величина константы равновесия воды при 20 о. С.

Так как К очень мала, то можно считать равновесную концентрацию воды величиной постоянной [H 2 O] ≈ const. Рассчитаем молярную концентрацию воды, зная, что один литр воды составляет 1000 миллиграммов, а молярная масса воды – 18 г/моль. [H 2 O] =55, 56 моль/л. Произведение постоянных величин К и [Н 2 О] есть величина постоянная и ее называют ионным произведением воды: К∙[H 2 O] = В соответствии с уравнением (1) запишем: [H+][OH-] = 10 -14. Поскольку один моль воды при диссоциации дает по одному молю ионов водорода и гидроксильной группы, то их равновесные концентрации равны: [H+] = [OH-] = 10 -7 моль/л.
 – это отрицательный десятичный логарифм равновесной концентрации")
ВОДОРОДНЫЙ ПОКАЗАТЕЛЬ PH Водородный показатель (p. H) – это отрицательный десятичный логарифм равновесной концентрации ионов водорода p. H = -lg[H+] (2) р. Н = -lg[10 -7] = 7 При р. Н = 7 – среда нейтральная. Если р. Н > 7, то среда щелочная, а при р. Н < 7 – среда кислая.

ЗНАЧЕНИЕ МЕТОДА НЕЙТРАЛИЗАЦИИ Данный метод позволяет анализировать кислоты, гидроксиды, соли, оксиды, то есть любые вещества, способные взаимодействовать с кислотой и со щелочью. Этот способ получил широкое применение в гидро- и электрометаллургии. Точность метода приблизительно 0, 1÷ 0, 2%.

ВЫБОР ИНДИКАТОРА В методе кислотно-основного титрования все вещества реакции не имеют окраски, поэтому для определения КТТ используют индикаторы кислотно-основного титрования. Каждый индикатор меняет свою окраску в определенном интервале p. H (интервал перехода окраски индикатора – ИПО). Название индикатора ИПО, p. H РТ*, p. Н фенолфталеин 8, 0 - 10, 0 лакмус Окраска p. H<7 p. H>7 9, 0 бесцветный малиновый 5, 0 - 8, 0 7, 0 красный синий метиловый оранжевый 3, 0 - 4, 4 4, 0 малиновый желтый метиловый красный 4, 4 - 6, 2 5, 0 красный желтый *РТ – показатель титрования

СТАНДАРТИЗАЦИИ РАБОЧИХ РАСТВОРОВ


РАСЧЁТЫ В МЕТОДЕ КИСЛОТНО-ОСНОВНОГО ТИТРОВАНИЯ

УСТАНОВКА ТИТРА И НОРМАЛЬНОСТИ РАСТВОРА НСI Бура подвергается гидролизу: Na 2 B 4 O 7+7 H 2 O=2 Na. OH+4 H 3 BO 3. 2 Na. OH+2 HCl=2 Na. Cl+2 H 2 O. Суммируя оба уравнения, получим общее уравнение реакции титрования: Na 2 B 4 O 7+2 HCl+5 H 2 O=2 Na. Cl+4 H 3 BO 3 Водный раствор буры обладает сильнощелочной реакцией и может титроваться кислотой.

v
, получаем уравнение для расчёта массы щёлочи")
Произведя сокращения и подставив численное значение M(f·Na. OH), получаем уравнение для расчёта массы щёлочи в растворе: m(Na. OH) = 40·C(f·HCl) V(HCl)· 10 -3, где m(Na. OH) – масса щелочи, г; С(f·HCl) – молярная концентрация эквивалента HCl, рассчитанная по формуле (6), моль/л; V(HCl) – объём соляной кислоты, пошедший на титрование задачи, мл. Для расчёта массы щёлочи в миллиграммах получим соотношение: m(Na. OH) = 40·C(f·HCl) V(HCl)

v

ТЕХНИКА БЕЗОПАСНОСТИ Запрещается начинать работу без точного знания методики опыта. На рабочем столе должен быть безупречный порядок. Работы с ядовитыми, дурнопахнущими, огне- и взрывоопасными веществами выполняются обязательно в вытяжном шкафу. При работе под тягой, голову необходимо держать вне вытяжного шкафа. При работе с горючими веществами недопустимо наличие поблизости открытого огня. Не пробуйте на вкус и не вдыхайте неизвестное вещество. Смешивая концентрированные кислоты с водой, лейте кислоту в воду. Наполнение пипеток кислотами, щелочами и другими ядовитыми путем засасывания ртом запрещается. Для наполнения пипеток следует пользоваться грушей. При порезах стеклом удаляют из раны осколки, промывают водой, либо протирают тампоном, смоченным спиртом, смазывают 5%-ным раствором йода и накладывают повязку.

Для количественных измерений потребуются следующая посуда и реактивы: Посуда и реактивы: установка для титрования, раствор соляной кислоты приблизительной концентрации, навески буры по 0, 2 г, коническая колба на 250 мл, дистиллированная вода, раствор индикатора метилового оранжевого, задача (анализируемая проба), раствор щёлочи.

ПОДГОТОВКА РАБОЧЕГО МЕСТА И ВИДЕОПРОЦЕСС https: //vk. com/video-69626081_456239039

Оксидиметрия – титранты являются окислителями Окислительно-восстановительное титрование – титранты это вещества с окислительно-восстановительными свойствами. z/1, z/2–эквивалентное число определяемого вещества и вещества рабочего раствора, соответственно; Точка эквивалентности –это момент титрования , когда количество эквивалентов добавляемого вещества равно количеству эквивалентов определяемого вещества В области точки эквивалентности при переходе от раствора, недатированного на 0, 1%, к раствору перетитрованному на 0, 1 %, потенциал изменяется больше, чем на 0, 5 В. Резкий скачок потенциала позволяет использовать для обнаружения точки эквивалентности непосредственно потенциометрические измерения или окислительно-восстановительные индикаторы, окраска которых изменяется при изменении потенциала.

В зависимости от свойств используемого титранта различают оксидиметрию и редуктометрию 1. Оксидиметрия 2. Редуктометрия Окислительно-восстановительное титрование Оксидиметрическое титрант - окислитель Редуктометрическое титрант - восстановитель

ИНДИКАТОРЫ ДЛЯ ОКСИДИМЕТРИИ ПО ИХ ДЕЙСТВИЮ МОЖНО РАЗДЕЛИТЬ НА ДВЕ ГРУППЫ Индикаторы, которые вступают в специфическую реакцию с окислителем или восстановителем. Индикаторы, у которых перемена окраски не зависит от специфических свойств окислителя или восстановителя, реагирующих между собой при титровании, а связана с достижением титруемым растровом определенного окислительно-восстановительного потенциала. Окислительно-восстановительные индикаторы представляют собой вещества, способные обратимо окисляться или восстанавливаться, причем окисленная и восстановленная формы их имеют различную окраску.

КРИВЫЕ ТИТРОВАНИЯ

ПРИГОТОВЛЕНИЕ 0, 1 Н. РАСТВОРА ОКСАЛАТА НАТРИЯ МЕТОДОМ ТОЧНЫХ НАВЕСОК
")
Готовим 100 мл точно 0, 1 н. Na 2 C 2 O 4 (fэкв=1/2) так: взвешивают на аналитических весах его точную навеску, равную 0, 1× 67, 00=0, 6700 г которую переносят без потерь (через воронку) в мерную колбу на 100 мл. растворяют в небольшом количестве дистиллированной воды, после чего доливают дистиллированной водой до метки и хорошо перемешивают. Полученный раствор будет иметь: c (1/2 Na 2 C 2 O 4) = 0, 1 моль/л

УСТАНОВЛЕНИЕ МОЛЯРНОЙ КОНЦЕНТРАЦИИ ЭКВИВАЛЕНТОВ ПЕРМАНГАНАТА И ЕГО ТИТРОВАНИЕ МЕТОДОМ ПИПЕТИРОВАНИЯ

Раствор перманганата наливают в бюретку, предварительно ополоснув ее дважды этим раствором, устанавливают уровень жидкости на нуле (по верхнему мениску). Затем из мерной колбы пипеткой берут аликвотную часть (равную 10 мл) приготовленного раствора оксалата натрия, переносят его в коническую колбу на 250 мл, прибавляют туда же 10 мл 2 н. серной кислоты примерно 50 мл (на два пальца) дистиллированной воды и нагревают почти до кипения (80– 900). Горячий раствор титруют раствором перманганата, приливая каждую следующую каплю раствора только после того, как исчезнет окраска предыдущей. Первые капли обесцвечиваются довольно медленно, но как только образуется немного двухвалентного марганца, являющегося катализатором данной реакции, дальнейшее обесцвечивание происходит практически мгновенно. Титрование производится до тех пор, пока одна капля перманганата окрасит весь раствор в неисчезающий в течение одной минуты бледно-розовый цвет.


