Семиотика и наследственные болезни.ppt
- Количество слайдов: 43



Семиотика и наследственные болезни ПЛАН 1. Классификация Наследственных болезней. 2. Семиотика наследственной патологии. . 3. Признаки дисморфозов. 4. Генные и хромосомные болезни.

Классификация наследственных патологий Врожденные болезни не всегда наследственные, но не все наследственные врожденные. Семейные болезни могут быть наследственными и ненаследственными. - Генетическая классификация: Генные болезни; Хромосомные (хромосомные аберрации, геномные мутации); Генетические болезни соматических клеток; Болезни генетической несовместимости матери и плода;

Болезни с наследственной предрасположенностью: моногенные и полигенные. Болезни соматических клеток. Болезни, возникающие при несовместимости матери и плода. Клиническая классификация наследственных болезней( в основе системный и органный принцип): Нервные, нервно-мышечные, психические, болезни кожи и др.

Болезни с наследственной предрасположенностью: 1 группу составляют наследственные болезни не зависящие от среды. 2 группу – болезни, проявляющиеся специфических условиях. 3 группа – болезни, этиологическим фактором является среда, тяжесть зависит от предрасположенности. 4 группа – наследственность не играет роли. Генетические факторы влияют только на течение патологических процессов.

Семиотика наследственных заболеваний Семиотика изучает признаки наследственных болезней и патологических состояний, вызванных воздействием наследственных и средовых факторов. Особенности клинических проявлений В основе лежат генетические закономерности действия и взаимодействия. 1. СЕМЕЙНЫЙ ХАРАКТЕР ЗАБОЛЕВАНИЯ. (клинико-генеалогический метод). 2. ХРОНИЧЕСКОЕ ТЕЧЕНИЕ С ПРОГРЕДИЕНТНОЙ КЛИНИЧЕСКОЙ КАРТИНОЙ. Хронический процесс развивается в рез-те постоянного действия мутантного гена. Степень хронизации объясняется взаимодействием генов.

Специфические симптомы Наличие у больного редко встречающихся симптомов или их сочетаний дает основание думать о врожденной или наследственной причине заболевания: Грубые черты лица для больных мукополисахаридозом
 Непропорциональное соотношение тела и конечностей, низкий")
Астеническое телосложение с измененной грудной клеткой (синдром Морфана) Непропорциональное соотношение тела и конечностей, низкий рост, ахондроплазия (ахондроплазия) Право- и левосторонняя ассиметрия лица и конечностей (гемигипертрофия)
 Врожденные ненаследственные( краснушный, сифилитический,")
- Врожденный характер заболевания: Наследственные (хромосомные синдромы и др. ) Врожденные ненаследственные( краснушный, сифилитический, алкогольный синдром и др «Резистентность» к наиболее распространенным методам терапии ПРИ ОБЩЕМ КЛИНИЧЕСКОМ АНАЛИЗЕ ЛЮБОГО БОЛЬНОГО ПОСТАНОВКА ДИАГНОЗА ДОЛЖНА ЗАВЕРШИТЬСЯ ОДНИМ ИЗ ТРЕХ ЗАКЛЮЧЕНИЙ: 1. Четко поставлен диагноз ненаследственного заболевания. 2. Четко поставлен диагноз наследственной болезни. 3. Имеется подозрение, что основная или сопутствующая болезнь наследственная.

