dcb3060de94e9cf16e473a96744ed88c.ppt
- Количество слайдов: 76



Recombinant DNA Technology

Recombinant DNA Technology combines DNA from different sources – usually different species Utility: this is done to study DNA sequences to mass-produce proteins to give recipient species new characteristics as a therapy/curative for genetic disorders (‘gene therapy’) Human insulin, created in bacteria Corn damaged by corn borer and fungi “bt-corn”, with a bacterial gene

Recombinant DNA Technology A. Overview: 1. Purify DNA 2. Cut it with restriction enzymes – create a DNA ‘library’ of the fragments 3. Insert it into a vector, creating a recombinant DNA molecule 4. Insert the vector into a host cell 5. Create a population of cells (clone) that have this new DNA. 6. The DNA or protein product can be isolated and purified, or the new organisms with this cell type can be used.

Recombinant DNA Technology A. Overview: B. Creating a DNA Library - the more you know about the location of your gene of interest, the better; the LESS DNA you will have to manipulate.

Recombinant DNA Technology A. Overview: B. Creating a DNA Library - the more you know about the location of your gene of interest, the better; the LESS DNA you will have to manipulate. - Best case – know the location of the gene and can pinpoint it

Recombinant DNA Technology A. Overview: B. Creating a DNA Library - the more you know about the location of your gene of interest, the better; the LESS DNA you will have to manipulate. - Best case – know the location of the gene and can pinpoint it - know the chromosome it is on - Worst case – have to screen the entire genome.

Recombinant DNA Technology A. Overview: B. Creating a DNA Library - the more you know about the location of your gene of interest, the better; the LESS DNA you will have to manipulate. - Best case – know the location of the gene and can pinpoint it - know the chromosome it is on - Worst case – have to screen the entire genome. Three Ways: - cut up DNA with restriction enzymes and isolate gene - make copies of gene with PCR - use m-RNA to make copies of gene with reverse transcriptase

Recombinant DNA Technology A. Overview: B. Creating a DNA Library - the more you know about the location of your gene of interest, the better; the LESS DNA you will have to manipulate. - Best case – know the location of the gene and can pinpoint it - know the chromosome it is on - Worst case – have to screen the entire genome. 1. - cut it with a restriction enzyme that cuts at specific sequences and leaves specific “tails” (the fewer fragments the better!!!)

Recombinant DNA Technology A. Overview: B. Creating a DNA Library - the more you know about the location of your gene of interest, the better; the LESS DNA you will have to manipulate. - Best case – know the location of the gene and can pinpoint it - know the chromosome it is on - Worst case – have to screen the entire genome. 1. - cut it with a restriction enzyme that cuts at specific sequences and leaves specific “tails” (the fewer fragments the better!!!) - cut a ‘vector’ with the same restriction enzyme and ligate them with ligase…
… Absorbed by inducing a state")
The vector could be plasmid (for placement in bacteria)… Absorbed by inducing a state of ‘competence’ with calcium salts, and transforming the bacteria (uptake of exogenous DNA).
 for the manipulation and study")
…or a ‘yeast artificial chromosome’ – YAC - (“yak”) for the manipulation and study of eukaryotic genes… Bacterial plasmid that has had yeast centromeres, telomeres, and replication origins inserted. Eukaryotic gene is inserted into the plasmid, then the chromosome is “linearized” with Bam. HI and inserted into a yeast colony. Why insert a eukarytoic gene into yeast, rather than bacteria?

…or a virus, capable of infecting other cells with their new gene.

Identify cells that have absorbed a recombinant plasmid. - grow them on an ampicillin plate – only cells that have accepted a plasmid can grow - since the fragments insert into a functional gene for lactose metabolism, cells that have accepted a recombinant plasmid will be white (no lactose metabolism). - WHICH of THESE WHITE cells absorbed the gene of interest? ? ? (small library is better!)