ПРОИЗВЕДЕНИЕ РАСЧЕТОВ

v Т

ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ХРОМА В РАСТВОРЕ БИХРОМАТА МЕТОДОМ ОБРАТНОГО ТИТРОВАНИЯ
, так как")
При определении хрома в бихромате применяют метод обратного титрования (титрование по остатку), так как бихромат не может титроваться перманганатом, так как оба являются окислителями. В исследуемый раствор вводят измеренный избыток восстановителя – раствора сульфата железа (II). Бихромат в кислой среде окисляет соли двухвалентного железа до трехвалентного: K 2 Cr 2 O 7+6 Fe. SO 4+7 H 2 SO 4=3 Fe 2(SO 4)3+K 2 SO 4+Cr 2(SO 4)3+H 2 O Избыток сульфата железа, не вошедший в реакцию, оттитровывают перманганатом и по результатам титрования рассчитывают количество хрома в растворе: 10 Fe. SO 4+2 KMn. O 4+8 H 2 SO 4=5 Fe 2(SO 4)3+K 2 SO 4+2 Mn. SO 4+8 H 2 O

В исследуемый раствор вводится избыток сульфата железа, поэтому сначала устанавливают соотношение между этими растворами: Установка соотношения избытка сульфата железа и пермангантата калия. В коническую колбу на 250 мл из бюретки берут 15 мл сульфата железа, прибавляют 10 мл серной кислоты (1 , )4׃ разбавляют водой до 50 мл (на два пальца) и титруют перманганатом до бледно-розового окрашивания. Количество перманганата пошедшего на титрование, обозначим V 1. Установка величины остатка. К исследуемому раствору приливают 10 мл серной кислоты (1 , )4׃ затем точно из бюретки 15 мл сульфата железа разбавляют водой примерно до 50 мл (на два пальца) и титруют перманганатом до изменения окраски из зеленого в сиреневый цвет. Количество перманганата, пошедшего на титрование, обозначим через V 2. ВИДЕО ПО ДАННОЙ ПРЕЗЕНТАЦИИ HTTPS: //VK. COM/VIDEOS-69626081? SECTION=ALL&Z=VIDEO 69626081_456239038%2 FCLUB 69626081%2 CALBUM-69626081%2 FPL_ -69626081

В данной работе определение содержания остаточного активного хлора в питьевой воде производится методом йодометрии по ГОСТ 18190 -72. Йодометрия – титрометрический (объёмный) метод определения веществ, основанный на реакциях окисления – восстановления (разновидность оксидиметрии), связанных с восстановлением I 2 до I – ионов и с окислением I- ионов до I 2 : Поэтому методом йодометрии можно определять как окислители, так и восстановители. Косвенное йодометрическое титрование используется для титрования окислителей и основан на окислении йодида активным хлором до йода, который титруют тиосульфатом натрия (рабочий раствор).

ПОСУДА И РЕАКТИВЫ

ПОДГОТОВКА К АНАЛИЗУ Приготовление 0, 5% раствора крахмала 0, 5 г растворимого крахмала смешивают с небольшим объемом дистиллированной воды, прибавляют к 100 см 3 кипящей дистиллированной воды и кипятят несколько минут, затем охлаждают.

ХОД ОПРЕДЕЛЕНИЯ В коническую колбу вместимостью 500 см 3 помещают 0, 5 г иодида калия, растворяют его в 12 см 3 дистиллированной воды, затем добавляют 5 см 3 буферного раствора, после чего добавляют 250 см 3 анализируемой воды и 1 см 3 0, 5%-ного раствора крахмала.

При наличии активного хлора в воде происходит окрашивание раствора в синий цвет, т. к. хлор вытесняет из иодида калия йод: 2 KI + Cl = 2 KCl + I 2

К этому раствору прибавляют по каплям из бюретки 0, 005 н раствор тиосульфата натрия до обесцвечивания раствора. Обесцвечивание наступает в тот момент, когда весь йод связывается с тиосульфатом натрия: I 2 + 2 Na 2 S 2 O 3 = 2 Na. I + Na 2 S 4 O 6

ИОДИД НАТРИЯ И ТЕТРАТИОНАТ НАТРИЯ ОКРАШИВАНИЯ С КРАХМАЛОМ НЕ ДАЮТ. ТИТРОВАНИЕ ПРОВОДЯТ ТРИ РАЗА И БЕРУТ СРЕДНИЙ РЕЗУЛЬТАТ

РАСЧЕТ Содержание остаточного хлора Х в мг/дм 3 вычисляют по формуле: , где - средний объем раствора тиосульфата натрия, пошедший на титрование, см 3; - молярная концентрация эквивалента тиосульфата натрия, моль/дм 3 ; -молярная масса эквивалента хлора, г/моль; V- объем пробы воды, взятой для титрования.

СУЩНОСТЬ ГРАВИМЕТРИЧЕСКОГО АНАЛИЗА И КЛАССИФИКАЦИЯ ЕГО МЕТОДОВ Гравиметрическим анализом называют метод количественного химического анализа, который базируется на точном измерении массы определяемого вещества или его составных частей, выделенных в химически чистом состоянии или в виде соответствующих соединений (точно известного постоянного состава). Гравиметрический анализ (весовой) один из важнейших методов количественного анализа. Он сыграл большую роль при установлении законов постоянности состава, кратных отношений, периодического закона. Его применяют при определении химического состава разнообразных естественных и технических объектов, горных пород и руды, минералов, металлов, сплавов, силикатов и других неорганических и органических веществ. Все многочисленные гравиметрические определения можно разделить на три большие группы: 1. Методы выделения; 2. Методы осаждения; 3. Методы отгонки.

МЕТОД ВЫДЕЛЕНИЯ В методе выделения определяемый компонент количественно выделяют в свободном состоянии из анализируемой смеси и взвешивают на аналитических весах. Так, например, количественно определяют золото и медь в сплаве. При растворении определенной навески сплава в царской водке получают раствор, который содержит ионы Au 3+ и Cu 2+. Добавлением к полученному раствору пероксида водорода, который восстанавливает ионы золота до элементного золота и не влияет на ионы Cu 2+, все золото выделяют в элементном состоянии. Золото, которое выделилось, отфильтровывают, промывают разбавленным раствором хлоридной кислоты от посторонних примесей, помещают вместе с фильтром в предварительно взвешенный фарфоровый тигель, высушивают, прожаривают для удаления летучих примесей и после охлаждения взвешивают. По массе золота, которое выделилось, рассчитывают его содержание в анализируемом сплаве.

Если через промывные воды и фильтр, который остался после отделения золота, пропустить при определенных условиях постоянный электрический ток, то на предварительно взвешенном инертном по отношению к раствору платиновом катоде количественно выделится металлическая медь. По увеличению массы катода рассчитывают массу меди и ее содержание в сплаве. Описанный метод определения золота в сплаве называют гравиметрическим, а меди – электрогравиметрическим. Другим примером подобного определения есть определение массовой доли золы в твердом топливе, которое базируется на сжигании и прожаривании до постоянной массы навески топлива в предварительно взвешенном тигле. Золу, которая остается в тигле, взвешивают. По массе золы рассчитывают ее массовую долю в данном образце твердого топлива.

МЕТОДЫ ОСАЖДЕНИЯ В методах осаждения определяемый компонент количественно осаждают химическими способами в виде малорастворимого химического соединения строго определенного состава. Осадок, который выделяется, промывают, высушивают или прожаривают. При этом осадок в большинстве случаев превращается в новое вещество точно известного состава, которое и взвешивают на аналитических весах. В анализе различают: осаждаемую форму, то есть форму, в виде которой осаждают определяемое вещество, и весовую форму, то есть форму, в виде которой определяемое вещество взвешивают. Весовая форма (гравиметрическая) может иметь ту же формулу, что и осаждаемая форма. Например, при определении сульфат – ионов гравиметрическим методом, путем осаждения их ионами бария, формула осаждаемой формы (осадка) и формула весовой формы при соблюдении всех необходимых условий анализа одна и та же. Ссылка на видео: https: //www. youtube. com/watch? v=BGLw. Lq. NDp. SM

МЕТОДЫ ОТГОНКИ В методах отгонки определяемый компонент количественно отгоняют в виде летучего соединения. Определяемую часть отгоняют путем нагревания анализируемого вещества или действием соответствующих реагентов, которое сопровождается выделением летучих продуктов. Методы отгонки бывают прямые и косвенные. Прямые методы отгонки. Определяемый летучий компонент поглощают специфическим поглотителем и по увеличению массы последнего рассчитывают массу определяемого компонента. Примером прямого гравиметрического определения летучего вещества является определение CO 2 в карбонатных породах, которое базируется на разложении карбонатов кислотами :

Преимущества и недостатки гравиметрического анализа Гравиметрические методы анализа разрешают с относительно высокой точностью определять в данном образце анализируемого вещества количественное содержание отдельных компонентов или (если дан раствор) концентрацию их в растворе. Гравиметрический анализ пригоден для определения многих металлов (катионов) и неметаллов (анионов), составных частей сплавов, руд, силикатов, органических соединений и т. д. Важным недостатком является продолжительность во времени определения, которое намного превышает продолжительность определений, выполняемых с помощью титриметрических методов. По этой причине гравиметрический анализ нечто утратил свое предшествующее значение; в практике их заменяют современными экспрессными химическими и физико-химическими методами. Однако гравиметрические методы, которые характеризуются высокой точностью, полностью сохранили свое значение при арбитражных анализах и широко используются в научно-исследовательских роботах для сравнения аналитическими данными, полученными разными методами. С помощью гравиметрического анализа определения проводят с точностью до 0, 010, 005%, что превышает точность титриметрических методов.