Врожденные пороки развития Морфологический дефект органа или большой части тела, ведущий к нарушению функций. - ВПР – являются следствием нарушения органогенеза. Морфогенез реализация генетической программы развития в трехмерном пространстве и во времени, осуществляемая под влиянием внешней среды. А. По степени распространенности в организме эмбриона: 1) изолированные, с поражением одного органа; 2) системные, с поражением нескольких органов одной сис темы; 3) множественные врожденные пороки развития, характери зуются поражением органов разных систем. В. По локализации: например, внутриутробные пороки разви тия центральной нервной системы, сердечно сосудистой системы, мочевыделительной системы и т. д. Врожденные пороки развития могут проявляться в виде: 1) отсутствия органа или части тела (агенезия, аплазия); 2) недоразвития органа (гипоплазия); 3) избыточного развития (гиперплазия), удвоения органа; 4) изменения формы органа (слияние, отрезая — заращение, стеноз — сужение} и его расположения (эктопия); 5) сохранения (персистенция) провизорных органов. -
 сформулировал и подтвердил теорию критических периодов развития. Каждый этап эмбриогенеза")
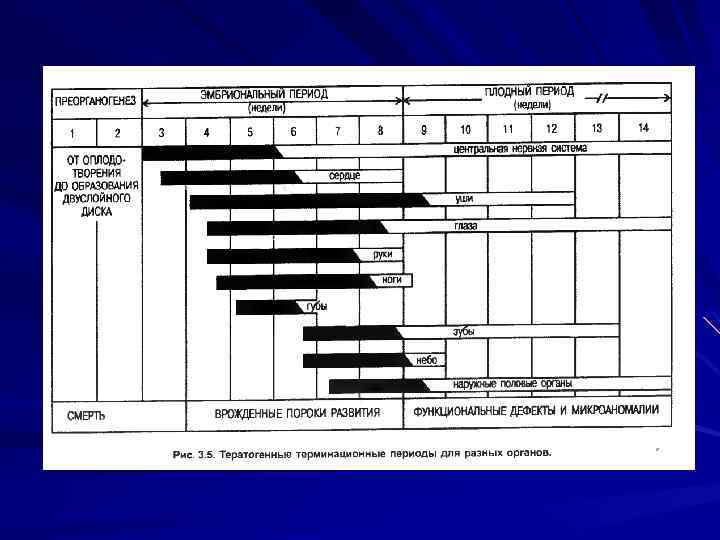
П. Г. Светлов (1960) сформулировал и подтвердил теорию критических периодов развития. Каждый этап эмбриогенеза начинается с периода качественной перестройки, сопровождающейся изменениями процессов детерминации, пролиферации и дифференцировки клеток. Выделяют периоды: устойчивости, максимальной чувствительности и снижения чувствительности к действию тератогена. Период устойчивости – 10 11 дней после оплодотворения. В этот период зародыш погибает или развивается нормально. Период максимальной чувствительности – 12 56 день развития (гисто органогенез); Период сниженной чувствительности (с 9 недели). В этот период в основном нарушаются темпы роста и дифференцировки тканей и органов плодов. Внутриутробный период развития человека имеет 6 критических периодов: 1. Оплодотворение; 2. Имплантация; 3. Развитие зачатков осевых органов зародыша и образование плаценты (3 8 недели); 4. Период интенсивного развития головного мозга (15 20 я неделя); 5. Период образования основных функциональных систем (20 24 я неделя); 6. Рождение.
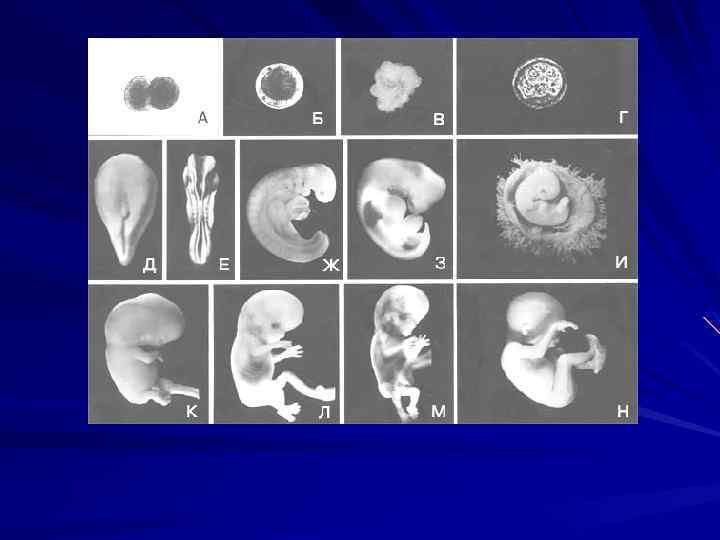

Аномалии в развитии бластулы

ВПР центральной нервной системы

Менингоцель
 1. Гаметопатии; 2. Бластопатии; 3. Эмбриопатии; 4.")
ПРЕНАТАЛЬНАЯ ПАТОЛОГИЯ( в зависимости от стадии онтогенеза) 1. Гаметопатии; 2. Бластопатии; 3. Эмбриопатии; 4. Фетопатии. Синдромы, обусловленные воздействием тератогенных факторов – заболевания обусловленные воздействием, как на эмбрион, так и на плод внешних, средовых патогенных факторов. Тератогенные факторы: Инфекционные агенты; Лекарства; Гормоны; Ионизирующая радиация; Механическое воздействие; Сосудистые расстройства; Антитела; Питание, возраст, количество родов; И другие факторы.