Recombinant DNA Technology A. Overview: B. Creating a DNA Library - the more you know about the location of your gene of interest, the better; the LESS DNA you will have to manipulate. - Best case – know the location of the gene and can pinpoint it - know the chromosome it is on - Worst case – have to screen the entire genome. 1. - cut it with a restriction enzyme that cuts at specific sequences and leaves specific “tails” (the fewer fragments the better!!!) - cut a ‘vector’ with the same restriction enzyme and ligate them with ligase. OR 2. - use the Polymerase Chain Reaction (‘PCR’) to clone DNA.
 - in preparation, you have purified the")
PCR (Mullis – 1993 Nobel in Chemistry) - in preparation, you have purified the DNA of interest… it may be in very small amounts – even one cell or a DNA fragment Step 1: Denaturation of DNA: Heating the sample to 90 -95 o. C breaks the H-bonds, denaturing the DNA (separating the helices). ~ 1 min.

PCR - in preparation, you have purified the DNA of interest… it may be in very small amounts – even one cell or a DNA fragment Step 1: Denaturation of DNA: Heating the sample to 90 -95 o. C breaks the H-bonds, denaturing the DNA (separating the helices). ~ 1 min. Step 2: Anneal Primers: The key to PCR is that you must know something about the DNA sequence… or be able to guess. You create ss-DNA primers that anneal (h-bond) to complementary sequences in DNA when it is cooled down (50 -70 o. C)

PCR - in preparation, you have purified the DNA of interest… it may be in very small amounts – even one cell or a DNA fragment Step 1: Denaturation of DNA: Heating the sample to 90 -95 o. C breaks the H-bonds, denaturing the DNA (separating the helices). ~ 1 min. Step 2: Anneal Primers: The key to PCR is that you must know something about the DNA sequence… or be able to guess. You create ss-DNA primers that anneal (h-bond) to complementary sequences in DNA when it is cooled down (50 -70 o. C) Step 3: Extension (Polymerization): Use a thermally stable polymerase (Taq) to polymerize DNA. Taq is from Thermus One aquaticus – an archaean Cycle (2 -5 min. ) from hot springs – stable at high temps

PCR 4. Repeat Cycle: Heat up and denature, anneal, polymerize 3 hrs, ~ 25 cycles = 225 copies 33, 554, 432

PCR Benefits: - with correct primer, it is very sensitive and amplifies very specific sequences from very small samples - genetic testing - forensics - molecular paleontology

PCR Benefits: - with correct primer, it is very sensitive and amplifies very specific sequences from very small samples - genetic testing - forensics - molecular paleontology - primers can be used as PROBES that recognize sequences differing by single bases (alleles)

Recombinant DNA Technology A. Overview: B. Creating a DNA Library - the more you know about the location of your gene of interest, the better; the LESS DNA you will have to manipulate. - Best case – know the location of the gene and can pinpoint it - know the chromosome it is on - Worst case – have to screen the entire genome. 1. - cut it with a restriction enzyme that cuts at specific sequences and leaves specific “tails” (the fewer fragments the better!!!) - cut a ‘vector’ with the same restriction enzyme and ligate them with ligase. OR 2. - use the Polymerase Chain Reaction (‘PCR’) to clone DNA. OR 3. - create c-DNA (‘complementary DNA’) from the isolated m-RNA transcript, using reverse transcriptase.

Isolate m-RNA’s in eukaryotic cell. Use a “poly-T primer” to recognize the Poly-A tails of the m-RNA. Use reverse transcriptase to polymerize the DNA.
, which cuts out rest of")
Partially digest with Rnase. Add DNA Polmerase I (repair), which cuts out rest of RNA and polymerizes DNA across template. Add ligase to complete ds-DNA

You can also use “reverse transcription PCR” to amplify a particular c-RNA directly.

Benefits of a c-DNA Library: 1. The absence of introns means that vectors and bacteria can handle the size and structure of the eukaryotic c-DNA gene. 2. If you can localize the cell that is producing the protein of interest, then the library will only contain DNA of active (translated) genes – not ALL genes like in a whole genome library. 3. If made from m-RNA, you can amplify genes that are very low in productivity, and can amplify genes at different times of development and get a picture of gene activation.