МЕТОД ОСАЖДЕНИЯ: ТРЕБОВАНИЯ К ОСАДКАМ, ТЕХНИКА ВЫПОЛНЕНИЯ Среди всех методов анализа наибольшее значение имеет метод осаждения. Он известный с того времени, как возникла аналитическая химия. Метод не утратил своего значения до этого времени, что обусловлено хорошим теоретическим обоснованием и его широкой экспериментальной проверкой. Схема анализа и главные операции метода осаждение такие: – расчет и взятие навески; – растворение навески; – выбор осадителя и осаждение; – фильтрование; – промывание осадка; – прожаривание или высушивание осадка. По массе осадка и его формуле вычисляют содержание определяемых ионов и их процентное соотношение к навеске.
 при осаждении кристаллических")
В общем виде при расчетах навесок анализируемых веществ пользуются формулами: а) при осаждении кристаллических осадков б) при осаждении аморфных осадков: где: а – масса навески анализируемого вещества, г; МА - молярная масса определяемого вещества; МА 1 – молярная масса весовой формы; 0, 5 – практически удобная масса весовой формы определяемого вещества для кристаллических осадков, г; 0, 1 – практически удобная масса весовой формы определяемого вещества для аморфных осадков, г; m, n – коэффициенты (m относится к определяемому веществу, а n – к весовой форме определяемого вещества).

Величина навески анализируемого вещества, вообще говоря, зависит от процентного содержания определяемого компонента, массы осаждаемой и гравиметрической формы, чувствительности весов и содержания определяемого компонента в гравиметрической форме. Расчет величины навески для выполнения одного определения можно провести, воспользовавшись формулой: где g – масса навески, г; m – масса гравиметрической (весовой) формы, г; р – процентное содержание определяемого компонента; F – гравиметрический фактор.

При выражении гравиметрического фактора в числителе указывают формулу определяемого вещества, а в знаменателе – его весовой формы. Например, для определения Fe через Fe 2 О 3: то есть, связывая с нашими предыдущими обозначениями где МА - молярная масса определяемого вещества; МА 1 – молярная масса весовой формы; m, n – коэффициенты (m относится к определяемому веществу, а n – к весовой форме определяемого вещества). Значение гравиметрического фактора находят по таблицам или рассчитывают по значениям молярных масс. Масса гравиметрической формы определяется: – погрешностью весов; – оптимальной массой осаждаемой формы.

Взятие навески Точное взятие навески играет решающую роль в количественном анализе. 1. Для взятия навесок твердых веществ пользуются часовым стеклом, бюксами, пробирками. 2. Для жидких веществ – капельницы, маленькие колбы вместительностью 1 -2 мл, желатиновые капсулы, подвесные пипетки с пришлифованными кранами. 3. Для взятия навесок легко летучих веществ применяют тонкостенные ампулы, из которых перед заполнением удаляют воздуха.
 с")
Перенесение навески: 1. Твердого вещества: Взвешенный с веществом бюкс (часовое стекло или пробирку) с навеской осторожно снимают из шальки весов. Содержимое бюкса осторожно высыпают в стакан. После перенесения навески в стакан бюкс с остатком навески снова взвешивают на аналитических весах. Масса навески определяется по разности масс бюкса с навеской и бюкса после высыпания навески. Взвешенный бюкс с навеской осторожно снимают с шальки весов и переносят содержимое бюкса в стакан. Потом остатки навески смачивают и смывают дистиллированной водой из промывалки в стакан. В этом случае массу навески определяют по разности масс бюкса с навеской и чистого. При перенесении навески в стакан следует не утратить даже незначительного количества вещества.

Требования к весовой форме 1. Важнейшее требование – точное соответствие состава химической формуле (осаждаемая Fe(OH)3 º Fe 2 O 3 × x. Н 2 O, а весовая! Fe 2 O 3). 2. Достаточная химическая устойчивость весовой формы. Очевидно, что работа очень усложнится, если весовая форма будет легко изменять свой состав, вследствие, например, поглощения водных паров или СО 2 из воздуха, окисления или восстановления, разложения и других процессов. Ведь при этом нарушается соответствие состава осадка формуле. Например, весовая форма Са. О плохая, так как поглощает Н 2 О и СО 2 из воздуха, поэтому лучше весовой формой будет Са. SО 4. 3. Содержание определяемого элемента в весовой форме должно быть по возможности меньшим, так как погрешности определения (например, погрешность взвешивания, потеря при растворимости или неполное перенесение осадка на фильтр) при этом будут меньше проявляться на конечном результате анализа. Например, одинаковая по абсолютной величине погрешность при определении массы осадков Ba. Cr. О 4, Cr 2 O 3 влияет на найденное содержание хрома в первом случае в 3, 5 разы меньше, чем в втором. Действительно, потеря 1 мг осадка при анализе отвечает следующим погрешностям при определении массы хрома:

Весовая форма Cr 2 O 3 Весовая форма Ba. Cr. О 4 В 152 г Cr 2 O 3 содержится 104 г Cr В 1 мг Cr 2 O 3 - х мг Cr Х = 104 × 1/152 = 0, 7 мг (Cr) В 253, 3 г Ba. Cr. О 4 содержится 52 г Cr В 1 мг Ba. Cr. О 4 - х мг Cr Х = 52 × 1/253, 3 = 0, 2 мг (Cr) ССЫЛКА НА ВИДЕО: HTTPS: //WWW. YOUTUBE. COM/WATCH? V=_VKYGS 7 XCLU

Количество осадителя Образование осадков происходит лишь при условии, если произведение концентраций соответствующих ионов превышает величину произведения растворимости осаждаемого соединения при данной температуре. В гравиметрическом анализе осаждение считается практически полным, если в растворе масса осаждаемого соединения находится за пределами точности взвешивания < 0, 0002 г. Для того, чтобы осаждение того или иного иона могло быть практически полным, необходимо, очевидно, взять достаточное количество осадителя. Сколько его необходимо, можно приблизительно рассчитать по уравнению реакции. Но для полного осаждения кристаллических и аморфных осадков количество раствора осадителя следует брать приблизительно в 1, 5 раза больше рассчитанного количества. Для осаждения кристаллических осадков пользуются разбавленными растворами осадителя, поэтому вымеренный объем или навеску осадителя разводят водой приблизительно до 50 мл. Для осаждения аморфных осадков пользуются концентрированными растворами осадителя.

Осаждение Наиболее часто осаждение ведут в той же посуде, в которой проводится растворение пробы. Если пробу растворяли в фарфоровой чашке, то содержимое чашки переливают в химический стакан емкостью 300– 400 мл и чашку тщательно обмывают водой, собирая воду в тот же стакан. Осаждение ведут при нагревании. Техника осаждения кристаллических и аморфных осадков разная. Рассмотрим процесс образования осадка. Он намного сложнее от: Ba 2+ + SO 42 - Ba. SO 4 Почти всегда наблюдается индукционный период, который длится с момента смешивания растворов реагентов, которые содержат реагирующие вещества, до появления видимого осадка. Ссылка на видео https: //www. youtube. com/watch? v=sverbqo 5 RAc

Условия осаждения кристаллических осадков: 1. Осаждение проводить из достаточно разбавленного раствора разбавленным раствором осадителя; 2. Прибавлять осадитель очень медленно, по каплям (в особенности в начале осаждения); 3. Непрерывно перемешивать раствор стеклянной палочкой, чтобы предотвратить сильных местных точек пересыщения при добавлении осадителя; 4. Вести осаждение из горячего раствора, а иногда нагревают и раствор осадителя (чтобы повысить растворимость); 5. Отфильтровывать осадок только после охлаждения раствора; 6. Прибавлять при осаждении вещества, которые повышают растворимость осадка. Почти всегда после добавления осадителя оставляют осадок на несколько часов, а наиболее часто на протяжении суток, чтобы осадок “созрел” (происходит укрупнение кристаллов). Причиной укрупнения кристаллов является большая растворимость очень маленьких кристаллов вещества по сравнению с растворимостью более крупных кристаллов при других одинаковых условиях.

Условия осаждения аморфных осадков 1. Осаждение ведут из концентрированных растворов концентрированными растворами осадителя; 2. Осаждения ведут в горячих растворах (повышение температуры оказывает содействие быстрой коагуляции осадка); 3. Осаждения ведут в присутствии какого-нибудь электролита коагулянта; 4. Осадки быстро фильтруют и не оставляют под маточным раствором. Кристаллы, которые оседают, захватывают с собой из раствора разные примеси. Например, если на раствор, который содержит смесь Ba. Cl 2 и Fe. Cl 3, подействовать H 2 SO 4, то будет осаждаться Ba. SO 4 и частично будет оседать Fe 2(SO 4)3, хотя эта соль растворима в воде.