Воздействия внешних факторов возможны в условиях генетической предрасположенности. Синдромы ВПР: синдромы краснухи, талидомидовый, диабетический, алкогольный, фетопатии, обусловленные возбудителями внутриутробного токсоплазмоза, и др.
 являются составной частью")
Признаки дизморфогенеза в диагностике наследственной и врожденной патологии Признаки дисморфогенеза (ДМ) являются составной частью наследственных и врожденных болезней. Они встречаются по всем системам и весьма разнообразны по проявлениям. Большинство признаков ДМ нарушают функцию того органа, к которому они относятся (кожа, глаза, небо, конечность и др. ) Морфологические отклонения в развитии , которые выходят за пределы нормальных реакций, но не нарушают функции органа, в отличие от ВПР). Они являются неспецифическими признаками Эмбрионального ДМ, которые отражают небольшие отклонения в организме, либо наследственную патологию, либо отклонения, вызванные тератогенными факторами. Врожденные морфогенетические варианты у здоровых людей, но наличие нескольких признаков указывает на необходимость более внимательного обследования больного на предмет врожденной или наследственной патологии. Поскольку любое нарушение морфогенеза имеет диагностическое значение, при обследовании больных необходим внимательный осмотр на предмет выявления признаков ДМ.

Признаки дисморфозов

КОЖА: ангиомы, венозная сеть, пигментные пятна, депигментация, гипертрихоз, липомы, фибромы, келлоидные рубцы, повышенная растяжимость( складчатая вялая кожа), нарушения потоотделения, гиперкератоз.

2. Подкожная жировая клетчатка: избыточное отложение, уменьшенное количество. 3. Волосы: сухие, редкие, шерстистые, микроцефалия, седая прядь надо лбом «мыс вдовы» и др. 4. Череп: гидроцефалия, акроцефалия, выступающий затылок, плоский затылок и др.

6. Ушные раковины: анотия, макротия, деформированные Низко расположенные, оттопыренные назад, предушные фистулы и др.
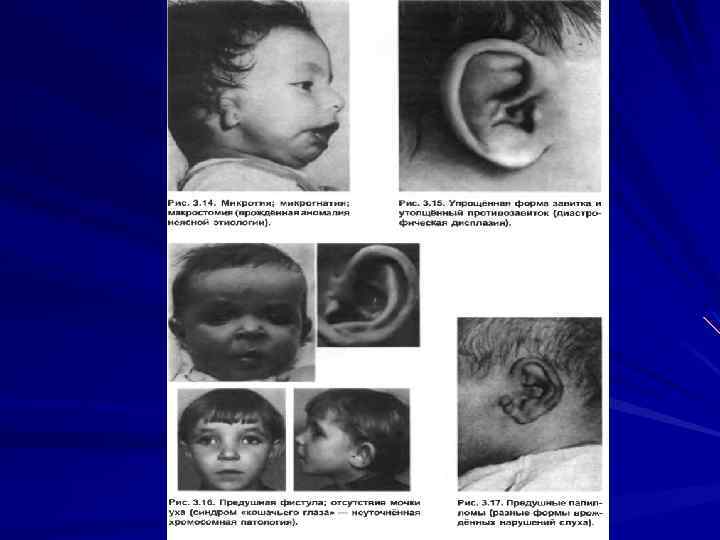

Лицо: круглое, плоское, треугольное, вытянутое, грубые черты.

8. Область глаз и глаза: антимонголоидный, монголоидный разрез глаз, эпикант, голубые склеры, косоглазие, короткая глазная щель, миопия и др.

9. Нос: коротки, клювовидный, седловидная переносица, широкая плоская переносица, плоские крылья носа, открытые вперед ноздри.

12. Губы и полость рта: макростомия, микростомия, губы тонкие , небо плоское, раздвоение язычка, короткая уздечка языка и др.
, или варусная деформация (О-образные), полидактилия, олигодактилия,")
16. Конечности: укороченные, удлиненные, вальгусная деформация (Х -образные), или варусная деформация (О-образные), полидактилия, олигодактилия, и т. д.

Генные болезни Фенилкетонурия Альбинизм Алкаптонурия Галактоземия Муковисцидоз Алкаптонурия Миодистрофия Дюшена Синдром фрагильной Х – хромосомы и др.

Генные болезни Нарушения аминокислотного обмена: ФЕНИЛКЕТОНУРИЯ и АЛЬБИНИЗМ Фенилкетонурия. Ген располагается 12 q 22 q 24. Аутосомно рецессивный тип наследования. Диагностика: Биохимический метод. Метод лечения: диетотерапия ( снижение содержания фенилаланина (ФА) с первых недель жизни, до 7 10 лет постоянно следить за содержанием ФА.