Recombinant DNA Technology A. Overview: B. Creating a DNA Library Summary

Recombinant DNA Technology A. Overview: B. Creating a DNA Library C. Recover the clone of interest The white cells have absorbed recombinant plasmids… but which one has absorbed the fragment we want – the one with our gene of interest?

Probes are short sequences of DNA that will bind to the gene of interest. They are either radioactive or colorimetric. Often the probe is a stretch of c-DNA.

Recombinant DNA Technology and Genomics A. B. C. D. Overview: Creating a DNA Library Recover the clone of interest Analyzing/characterizing the DNA - create a restriction map Just know WHY a map of resitrction sites would be useful… So you can pick a restriction enzyme that cuts OUTSIDE the gen, and not WITHIN the gene….

Recombinant DNA Technology A. B. C. D. Overview: Creating a DNA Library Recover the clone of interest Analyzing/characterizing the DNA - create a restriction map - combine restriction mapping and clone isolation – Southern Blot - assessing gene activation – Northern Blot - DNA sequencing

Recombinant DNA Technology A. B. C. D. Overview: Creating a DNA Library Recover the clone of interest Analyzing/characterizing the DNA - create a restriction map - combine restriction mapping and clone isolation – Southern Blot - assessing gene activation – Northern Blot - DNA sequencing Sanger Method: Logic – DNA to be sequenced is denatured, and used as a template for the formation of new DNA. A Primer is used to begin synthesis. Synthesis is terminated by the incorporation of di-deoxy bases that lack a –OH on 3’ end. So, when they are incorporated, synthesis stops. Electrophoresis separates the fragments
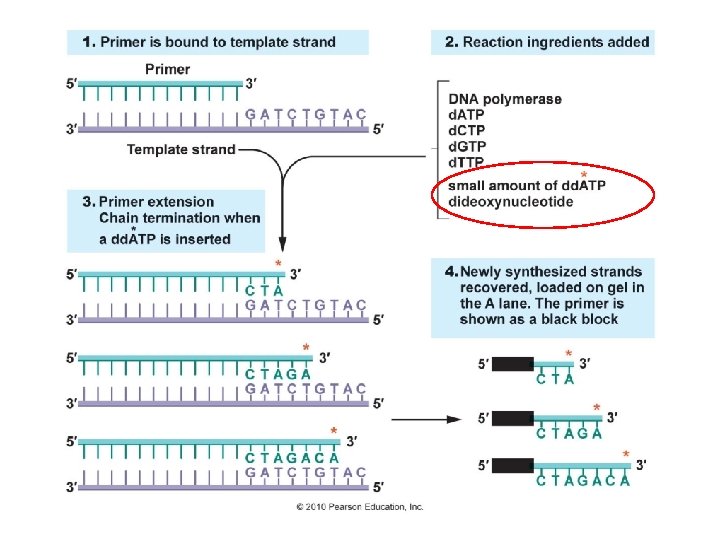

“bottom” of gel Different reaction tubes I know this was a bit confusing… there are two methods at work here. FIRST, you could do four separate reaction tubes, with a different dideoxy base in each one. Then, you run each sample in a different lane… reading the sequences off from the bottom of the gel (shortest fragment with one base beyond the primer) to the top (longest fragment). Or, in the next slide…. G A T C

One reaction tube “bottom” of gel You use ONE reaction vessel with all 4 dideoxy bases (in addition to all four regular bases, of course…). Each dideoxy bas has a different fluorescent tag on it, so that the fragments terminated by different dideoxy bases fluoresce a different color. There is just one reaction, and the fragments are all run in one ‘lane’ of a gel… discriminated by the different colors they fluoresce. G, A, T, C

Finally, in an additional modification described later in “contig analysis”, the gel can be made in long capillary tubes so longer sequences can be sequenced. A laser ‘reads’ the bands in the gel, recording the wavelengths of the reflected flourescent light - which indicates the last base added in the fragment. This is a readout from such an analysis. Each peak shows the fluorescence of a band in the gel that travelled to that position… the color denotes the fluorescent dideoxy that was placed in that position and terminated replication.