Окклюзия При окклюзии загрязняющее вещество находится внутри частичек осадка. Причины: 1. Адсорбция в процессе кристаллизации; 2. Захватывание загрязненного вещества при кристаллизации; 3. Образование химического соединения между осадком и соосадительной примесью. Изоморфизм – образование смешанных кристаллов. Изоморфными называются такие вещества, которые способны кристаллизоваться, образуя совместную кристаллическую решетку, причем образуются так называемые смешанные кристаллы. Значение изоморфизма и количественные закономерности, которые наблюдаются при явлениях соосаждения, были впервые установлены Хлопиным и Ганом.

После осаждения – формирование нового осадка на поверхности основного при стоянии. Уменьшение соосаждения: 1. Рациональный выбор хода анализа; 2. Рациональный выбор осадителя: органические осадители дают меньше соосаждение посторонних веществ, чем неорганические; 3. Осаждение крупнокристаллических осадков; 4. Выдерживание осадка под маточным раствором; 5. Промывание осадка; 6. Переосаждение. Промывание. Для освобождения осадка от адсорбированных примесей применяют промывание. Кристаллические осадки с низкой растворимостью можно промывать водой. Для очистки аморфных осадков следует использовать промывные жидкости; чаще всего это растворы летучих электролитов, которые разрешают избегнуть пептизации осадка. Осадки с высокой растворимостью промывают растворами электролитов, которые содержат одноименный с осадком ион.
. Масса")
Фильтрование В количественном анализе применяют беззольные фильтры, которые почти полностью сгорают (без остатка). Масса золы, которая остается составляет 0, 00003– 0, 00007 г и, обычно ею пренебрегают. При проведении очень точных анализов массу золы, указанную на упаковке пачки фильтров, учитывают при расчетах. Иногда при фильтровании применяют так называемую фильтровальную массу (измельченная беззольная фильтровальная бумага). Осадки, которые легко восстанавливаются при сгорании бумаги, следует фильтровать через пористые стеклянные или фарфоровые тигли или через специальные воронки с пластинками пористого стекла. Ссылка на видео : https: //www. youtube. com/watch? v=EHk. No. GVVp. Uw https: //www. youtube. com/watch? v=cl. WQ_U 8 X 1 KQ

Прокаливание или высушивание осадков. Осадки высушивают, как правило, в стеклянных тиглях – тигель Шота – в сушильных шкафах. Осадки прокаливают в фарфоровых, кварцевых и платиновых тиглях, предварительно доведенных до постоянной массы. Охлаждения тигля ждут, помещая его до взвешивания в эксикаторах. Прожаривание и высушивание гравиметрической формы проводят до постоянной массы, то есть разность между параллельными взвешиваниями не должна превышать ± 0, 0002 г.
. 2.")
ПРИМЕНЕНИЕ ГРАВИМЕТРИИ В ФАРМАЦЕВТИЧЕСКОМ АНАЛИЗЕ 1. Определение влажности в препаратах (косвенная отгонка). 2. Определение сухого остатка в экстрактах, настойках (косвенная отгонка). 3. Определение сульфатной золы и золы (метод выделения). 4. Определение в лекарственных средства (пиперазина хромат – метод осаждения).

РАЗДЕЛ 4. ИНСТРУМЕНТАЛЬНЫЕ МЕТОДЫ КОНТРОЛЯ И АНАЛИЗА 4. 1. СПЕКТРОСКОПИЧЕСКИЕ МЕТОДЫ В инструментальных методах анализа устанавливается связь между физико-химическим параметром и концентрацией анализируемого вещества. Спектральные и другие оптические методы анализа основаны на использовании различных явлений и эффектов, возникающих при взаимодействии вещества и электромагнитного излучения.

Основные характеристики электромагнитного излучения. Поскольку свет имеет двойственную природу – волновую и корпускулярную, для его описания используют два вида характеристик – волновые и квантовые. Волновые характеристики – частота колебаний, длина волны и волновое число. Квантовая характеристика – энергия квантов. Частота колебаний ν показывает число колебаний в 1 сек. , измеряется в герцах (Гц). Высокие частоты измеряются в килогерцах (1 к. Гц = 103 Гц), мегагерцах (1 м. Гц = 106 Гц). Длина волны (λ) показывает наименьшее расстояние между точками, колеблющимися в одинаковых фазах. Это линейная единица, измеряется в СИ в метрах (м) и его долях – сантиметрах (см), миллиметрах (мм), нанометрах (1 нм = 10 -9 м).

Таблица 4. 1. 1. Характеристика электромагнитного спектра Интервал длин волн 10 -4 – 0, 1 нм 10 -2 – 10 нм 10 – 400 нм 400 – 760 нм 760 – 106 нм 10 -3 – 1 м >1 м Участок спектра γ- Излучение Рентгеновское излучение Ультрафиолетовое излучение Видимый свет Инфракрасное излучение Микроволновое излучение Радиоизлучение

Таблица 4. 1. 2. Характеристика спектра видимого света Длина 400 -500 500 -550 550 -600 600 -650 650 -700 Поглощаемый Свет Фиолетовый Синий Зеленый Желтый Оранжевый Красный Наблюдаемый свет Желтый Красный Фиолетовы й Синий Зеленый волны, нм

Длина волны и частота колебаний связаны между собой соотношением: ν = с / λ, где с – скорость света. Если скорость света выражены в см/с, а дина волны – в см, то ν = 3∙ 1010/ λ, где ν выражена в Гц. Пример. Для зеленого света λ=500 нм=5∙ 10 -5 см, частота ν = 3∙ 1010/ 5∙ 10 -5= 6∙ 1014 Гц. Величину, обратную длине волны, называют волновым числом (ν, ') и обычно выражают в обратных сантиметрах (см-1). Пример. Для зеленого света ν, '= 1 / 5∙ 10 -5= 2∙ 104 (см-1). Энергия электромагнитного излучения Е определяется соотношением: Е = h ν, где h – постоянная Планка, равная 6, 62 10 -34 Дж∙с; Е – энергия электромагнитного излучения, Дж/моль.
;")
Спектральные методы анализа делят на: Методы, основанные на спектрах излучения: атомно-эмиссионная спектроскопия (лабораторная работа); плазменная спектроскопия. Методы, основанные на спектрах поглощения: фотоколориметрия в видимой части спектра (лабораторная работа); спектрофотометрия в ультрафиолетовой и видимой частях спектра.

4. 1. 1. АТОМНО-ЭМИССИОННАЯ СПЕКТРОСКОПИЯ. СПЕКТРАЛЬНЫЙ АНАЛИЗ ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ ЛЕГИРУЮЩИХ КОМПОНЕНТОВ В КОНСТРУКЦИОННОЙ СТАЛИ Для определения содержания легирующих компонентов в конструкционной стали одним из основных является метод атомной эмиссионной спектроскопии, основанный на атомарных спектрах излучения. Испускание света атомами происходит за счет изменения энергии атомов. Атомы могут обладать только строго определенными дискретными запасами внутренней энергии: Ео, Е 1, Е 2 и т. д. Это означает, что атомы не могут иметь энергию, промежуточную между Ео и Е 1 или между Е 2 и Е 1 и так далее. В невозбужденном, нормальном состоянии атомы обладают минимальной энергией Ео. Если атому сообщить дополнительную энергию, то атомы возбуждаются, то есть переходят на более высокий энергетический уровень: Е 1, Е 2 и так далее.
 АТОМ САМОПРОИЗВОЛЬНО ВОЗВРАЩАЕТСЯ В НОРМАЛЬНОЕ ИЛИ")
ЧЕРЕЗ ОЧЕНЬ КОРОТКОЕ ВРЕМЯ (~ 10 -8 С) АТОМ САМОПРОИЗВОЛЬНО ВОЗВРАЩАЕТСЯ В НОРМАЛЬНОЕ ИЛИ КАКОЕ-ЛИБО БОЛЕЕ НИЗКОЕ ВОЗБУЖДЕННОЕ СОСТОЯНИЕ. ОСВОБОЖДАЮЩАЯСЯ ПРИ ЭТОМ ЭНЕРГИЯ ΔЕ ИЗЛУЧАЕТСЯ В ВИДЕ СВЕТОВОГО КВАНТА: ГДЕ H – ПОСТОЯННАЯ ПЛАНКА; – ЧАСТОТА ЭЛЕКТРОМАГНИТНОГО ИЗЛУЧЕНИЯ. КАЖДОМУ ЕДИНИЧНОМУ ПЕРЕХОДУ ЭЛЕКТРОНА (ЕДИНИЧНОМУ ИЗЛУЧЕНИЮ КВАНТА ЭНЕРГИИ) СООТВЕТСТВУЕТ ОДНА СПЕКТРАЛЬНАЯ ЛИНИЯ В СПЕКТРЕ АТОМА.