Ахондроплазия

Миодистрофия ДЮШЕНАБЕККЕРА Ген Хр21, Частота встречаемости: 1: 3500 Наследование сцеплено с полом. Диагностика: клиническая картина, биохимический метод.

Синдром умственной отсталости с ломкой Х . хромосомой Ген расположен в Х-q. Частота встречаемости 1: 1500 новорожденных мальчиков. Диагностика: цитогенетические методы. Описан терапевтический эффект

Мутантные аллели различных генов, которые контролируют синтез коллагена и фибриллина, нарушение свойств соединительной ткани, наблюдаются множественные влияния этих мутаций на клиническую картину (фенотип)

Синдром Элерса-Данло
 Частота встречаемости 1: 6000. Умеренная микроцефалия, низкий скошенный")
Хромосомные болезни Синдром ПАТАУ (трисомия 13) Частота встречаемости 1: 6000. Умеренная микроцефалия, низкий скошенный лоб, суженные глазные щели, помутнение роговицы, запавшее переносье, двустронняя расщелина губы, и т. д. Живут недолго, умирают до 3 лет.

Синдром Эдварса – трисомия 18. Частота встречаемости 1: 7000. Аномалии мозгового черепа, западение лобной кости, нижняя челюсть и отверстие рта маленькие, ушные раковины деформированы, наружный слуховой проход, мочка и козелка нет и др. Продолжительность жизни детей: до 60 % умирают до 3 мес. , до 1 года доживает 1 из 10.
 Частота встречаемости 1: 900. Округлая голова, уплощенный затылок, лоб скошен,")
Синдром Дауна (трисомия 21) Частота встречаемости 1: 900. Округлая голова, уплощенный затылок, лоб скошен, узкий, лицо плоское, плоская стенка носа, косой разрез глаз, и др. Умственная отсталость дебильность, идиотия. Продолжительность жизни ниже 36 лет.
. Делеция короткого плеча 5 хромосомы. Частота встречаемости")
Синдром «кошачьего крика» ( 5 q ). Делеция короткого плеча 5 хромосомы. Частота встречаемости 1: 45 000. Специфический плач, умственное и фичическое недоразвитие, деформированные ушные раковины, косоглазие, гипотония мышц и др. Продолжительность жизни снижена, около 14% переживает 10 лет.

Синдром Шерешевского Тернера Кариотип: 45, Х. Фенотип женский. Частота встречаемости 1: 800, 1: 1000. Рост 135 145 см. Крыловидная складка на шее, низкое расположение ушей, недоразвитие первичных и вторичных половых признаков, пороки развития сердца и др. Интеллект страдает редко.
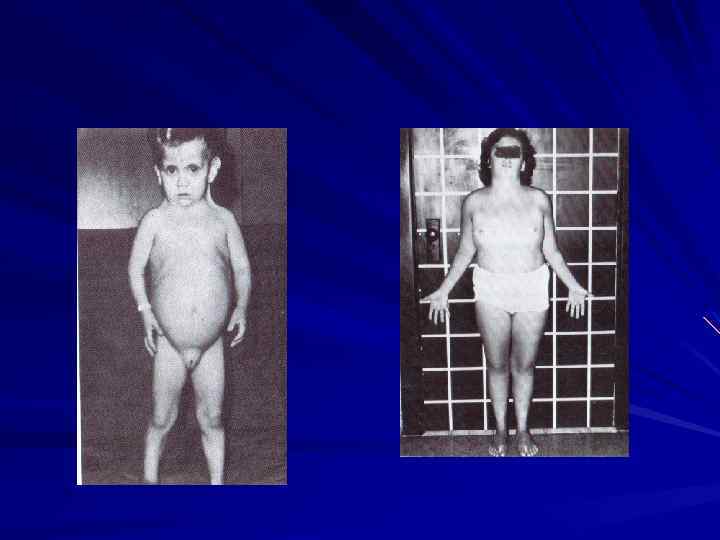

Синдром Клайнфельтера Кариотип – 47, ХХУ, 48, ХХХУ и др. Фенотип мужской, женский тип телосложения, гинекомастия, высокий рост , интеллект снижен, недоразвиты первичные и вторичные половые признаки, половые рефлексы сохранены. Иногда эффективно лечение гормонами. Зиготы УО, и ОО нежизнеспособны. ХУУ – больные имеют признаки с. Клайнфельтера, высокий рост, агрессивное поведение. Аномалии зубов, костной ткани. Половые железы развиты нормально.
Семиотика и наследственные болезни.ppt