Genomics A. Overview: PHENOTYPE The history of understanding genome structure and function has been largely a “top-down” process, by correlating changes in the phenotype with the inheritance of a particular gene. - the down sides are: 1) can’t ‘cross’ most species 2) mutants can be very rare 3) mutations may be silent 4) building recombination maps is laborious GENE

Genomics A. Overview: PHENOTYPE The ability to sequence DNA allows for a bottom up approach – sequencing genes and then trying and figure out what they do at a phenotypic level. - the benefits are: 1) All species can be studied 2) The techniques are not difficult GENE

Genomics A. Overview: PHENOTYPE The ability to sequence DNA allows for a bottom up approach – sequencing genes and then trying and figure out what they do at a phenotypic level. - the benefits are: 1) All species can be studied 2) The techniques are not difficult - there are still significant hurdles: 1) most DNA is non-coding; finding genes is hard 2) linking a coding sequence to a function is difficult Knowing the sequence of A, T, C, G in a genome is just the beginning, and does not answer the fundamental question of how a genome encodes a phenotype. GENE

Genomics A. Overview: B. Sequencing: - Basically, you sequence the longest fragments of DNA that you can, by the methods we have described already. - Then, you enter the sequence in a computer, and you group together “contiguous sequences” (contigs) based on regions of overlap. Eventually, you cover the entire map.

Genomics A. Overview: B. Sequencing: - Clone-by-Clone Method: Once you have a restriction map of a chromosome, you can partially digest with different combinations of enzymes to make overlapping clones that are inserted into BAC’s or YAC’s or cosmids or plasmids and then replicated in cell clones and sequenced by the Sanger Method.

Genomics A. Overview: B. Sequencing: - Clone-by-Clone Method: Once you have a restriction map of a chromosome, you can partially digest with different combinations of enzymes to make overlapping clones that are inserted into BAC’s or YAC’s or cosmids or plasmids and then replicated in cell clones and sequenced by the Sanger Method. Reconstruction occurs by matching overlapping ‘contiguous sequences’. LABOR INTENSIVE – can only sequence ~300 bases/gel

Genomics A. Overview: B. Sequencing: - Whole Genome Shotgun: Initially the same approach; partial digests create overlapping fragments. High-throughput Automated Sequencers use dd-NTP’s and capillary tubes several feet long as the single lane ‘gels’, able to sequence 900 base fragments. The sequencer may run 96 capillary tubes at a time, sequencing nearly 2 million bases/day. Contig sequences analyzed by computer, and a sequence map is created.

Genomics A. Overview: B. Sequencing: C. Finding Genes – structural genomics and ‘annotation’: - once you have the sequence data, you really have just started. - The goals are then: - identify where genes are (Open Reading Frames) - find promoters and regulatory elements to confirm this is a gene (and not a pseudogene). - in eukaryotes, find splice sites, introns and exons - identify structural sequences like telomeres and centromeres - convert the DNA sequence into the predicted AA sequence of the protein - predict protein structure and function by identifying ‘domains’ and ‘motifs’ - These goals are attained by computer analyses of gene/AA sequence data, and comparison with known described genes. This is: BIOINFORMATICS