ЭТАПЫ АНАЛИЗА Термообработка пробы. Чтобы атом излучал энергию необходимо вещество перевести в газообразное состояние и возбудить. Для щелочных и щелочно-земельных металлов необходима температура ~ 4000 о. С, для черных сплавов – 5000 -7000 о. С. Такие температуры достигаются в электрической дуге или в искре. Разложение электромагнитного излучения. Для этого применяют дифракционную решётку или призму Карно. Регистрация спектра. Различают три вида регистрации спектров: визуальную в видимой части спектра (в приборах стилоскопах); фотографическую (в приборах спектрографах); фотоэлектрическую (в приборах квантометрах). Для испарения пробы, возбуждения свечения и фотографирования спектра конструкционной стали используют спектрограф ИСП-28. На рисунке 1 показана оптическая схема спектрографа.

Спектрограф снабжён фотокамерой с кассетой. Конструкция прибора обеспечивает совмещение светочувствительного слоя фотопластинки с фокальной плоскостью объектива. Кассету можно перемещать по вертикали. Это даёт возможность снять несколько спектров на одной фотопластинке Оптическая схема спектрографа: 1 – электрическая дуга, 2 – щель прибора, 3 – коллиматор (поворотное зеркало), 4 – призма Карно, 5 – система оптических стёкол, 6 – фотопластинка
 открывают затвор щели и производят фотографирование спектра (время экспозиции 25")
После обжига (5 с) открывают затвор щели и производят фотографирование спектра (время экспозиции 25 с). Затем закрывают затвор щели и выключают генератор кнопкой «Стоп» . С помощью маховика переводят кассету на два деления. Меняют медный электрод, изменяют положение анализируемого образца и фотографируют второй раз. Чтобы на фотопластинке получить некоторое расстояние между спектрами различных образцов, кассету следует передвинуть на три деления (таблица 1).

Таблица 1 – Порядок съёмки спектров № пп Наименование образца Положение кассеты, мм 1 Стандартный образец 281 10 2 Стандартный образец 281 12 3 Стандартный образец 282 15 4 Стандартный образец 282 17 5 Стандартный образец 283 20 6 Стандартный образец 283 22 7 Исследуемый образец 25 8 Исследуемый образец 27 По окончании фотографирования спектров всех эталонов и анализируемых образцов кассету закрывают и вынимают. В фотокомнате её проявляют, закрепляют, промывают водой и просушивают

КАЧЕСТВЕННЫЙ АНАЛИЗ ПО ЭМИССИОННЫМ СПЕКТРАМ Задача качественного анализа сводится к отысканию линий определяемого элемента в спектре пробы. Принадлежность линии данному элементу устанавливается по длине волны и интенсивности линии. Однако общее число линий в спектре многих элементов очень велико: например, спектр тория насчитывает свыше 2500 линий, а спектр урана – более 5000. Нет необходимости определять длины волн всех спектральных линий в спектре пробы. Для целей качественного анализа необходимо установить наличие или отсутствие в спектре так называемых аналитических или последних линий. При уменьшении содержания элемента в пробе интенсивность линий элемента в спектре будет уменьшаться. При какой-то очень малой концентрации останется всего несколько линий. Это аналитические, последние линии, по которым обычно проводится качественный анализ. Последние линии хорошо изучены (таблица 2). Их длины волн и характеристику интенсивности можно найти в специальных таблицах и атласах спектральных линий.

Таблица 2 – Аналитические линии легирующих элементов в спектре конструкционной стали № пп Элемент Длина волны, Å Элемент Длина волны Å 1 Si 2506, 9 Fe 2507, 9 2 Cr 2667, 1 Fe 2684, 7 3 Mn 2933, 1 Fe 2926, 6 4 Ni 3050, 8 Fe 3055, 3 5 V 3110, 7 Fe 3116, 6 6 Mo 3170, 3 Fe 3175, 0 Качественный анализ проводят по фотографиям спектров, снятых на стеклянных пластинах. Для расшифровки спектров применяют прибор спектропроектор. В таблицах их часто отмечают индексами U 1, U 2, и т. д. Индекс U 1 показывает, что при возбуждении спектра в дуге эта линия исчезает последней, линия с индексом U 2 исчезает предпоследней и так далее.

ОБЩИЙ ВИД СПЕКТРОПРОЕКТОРА Спектропроектор предназначен для получения на экране увеличенного изображение спектра, зафиксированного на фотопластинке.

НА РИСУНКЕ 2 ПРЕДСТАВЛЕНА ОПТИЧЕСКАЯ СХЕМА СПЕКТРОПРОЕКТОРА Рисунок 2 – Оптическая схема спектропроектора 1 – источник света, 2 – конденсоры, 3 – зеркальная призма (поворотное зеркало), 4 - объективы, 5 – спектрограмма, 6 –стеклянный столик, 7 – экран с проекцией спектра

Атлас спектральных линий представляет собой набор планшеток. На каждой из планшеток приведена спектрограмма части спектра чистого железа, шкала длин волн, соответствующих этой части спектра и отмечены положения отдельных наиболее характерных линий различных элементов в этой области спектра (рисунок 3). В совокупности все планшетки атласа составляют полную спектрограмму железа. Атлас является своеобразной шкалой длин волн, с помощью которой определяют длины волн линий спектра анализируемого образца. Рисунок 3 – Вид спектра железа (планшетка № 15) в области 2990 – 3140 Ǻ
, в")
ВИДЕО ПРОЦЕССА vk. com/video-69626081_456239078 Рисунок 4 – Область спектра железа (планшетка № 15), в которой обнаруживают линию легирующего элемента никеля
 зависит от интенсивности спектральной")
КОЛИЧЕСТВЕННЫЙ АНАЛИЗ ПО ЭМИССИОННЫМ СПЕКТРАМ Концентрация компонента в пробе (С) зависит от интенсивности спектральной линии (I) в соответствии с уравнением Ломакина-Шайбе: I = а. Cβ или lg I = lg a + β lg C, где а и β – постоянные величины, не зависящие от концентрации. При фотографической регистрации эмиссионных спектров измеряемой величиной (количественной характеристикой интенсивности спектральной линии) является почернение фотоэмульсии S.

В определённом интервале почернений фотоэмульсии величина S линейно зависит от логарифма отношения интенсивностей спектральной линии. где I 0 и I – интенсивности световых потоков одного и того же источника света, прошедших через не засвеченные и засвеченные участки фотоэмульсии. Для аналитических целей используют линейную функцию, связывающую почернение фотоэмульсии и логарифм концентрации определяемого элемента: S = lg a + γ·β lg C + j, где γ и j – постоянные для данной фотоэмульсии. Видео процесса: vk. com/video-69626081_456239078

ОБЩИЙ ВИД МИКРОФОТОМЕТРА

В расчётах используют относительную интенсивность почернения гомологической пары ∆S: ∆S = Sопр – Sосновы, где Sопр – оптическая плотность почернения линии определяемого элемента; Sосновы – оптическая плотность почернения линии основы. Рисунок 5 – Оптическая схема микрофотометра: 1 – источник света; 2 – конденсор, 3 – микро объективы, 4 – щель прибора, 5 – фотопластинка со спектрограммой, 6 – поворотные зеркальные призмы, 7 – фотоэлемент, 8 – микро гальванометр

ОТЧЕТ ПО ЛАБОРАТОРНОЙ № Спектра № Образцов РАБОТЕ Результаты Sопр. Sсрав ΔS % Сод-е опр. 281 7 3 4 0. 11 321 0 -3 3 0. 08 1 2 № ст. Обр Mn, % Si, % Ni, % Mo, % Cr, % V, % 281 1, 8 0, 26 4, 47 0, 75 0, 18 0. 11 282 0, 4 0. 44 2, 93 0, 47 0, 33 0, 25 283 0, 63 0, 3 1, 72 0, 3 0, 75 0, 44

ВЫВОД Проведен качественный анализ атомно-эмиссионной спектроскопии выявления линии ванадия V. Проведен количественный анализ атомно-эмиссионной спектроскопии процентное содержание V= 0, 11% с ошибкой 0, 08% Атомно-эмиссионная спектроскопия или атомно-эмиссионный спектральный анализ — совокупность методов элементного анализа, основанных на изучении спектров испускания свободных атомов и ионов в газовой фазе. АЭС — самый распространённый экспрессный высокочувствительный метод идентификации и количественного определения элементов примесей в газообразных, жидких и твердых веществах, в том числе и в высокочистых. Он широко применяется в различных областях науки и техники для контроля промышленного производства, поисках и переработке полезных ископаемых, в биологических, медицинских и экологических исследованиях и т. д. Важным достоинством АЭС по сравнению с другими оптическими спектральными, а также многими химическими и физико-химическими методами анализа, являются возможности бесконтактного, экспрессного, одновременного количественного определения большого числа элементов в широком интервале концентраций с приемлемой точностью при использовании малой массы пробы.

4. 1. 2. МОЛЕКУЛЯРНО-АБСОРБЦИОННЫЙ МЕТОД АНАЛИЗА. СПЕКТРОФОТОМЕРИЯ Спектрофотомерия — оптический метод исследования газообразных, жидких и твердых веществ, основанный на определении интенсивности поглощения света веществом (абсорбционная спектрофотомерия) или интенсивности излучения им света (эмиссионная спектрофотомерия) в зависимости от длины волны. Спектрофотомерия – метод качественного или количественного определения состава вещества по его спектру.