Genomics A. Overview: B. Sequencing: C. Finding Genes – structural genomics and ‘annotation’: 1. NCBI – BLAST search compares sequence to other sequences in the database 1 ggggcacccc tacccactgg ttagcccacg ccatcctgag gacccagctg cacccctacc 61 acagcacctc gggcctaggc tgggcggggg gctggggagg cagagctgcg aagaggggag 121 atgtggggtg gactcccttc cctcctcctc cccctctcca ttccaactcc caaattgggg 181 gccgggccag gcagctctga ttggctgggg cacgggcggctccccc tctccgaggg 241 gcagggttcc tccctgctct ccatcaggac agtataaaag gggcccgggc cagtcgtcgg 301 agcagacggg agtttctcct cggggtcgga gcaggaggca cgcggagtgt gaggccacgc 361 atgagcggac gctaaccccc tccccagcca caaagagtct acatgtctag ggtctagaca 421 tgttcagctt tgtggacctc cggctcctgc tcctcttagc ggccaccgcc ctcctgacgc 481 acggccaaga ggaaggccaa gtcgagggcc aagacgaaga cagtaagtcc caaacttttg 541 ggagtgcaag gatactctat atcgcgcctt gcgcttggtc ccgggggccg cggcttaaaa 601 cgagacgtgg atgatccgga gactcgggaa tggaagggag atgatgaggg ctcttcctcg 661 gcgccctgag acaggaggga gctcaccctg gggcgaggtt ggggttgaac gcgccccggg 721 agcgggaggt gagggtggag cgccccgtga gttggtgcaa gagagaatcc cgagagcgca 781 accggggaag tggggatcag ggtgcagagt gaggaaagta cgtcgaagat gggatggggg 841 cgccgagcgg ggcatttgaa gcccaagatg tagaagcaat caggaaggcc gtgggatgat 901 tcataaggaa agattgccct ctctgcgggc tagagtgttg ctgggccgtg ggggtgctgg 961 gcagccgcgg gaagggggtg cggagcgtgg gcgggtggag gatgagaaac tttggcgcgg 1021 actcggcggggtcct tgcgccccct gctgaccgat gctgagcact gcgtctcccg

Genomics A. Overview: B. Sequencing: C. Finding Genes – structural genomics and ‘annotation’: 1. NCBI – BLAST search compares sequence to other sequences in the database 2. Open Reading Frames: base sequences which would code for long stretches of AA’s before a stop codon would be reached. Typically, these are found by looking for [5’ – ATG…-3’] sequences that follow a promoter (TATA, CAAT, GGGCGG). The complement would be [3’ – TAC. . -5’], which would encode a start codon in RNA [5’- AUG… 3’]

Genomics A. Overview: B. Sequencing: C. Finding Genes – structural genomics and ‘annotation’: 1. NCBI – BLAST search compares sequence to other sequences in the database 2. Open Reading Frames: base sequences which would code for long stretches of AA’s before a stop codon would be reached. Typically, these are found by looking for [5’ – ATG…-3’] sequences that follow a promoter (TATA, CAAT, GGGCGG). The complement would be [3’ – TAC. . -5’], which would encode a start codon in RNA [5’- AUG… 3’] 3. Regulatory regions and splicing sites (GT-AG):
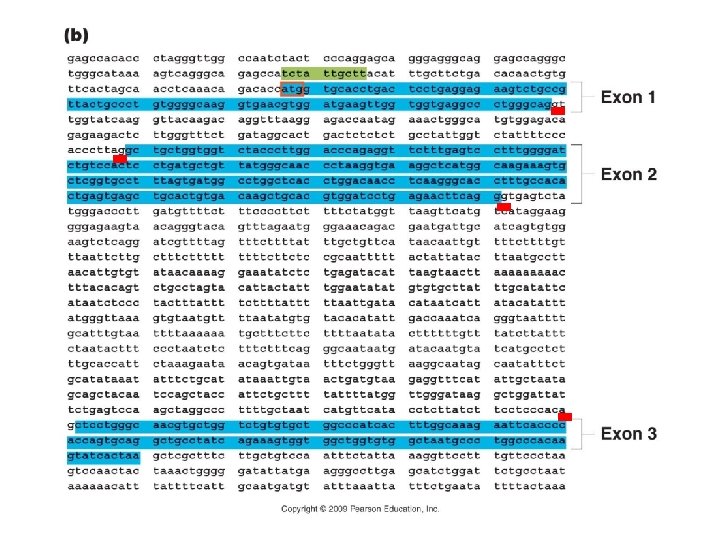

Genomics A. Overview: B. Sequencing: C. Finding Genes – structural genomics and ‘annotation’: D. Identifying Gene Function – functional genomics:

Genomics A. Overview: B. Sequencing: C. Finding Genes – structural genomics and ‘annotation’: D. Identifying Gene Function – functional genomics: - Sequence Homology: search libraries for similar sequences already described in other proteins with known function, in other species…. . Arabidopsis thalia