Спектрофотомерию широко применяют для изучения строения и состава различных соединений (комплексов, красителей, аналитических реагентов и т. д. ), для качественного и количественного определения веществ (открытия следов элементов в металлах и сплавах). Приборы, которыми пользуются в Спектрофотомерии, называют спектрофотомер-метрами.

ЦЕЛИ И ЗАДАЧИ МЕТОДА Спектрофотомерия основана на измерении оптической плотности и процента пропускания световых потоков определенной длины волны через исследуемый раствор и эталон на спектрофотометре. Спектрофотометры применимы для анализа как одного вещества, так и систем, содержащих несколько компонентов. Кроме того, они позволяют работать как с окрашенными растворами, так и с бесцветными.

КЛАССИФИКАЦИЯ , СТРУКТУРА МЕТОДА Все спектрофотометрические методы основаны на взаимодействии атомов, молекул или ионов, входящих в состав анализируемого вещества, с электромагнитным излучением. Это взаимодействие проявляется в поглощении или испускании фотонов (квантов). В зависимости от характера взаимодействия пробы с электромагнитным излучением выделяют две группы методов - эмиссионные и абсорбционные. В зависимости от того, какие частицы формируют аналитический сигнал, различают методы атомной спектроскопии и методы молекулярной спектроскопии.
, видимой и УФ (Ультрафиолетовая спектроскопия) областях спектра. В")
Различают спектрофотомерию в ИК (Инфра-красная спектроскопия), видимой и УФ (Ультрафиолетовая спектроскопия) областях спектра. В качестве источников излучения применяют дейтериевую (или водородную) лампу (в УФ области) и вольфрамовую лампу накаливания или галогенную лампу (в видимой и ближней ИК областях). Применение Спектрофотомерии в УФ и видимой областях спектра основано на поглощении электромагнитного излучения соединениями, содержащими хромофорные и ауксохромные группы. Поглощение излучения в этих областях связано с возбуждением электронов.

В ИК области проявляются переходы между колебательными и вращательными уровнями. Среди частот колебаний молекул выделяют так называемые характеристические, которые практически постоянны по величине и всегда проявляются в спектрах химических соединений содержащих определенные функции группы (вследствие чего эти частоты иногда называют групповыми). Теория колебаний сложных молекул позволяет расчетным путем предсказать колебательный спектр соединений, то есть определить частоты и интенсивности полос поглощения.

ОСНОВНОЙ ЗАКОН Основным законом, на котором основан количественный спектрофотометрический анализ, является закон Бугера-Ламберта-Бера. Закон Бугера-Ламберта: говорит о том, что каждый слой однородного вещества поглощает равную долю падающего на него монохроматического излучения. Закон Бера: устанавливает связь между поглощением и концентрацией: поглощение моно-хроматического излучения прямо пропорционально концентрации поглощающего вещества.
. где -интенсивность света, прошедшего через")
Закон Бугера-Ламберта-Бера и понятие об оптической плотности раствора (D). где -интенсивность света, прошедшего через раствор. -интенсивность падающего на раствор свет. -молярный коэффициент поглощения. -концентрация окрашенного вещества, моль/л. -толщина слоя светопоглащающего раствора, см.

Закон Бугера-Ламберта-Бера — основной закон, описывающий поглощение света средой. Он связывает между собой интен-сивности Il света, прошедшего слой среды толщиной l, и исходного светового потока I 0. где показатель поглощения вещества.

ОБОРУДОВАНИЕ, ПРИБОРЫ кюветы

СПЕКТРОФОТОМЕТР Спектрофотометр - это прибор, который регистрирует и записывает (на бумаге или на экране компьютера) спектры поглощения веществ. Спектрофотометр Кюветы для Спектрофометра Спектрофотометр

КЛАССИФИКАЦИЯ СПЕКТРОФОМЕТРОВ Все спектрофотометры содержат следующие элементы: 1. Источник света. 2. Оптические элементы, такие как сфера, зеркала, линзы, световоды. 3. Устройство разложения отраженного от образца света в спектр. 4. Фотоэлектрический приемник. 5. Эти элементы объединены оптической схемой прибора, определяющей характер прохожде-ния света от источника до приемника.

ОБЩАЯ СХЕМА СПЕКТРОФОМЕТРА

ЭТАПЫ АНАЛИЗА 1. 2. 3. 4. 5. Подготовка спектрофотометра к работе. Подготовка исследуемого раствора. Подготовка чистого кювет. Выливаем в кювет исследуемый раствор. Затем ставим кювет с исследуемым раствором в спектрофометр, закрываем крышку у спектрофотометра и исследуем наш раствор.

ОШИБКИ МЕТОДА Большое значение в спектрофотометрии имеет чистота рабочих стенок кювет. Многие вещества обладают способностью адсорбироваться на поверхности и загрязнять стенки кювет. Это может вызвать большие ошибки при измерении оптических плотностей исследуемых растворов. Поэтому не следует оставлять надолго кюветы с испытуемыми растворами.

ПРЕИМУЩЕСТВА СПЕКТРОФОТОМЕТРОВ Лучшие результаты получают на спектрофотометрах. Спектрофотометрические методы позволяют работать в узкой области оптимального светопоглощения, а это значительно увеличивает чувствительность и точность количественного определения. Эти методы применимы как для анализа одного вещества, так и для анализа систем.

ПРИМЕНЕНИЕ МЕТОДА Спектрофотометрия широко применяется в клинических, биохимических, санитарногигиенических, судебно-медицинских и фармацевтических лабораториях для качественного и количественного анализа различного рода объектов биологического происхождения (сыворотка крови, спинномозговая жидкость, моча и др. ), лекарственных средств, продуктов питания и т. д. Измерение концентрации белков и нуклеиновых кислот. Определение концентрации различных лекарственных средств, имеющих характерные спектры поглощения.

ФОТОКОЛОРИМЕТРИЯ. ОПРЕДЕЛЕНИЕ СОДЕРЖАНИЯ НИКЕЛЯ В ЧЁРНЫХ СПЛАВАХ Для определения содержания никеля в черных сплавах применяют метод фотоколориметрии, который основан на изучении молекулярных спектров поглощения в видимой части спектра (380 -700 нм).

КОЛИЧЕСТВЕННЫЕ ЗАКОНЫ ПОГЛОЩЕНИЯ СВЕТА Если на светопоглощающий раствор направить световой поток интенсивностью Iо, то интенсивность светового потока, прошедшего через слой вещества, составит величину I (рис 1).

ОПТИЧЕСКАЯ ПЛОТНОСТЬ Величину, равную , называют оптической плотностью раствора D. Зависимость оптической плотности раствора от концентрации растворённого вещества определяется законом Бугера-Ламберта-Бера, который справедлив при монохроматическом излучении, соответствующем абсорбционному максимуму .

ЗАКОН БУГЕРА-ЛАМБЕРТА-БЕРА где молярный коэффициент поглощения света, соответствующий абсорбционному максимуму СМ – концентрация светопоглощающего раствора, моль/л; b – толщина светопоглощающего слоя, см. Молярный коэффициент поглощения равен оптической плотности одномолярного раствора при толщине поглощающего слоя 1 см.

ЭТАПЫ АНАЛИЗА 1. Проведение фотоколориметрической реакции. 2. Измерение оптической плотности раствора. 3. Расчёт концентрации никеля в анализируемой пробе сплава.

ПРОВЕДЕНИЕ ФОТОКАЛАМЕТРИЧЕСКОЙ РЕАКЦИИ Никель в чёрных сплавах находится в виде компонента твёрдого раствора. Чтобы определить концентрацию никеля методом фотоколориметрии, необходимо анализируемую пробу растворить и получить окрашенное соединение. При растворении в азотной кислоте никель переходит в раствор в виде катиона Ni + 4 HNO 3 Ni(NO 3)2 + 2 NO 2 + 2 H 2 O; Ni + 4 H+ + 2 N Ni 2+ + 2 NO 2 + 2 H 2 O.

Ионы никеля образуют с диметилглиоксимом в присутствии сильных окислителей растворимый комплекс красного цвета.

Фотоколориметрическую реакцию проводят в щелочной среде, и для предупреждения осаждения щелочью гидроксидов железа, марганца, алюминия и других металлов, входящих в состав пробы, прибавляют комплексообразователь – сегнетовую соль, в присутствии которой перечисленные компоненты не осаждаются.
 помещают в коническую колбу на")
ХОД АНАЛИЗА Навеску стали 0, 1 г (рис. 1) помещают в коническую колбу на 100 мл и растворяют при нагревании в 10 мл азотной кислоты (1: 3), затем разбавляют небольшим количеством воды (5 -10 мл). Полученный раствор переводят в мерную колбу на 100 мл, тщательно ополаскивая колбочку, в которой велось растворение, небольшими порциями дистиллированной воды (4 -5 раз). Затем доводят содержимое мерной колбы водой до метки. Полученный раствор в мерной колбе тщательно перемешивают. Если при растворении выпадает осадок угля, то содержимое мерной колбы отфильтровывают. Рисунок 1 – Навеска стали

ПОДГОТОВКА К КОЛОРИМЕТРИРОВАНИЮ Для колориметрирования одновременно готовят два раствора. Один будет называться рабочим, другой – холостым (раствором сравнения). Из мерной колбы на 100 мл, отбирают пипеткой по 5 мл в две мерные колбы на 50 мл. В каждую колбу прибавляют 10 мл 20 % раствора сегнетовой соли, 10 мл 5 % раствора щѐлочи и хорошо взбалтывают. При этом раствор должен обесцветиться (допускается слабожѐлтая окраска, которая не будет мешать колориметрированию). Затем прибавляют 10 мл 3 % раствора персульфата аммония (NH 4)2 S 2 O 8 в качестве окислителя и взбалтывают.
.")
В одну из колб прибавляют 10 мл щелочного раствора диметилглиоксима (это будет рабочий раствор). Обе колбы доводят до метки 5 % раствором щѐлочи и тщательно перемешивают.

ИЗМЕРЕНИЕ ОПТИЧЕСКОЙ ПЛОТНОСТИ РАСТВОРА Оптическую плотность окрашенного раствора измеряют на приборе фотоколориметре. Внешний вид прибора КФК-2 МП показан на рисунке 2. 1 - колориметрический блок; 2 1 2 - вычислительный блок; 3 - маховик для замены 4 фотоприемника; 4 - крышка кюветного 5 отделения; 5 - рычаг перевода кювет; 3 6 - маховик для замены 6 светофильтра. Рисунок 2 – Фотоколориметр КФК – 2 МП
 и")
Подсоединить фотоколориметр к сети 220 В, открыть крышку кюветного отделения 4 (рис. 2) и включить тумблер «Сеть» . При этом должна загореться сигнальная лампа. Нажать клавишу «Пуск» – на цифровом табло появляется мигающая запятая и горит индикатор «Р» . Если запятая не появилась, повторно нажать клавишу «Пуск» . Выдержать колориметр во включенном состоянии в течении 15 минут при открытой крышке кюветного отделения, затем не менее 5 минут при закрытой крышке. Х Рисунок 3 – фотоколориметр КФК-2 МП с открытой крышкой Р Рисунок 4 – Расположение кювет в кюветном отделении фотоколориметра: Р - рабочий раствор, Х - холостой раствор
.")
Порядок работы на фотоколориметре КФК-2 МП 1. Открыть крышку кюветного отделения 4 (рис. 2). В него установить кюветы (рис. 4) с рабочим исследуемым раствором и холостым раствором сравнения. 2. Установить необходимый светофильтр 6 (рис. 2) и нужный фотоприемник (сине-зелёный, λ=490 нм). 3. Рычаг 5 (рис. 2) установить в крайнее левое положение. При этом в световой пучок вводится кювета с холостым раствором сравнения. 4. При открытой крышке кюветного отделения нажать «Ш(0)» . На цифровом табло справа от мигающей запятой высвечивается значение N 0 (начало измерений), а слева – символ « 0» . 5. Закрыть крышку кюветного отделения, нажать «К(1)» – появится символ « 1» . 6. Рычаг 5 (рис. 2) установить в крайнее правое положение. В световой пучок вводится кювета с рабочим исследуемым раствором). 7. Нажать клавишу «Д(5)» . На цифровом табло слева от мигающей запятой появляется символ « 5» , означающий, что произошло измерение оптической плотности. Отсчёт на цифровом табло справа от мигающей запятой соответствует оптической плотности исследуемого раствора. Для сброса показаний нужно нажать клавишу «Пуск» , появится мигающая запятая.
 установить в крайнее левое положение.")
При повторении опыта необходимо: 1. Рычаг 5 (рис. 2) установить в крайнее левое положение. 2. Открыть крышку кюветного отделения 4 (рис. 2). 3. По истечении 5 секунд нажать «Ш (0)» . На цифровом табло справа от мигающей запятой высвечивается значение N 0 (начало измерений), а слева – символ « 0» . 4. Закрыть крышку кюветного отделения, нажать «К(1)» – появится символ « 1» . 5. Рычаг 5 (рис. 2) установить в крайнее правое положение. 6. Нажать «Д(5)» – появится результат оптической плотности. 7. Сбросить показания, нажав клавишу «Пуск» . Появится мигающая запятая. Опыт повторяют 2 -3 раза. 1) 0, 169 2) 0, 165 Сср = 0, 167 3) 0, 168 С ист - истинное значение

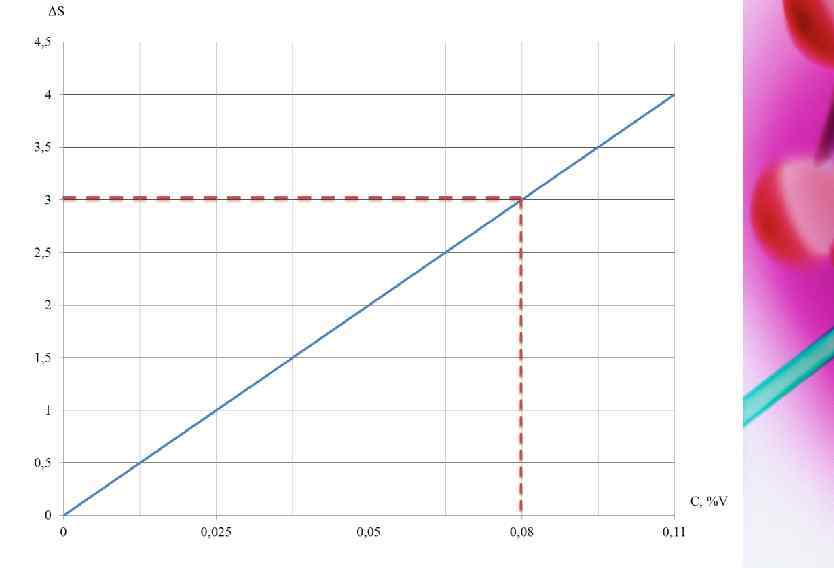
3. РАСЧЕТ КОНЦЕНТРАЦИИ НИКЕЛЯ В АНАЛИЗИРУЕМОЙ ПРОБЕ СПЛАВА Для этого измеряют оптическую плотность растворов минимум трёх стандартных образцов исследуемого сплава (стали, чугуна) строят градуировочный график в координатах оптическая плотность раствора D – массовая доля никеля (%) С (рис. 5). Измерение оптической плотности стандартных образцов анализируемой пробы проводят в одинаковых условиях. Рассчитав среднее значение D исследуемого образца из нескольких определений, по градуировочному графику находя массовую долю никеля в чѐрном сплаве Сх. Рисунок 5 – Градуировочный график D = f(C)


ВЫВОД Опытным путем определено содержание никеля в черных сплавах = 0, 167 с ошибкой 3 % используемый метод фотоколориметрии основан на изучении молекулярных спектров поглощения в видимой части спектра 490 нм.

4. 2. ЭЛЕКТРОХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА, ИХ КЛАССИФИКАЦИИ Классификация электрохимических методов анализа Электрохимические методы анализа и исследования основаны на изучении и использовании процессов, протекающих на поверхности электрода или в Прямые приэлектродном пространстве. Любой электрический параметр (потенциал, В прямых методах используют сила тока, сопротивление и др. ), зависимость силы функционально связанный с тока (потенциала концентрацией анализируемого раствора и т. д. ) от концентрации и поддающийся правильному определяемого измерению, может служить компонента. аналитическим сигналом. Методы анализа Косвенные В косвенных методах силу тока (потенциал и т. д. ) измеряют с целью нахождения конечной точки титрования определяемого компонента подходя щим титрантом, т. е. используют зависимость измеряемого параметра от объема титранта

ПОТЕНЦИОМЕТРИЯ Потенциометрические методы основаны на измерении разности потенциалов индикаторного электрода и электрода сравнения или, точнее, электродвижущих сил (ЭДС) различных цепей, поскольку экспериментально измеряется именно ЭДС, являющаяся разностью потенциалов. Равновесный потенциал индикаторного электрода связан с активностью и концентрацией веществ, участвующих в электродном процессе, уравнением Нернста: Е = Е° + R T/(n F) ln (аокис/авосст) Е = Е° + R T /(n F) ln ([окисл] үокисл /([восст] үвосст)), R - универсальная газовая постоянная, равная 8, 31 Дж/(моль. К); Т - абсолютная температура; F- постоянная Фарадея (96500 Кл/моль); n - число электронов, принимающих участие в электродной реакции; аокис, авосст - активности соответственно окисленной и восстановленной форм редокс-системы; [окисл] и [восст] - их молярные концентрации; үокис, үвосст - коэффициенты активности; Е° - стандартный потенциал редокс-системы. Подставляя Т = 298, 15 К и числовые значения констант в уравнение, получаем: Е = Е° + (0, 059 / n) lg (аокис/авосст) Е = Е° + (0, 059 / n) lg ([окисл] үокисл/([восст] үвосст))

Методы прямой потенциометрии основаны на применении уравнения Нернста для нахождения активности или концентрации участника электродной реакции по экспериментально измеренной ЭДС цепи или потенциалу электрода. Наибольшее распространение среди прямых потенциометрических методов получил метод определения р. Н, но создание в последнее время надежно работающих ионоселективных электродов значительно расширило практические возможности прямых методов. Показатель р. Н измеряют и методом потенциометрического титрования. Для определения р. Н чаще всего используют стеклянный электрод. Основными достоинствами стеклянного электрода являются простота работы, быстрое установление равновесия и возможность определения р. Н в окислительно-восстановительных системах. К недостаткам относятся хрупкость материала электрода и сложность работы при переходе к сильнощелочным и сильнокислым растворам. Кроме концентрации ионов водорода, прямым потенциометрическим методом с ионоселективными электродами можно определить содержание нескольких десятков различных ионов.