Genomics A. Overview: B. Sequencing: C. Finding Genes – structural genomics and ‘annotation’: D. Identifying Gene Function – functional genomics: - Sequence Homology: search libraries for similar sequences already described in other proteins with known function, in other species… or the same species

Genomics A. Overview: B. Sequencing: C. Finding Genes – structural genomics and ‘annotation’: D. Identifying Gene Function – functional genomics: - Sequence Homology: search libraries for similar sequences already described in other proteins with known function, even in other species. - Domain / Motif Analysis: Certain AA sequences are known to have a certain structure (‘motif’ like “helix-turn-helix”) or function (‘domain’ like an ion channel sequence, DNA binding region).

Genomics A. Overview: B. Sequencing: C. Finding Genes – structural genomics and ‘annotation’: D. Identifying Gene Function – functional genomics: - Sequence Homology: search libraries for similar sequences already described in other proteins with known function, even in other species. - Domain / Motif Analysis: Certain AA sequences are known to have a certain structure (‘motif’ like “helix-turn-helix”) or function (‘domain’ like an ion channel sequence, DNA binding region). - Mutant Analysis: Mutate the gene (insert a non-functional sequence) in vitro, then insert in cells and observe effects of “knocking out” function in different tissues or the whole organism.

Capecchi, Evans, and Smithies were awarded the 2007 Nobel Prize for their technique for inserting a gene into embryonic cells…this gene can be a mutant, non-functional gene (“knock-out”) or a functional gene (“knockin”). The goal here is to create an individual in which a particular gene has been ‘knocked-out’ in EVERY CELL of the body… and then see the effect on some aspect of the phenotype. This is an involved process that will be MUCH easier using CRISPR.

STEP 1: Extract some stem cells from an embryo, and transform them by inserting a mutant, screwed-up gene. The injected DNA will ‘cross-over’ with the normal, homologous gene in some cells. So, some cells will have a heterozygous genotype at this locus.

STEP 2: Inject transformed cells into embryos, and implant in a ‘surrogate’ mother. Offspring will have these transformed cells in different tissues, just dependent on where, randomly, the stem cells were during early development. These offspring are genetic mosaics, composed of cells with different genomes.

STEP 3: You screen the offspring, looking for those who, by luck, have transformed cells in their gonads. These mice - who are heterozygous for the transformed gene - will pass the mutation to ½ their gametes; so if you mate a male and female with transformed cells in their gonads, you will create some offspring that are homozygous for this mutation across their entire genome…. and you can see it’s effects. THAT WAS THE GOAL.

Genomics A. Overview: B. Sequencing: C. Finding Genes – structural genomics and ‘annotation’: D. Identifying Gene Function – functional genomics: E. General conclusions from these studies - there is remarkable homology in protein/gene sequence between species - physiological/developmental complexity is not correlated with genome size - only 2 -5% of human genome codes for proteins - although there are 100, 000 proteins, there are only 20, 000 genes… suggesting that most genes encode multiple proteins, produced through transcript and post-translational processing. - Most of the genome does NOT encode protein. However, large fractions of DNA do encode nc-RNA’s… “non-coding RNA’s” which are not translated but are produced by transcription and then exert a regulatory function (mi-RNA’s and others). - So, organisms with similarities in coding genes can be remarkably different…as a consequence of how the production of those proteins is regulated in different cell types and at different developmental periods.

PHEW!!!!

Recombinant DNA Technology combines DNA from different sources – usually different species Utility: this is done to study DNA sequences to mass-produce proteins to give recipient species new characteristics as a therapy/curative for genetic disorders (‘gene therapy’) Human insulin, created in bacteria Corn damaged by corn borer and fungi “bt-corn”, with a bacterial gene

Genomics Genetic Engineering A. To mass-produce proteins

Genomics Genetic Engineering A. To mass-produce proteins Making human insulin

Eukaryote genes may not be read properly by bacterial hosts because of introns and regulatory elements. In addition, the protein may not be processed correctly or fold correctly. Using a eukaryotic host solves these A. To mass-produce proteins problems… but tissue expression is the problem. Genomics Genetic Engineering A 1 -antitrypsin was the first; antithrombin is the first transgenic protein produced in animals to be approved by FDA for human use.