Потенциометрическое титрование основано на определении точки эквивалентности по результатам потенциометрических измерений. Вблизи точки эквивалетности происходит резкое изменение (скачок) потенциала индикаторного электрода. Так же, как и в других титриметрических методах, реакции потенциометрического титрования должны протекать строго стехиометрически, иметь высокую скорость и идти до конца. Для потенциометрического титрования собирают цепь из индикаторного электрода в анализируемом растворе и электрода сравнения. В качестве электродов сравнения чаще всего используют каломельный или хлорсеребряный электроды. Тип применяемого индикаторного электрода при потенциометрическом титровании зависит от свойств титриметрической смеси и ее взаимодействия с электродом. В кислотно-основном титровании используют стеклянный электрод, в окислительно-восстановительном - инертный (платиновый) электрод или электрод, обратимый по отношению к одному из ионов, содержащихся в титриметрической смеси; в осадительном – серебряный электрод; в комплексон метрическом - металлический электрод, обратимый к титруемому иону металла.

Для нахождения точки эквивалентности часто строят дифференциальную кривую в координатах DЕ/DV – V. На точку эквивалентности указывает максимум полученной кривой, а отсчет по оси абсцисс, соответствующий этому максимуму, дает объем титранта, израсходованн ого на титрование до точки эквивалентности. Определение точки эквивалентности до дифференциальной кривой значительно точнее, чем по простой зависимости Е – V.

К плюсам: Основными достоинствами метода потенциометрического титрования являются высокая точность и возможность проводить определения в разбавленных растворах, в мутных и окрашенных средах, а также определять несколько веществ в одном растворе без предварительного разделения. Значительно расширяется область практического применения потенциометрического титрования при использовании неводных растворителей. Они позволяют анализировать многокомпонентные системы, которые в водном растворе определить не удается, провести анализ веществ, нерастворимых или разлагающихся в воде, и т. д. Потенциометрическое титрование легко может быть автоматизировано. Промышленность выпускает несколько типов автотитраторов, использующих потенциометрические датчики. К недостаткам: потенциометрического титрования можно отнести не всегда быстрое установление потенциала после добавления титранта и необходимость во многих случаях проводить при титровании большое количество отсчетов.
 величина, характеризующая активность или концентрацию ионов водорода")
ОПРЕДЕЛЕНИЕ РН РАСТВОРА Водородный показатель (р. Н) величина, характеризующая активность или концентрацию ионов водорода в растворах. Водородный показатель численно равен отрицательному десятичному логарифму активности или концентрации ионов водорода, выраженной в молях на литр: p. H=-lg[ H+ ] В воде концентрация ионов водорода определяется электролитической диссоциацией воды по уравнению H 2 O=H++OH- Пренебрегая незначительной долей распавшихся молекул, можно концентрацию недиссоциированной части воды принять равной обшей концентрации воды, которая составляет: С[H 2 O ]=1000/18=55, 55 моль/л. Тогда: C[ H+ ] ·C[ OH- ]=K·C[H 2 O]=1, 8· 10 -16· 55, 55=10 -14

Для воды и ее растворов произведение концентраций ионов Н+ и ОН- величина постоянная при данной температуре. Она называется ионным произведением воды КВ и при 25° С составляет 10 -14. Постоянство ионного произведения воды дает возможность вычислить концентрацию ионов H+ если известна концентрация ионов OH- и наоборот: Понятия кислая, нейтральная и щелочная среда приобретают количественный смысл. В случае, если [ H+ ] =[ OH- ]эти концентрации (каждая из них) равны формула моль/л, т. е [ H+ ] =[ OH- ]=10 -7 моль/л и среда нейтральная, в этих растворах p. H=-lg[ H+ ]=7 и р. ОН=-lg[ OH-]=7 Если [ H+ ]>10 -7 моль/л, [ OH-]<10 -7 моль/л -среда кислая; р. Н<7. Если [ H+ ]<10 -7 моль/л, [ OH-]>10 -7 моль/л -среда щелочная; р. Н>7. В любом водном растворе р. Н + р. ОН =14, где р. ОН=-lg[ OH-]

Шрифт p. H и окраска универсального индикатора в разных средах.

ОПРЕДЕЛЕНИЕ РН РАСТВОРА

ОПРЕДЕЛЕНИЕ КОНЦЕНТРАЦИИ ИОНОВ МЕТАЛЛОВ Для вычисления р. Н раствор кислот и оснований следует предварительно вычислить молярную концентрацию свободных ионов водорода (Н+ ) или свободных гидроксил (ОН- ) , а затем воспользоваться формулами: p. H= -lg [Н+ ] ; p. OH= lg [ОН- ] ; p. H + p. OH =14 Концентрация любого иона в моль/л в растворе электролита можно вычислить по уровнению;

ЗАКЛЮЧЕНИЕ Дисциплина , , Методы контроля и анализа веществ” знакомит нас с аналитическим контролем и его методами. Рассмотрели электрические, тепловые, оптические и другие свойства веществ, а также методы их исследования. Основное внимание уделено их взаимосвязи и зависимости свойств от состава и структуры веществ. Приведены основные принципы устройств, позволяющие изучать изменение структуры и свойств материалов при изменении внешних параметров. Ознакомились с такими методами анализа, как титрометрический и гравиметрический, спектроскопические и атомно-эмиссионный, а так же молекулярно-абсорбционный и электрохимические.

СПИСОК ИСПОЛЬЗОВАННЫХ ИСТОЧНИКОВ 1. Васильев В. П. Аналитическая химия : учебник для вузов : в 2 кн. Кн. 1 : Титриметрические и гравиметрический методы анализа / В. П. Васильев. – 5 -е изд. , стер. – М. : Дрофа, 2005. – 367 с. : ил. – (Высшее образование). 2. Васильев В. П. Аналитическая химия : учебник для вузов : в 2 кн. Кн. 2 : Физико-химические методы анализа / В. П. Васильев. – 5 е изд. , стер. – М. : Дрофа, 2005. – 383 с. : ил. – (Высшее образование) 3. Атомно-эмиссионная спектроскопия. Определение легирующих компонентов в конструкционной стали [Электронный ресурс] : методические указания к выполнению лабораторной работы по дисциплинам : "Методы контроля и анализа веществ", "Аналитическая химия" и "Аналитическая химия и физикохимические методы анализа" / Сиб. гос. индустр. ун-т ; сост. : Ж. М. Шулина, О. Р. Глухова. – Электронные данные (1 файл). – Новокузнецк : Сиб. ГИУ, 2013. – Режим доступа: http: //library. sibsiu. ru.
![1. Определение содержания никеля в черных сплавах [Электронный ресурс] : практикум для проведения лабораторной](https://present5.com/presentation/-69626081_456382206/image-176.jpg "1. Определение содержания никеля в черных сплавах [Электронный ресурс] : практикум для проведения лабораторной")
1. Определение содержания никеля в черных сплавах [Электронный ресурс] : практикум для проведения лабораторной работы по дисциплинам : "Методы контроля и анализа веществ", "Аналитическая химия и физико-химические методы анализа" / Сиб. гос. индустр. ун-т ; сост. : Ж. М. Шулина, О. Р. Глухова, Р. М. Белкина. – Электронные данные (1 файл). – Новокузнецк : Сиб. ГИУ, 2014. – Режим доступа: http: //library. sibsiu. ru. 2. Кислотно-основное титрование. Определение щелочи в растворе [Электронный ресурс] : лабораторный практикум по дисциплинам «Методы контроля и анализа веществ» , «Аналитическая химия и физико-химические методы анализа» / Сиб. гос. индустр. ун-т ; сост. : Ж. М. Шулина, О. Р. Глухова, Р. М. Белкина, С. В. Зенцова. – Электронные данные (1 файл). – Новокузнецк : Сиб. ГИУ, 2015. – Режим доступа: http: //library. sibsiu. ru. 3. Окислительно-восстановительное титрование. Определение хрома в растворе дихромата [Электронный ресурс] : методические указания к выполнению лабораторной работы по дисциплинам «Методы контроля и анализа веществ» , «Аналитическая химия и физико-химические методы анализа» , «Аналитическая химия» / Сиб. гос. индустр. ун-т ; сост. : Ю. В. Бендре, С. В. Зенцова. – Электронные данные (1 файл). – Новокузнецк : Сиб. ГИУ, 2017. – Режим доступа: http: //library. sibsiu. ru

СПАСИБО ЗА ВНИМАНИЕ!
Obschee.ppt