Genomics Genetic Engineering A. To mass-produce proteins B. To give species new characteristics The EPSP synthase gene in E. coli confers resistance to glyphosate – the primary ingredient in herbicides like Round-Up©.

Genomics Genetic Engineering A. To mass-produce proteins B. To give species new characteristics Agrobacterium is a plant pathogen that inserts Ti plasmids into host cells. These plasmids have been used as vectors for introducing the gene into plant tissues, which grow into new plants.

Genomics Genetic Engineering A. To mass-produce proteins B. To give species new characteristics Bacillus thuringiensis is a bacterium that produces a protein that crystallizes in insect guts, killing the insect. Since the 1930’s, the bacteria were sprayed on crops to reduce insect damage. The treatment was very short term, as the bacteria died quickly.

Genomics Genetic Engineering A. To mass-produce proteins B. To give species new characteristics Same process – splice to an Agrobacterium plasmid, with tissue-specific promoters.

Genomics Genetic Engineering A. To mass-produce proteins B. To give species new characteristics Issues: - genetic homogeneity of crop plants - 2011 study – toxin present in 93% of pregnant women in a town in Canada, and increases in immunological responses. - used as feed for animal stock - patterns of use and the evolution of resistance

Genomics Genetic Engineering Place gene for growth hormone from chinook salmon into Atlantic salmon, next to a constitutive promoter (gene always on, right? ) A. To mass-produce proteins B. To give species new characteristics Grow 10 x faster, to same mature size Models suggest it would outcompete native species if released into the wild

Genomics Genetic Engineering A. To mass-produce proteins B. To give species new characteristics C. Gene Therapy - Create a viral vector with a functional human allele – adenosine deaminase - Infect target tissue - Probably need to repeat unless you can transform stem cells

1990 -first trial of gene therapy – Ashanti De. Silva. 40 treated since then with 100% efficacy.

Genomics Genetic Engineering A. B. C. D. To mass-produce proteins To give species new characteristics Gene Therapy Gene Editing: CRISPR technology (pp. 527 -529) Bhaya, et al. Annu Rev Genet. 2011; 45: 273 -97. doi: 10. 1146/annurev-genet 110410 -132430. Bacteria and archaea have evolved defense and regulatory mechanisms to cope with various environmental stressors, including virus attack. This arsenal has been expanded by the recent discovery of the versatile CRISPR-Cas system, which has two novel features. First, the host can specifically incorporate short sequences from invading genetic elements (virus or plasmid) into a region of its genome that is distinguished by clustered regularly interspaced short palindromic repeats (CRISPRs). Second, when these sequences are transcribed and precisely processed into small RNAs, they guide a multifunctional protein complex (Cas proteins) to recognize and cleave incoming foreign genetic material. This adaptive immunity system, which uses a library of small noncoding RNAs as a potent weapon against fast-evolving viruses, is also used as a regulatory system by the host. Exciting breakthroughs in understanding the mechanisms of the CRISPR-Cas system and its potential for biotechnological applications and understanding evolutionary dynamics are discussed.

CRISPR – cas 9 system A. B. C. D. To mass-produce proteins To give species new characteristics Gene Therapy Gene Editing: CRISPR technology (pp. 527 -529)

USES: 1 – target specific gene sequences for cleavage with complementary ‘guide-RNA’ 2 – Replace cut sequence with another sequence… which may differ by just one base
 doi: 10. 1038/nature 23305")
H Ma et al. Nature 1– 7 (2017) doi: 10. 1038/nature 23305
")
Gene correction in M-phase-injected human embryos H Ma et al. Nature 1– 7 (2017) doi: 10. 1038/nature 23305

Oddly, the Cas-9 protein did not use the inserted DNA template for repair; rather, it used the homologous chromosome of the female. This type of repair occurs naturally in cells when mutations occur. https: //www. nytimes. com/2017/08/02/science/gene-editing-human-embryos. html
dcb3060de94e9cf16e473a96744ed88c.ppt