Молекулярные основы болезней обмена веществ.ppt
- Количество слайдов: 74




РАЗДЕЛ I ГЕННЫЕ МУТАЦИИ. МОЛЕКУЛЯРНЫЕ БОЛЕЗНИ
 уровне) ХРОМОСОМНЫЕ БОЛЕЗНИ")
МУТАЦИИ ГЕНОМНЫЕ ХРОМОСОМНЫЕ ГЕННЫЕ (изменение (мутации на количества структуры молекулярном хромосом) уровне) ХРОМОСОМНЫЕ БОЛЕЗНИ ГЕННЫЕ (МОЛЕКУЛЯРНЫЕ) БОЛЕЗНИ
- изменение химической структуры генов в результате нарушения последовательности нуклеотидов в")
Генные мутации (точковые, истинные)- изменение химической структуры генов в результате нарушения последовательности нуклеотидов в ДНК, что ведет к изменению наследственной информации в соответствующих фрагментах молекул нуклеиновых кислот
 ДЕЛЕЦИЯ; 2) ДУПЛИКАЦИЯ; 3) ИНВЕРСИЯ; 4) ИНСЕРЦИЯ; 5) ЗАМЕНЫ;")
Виды генных мутаций: 1) ДЕЛЕЦИЯ; 2) ДУПЛИКАЦИЯ; 3) ИНВЕРСИЯ; 4) ИНСЕРЦИЯ; 5) ЗАМЕНЫ;

ДЕЛЕЦИЯ – нормальная последовательность нуклеотидов делеция кратная трём выпадение одного кодона выпадение одной аминокислоты синтез нового белка выпадение 6 нуклеотидов

ДЕЛЕЦИЯ – выпадение отдельного нуклеотида делеция некратная трём сдвиг рамки считывания замена аминокислоты синтез нового белка

ДУПЛИКАЦИЯ – удвоение участка гена нормальная последовательность нуклеотидов удвоение участка гена

ИНВЕРСИЯ – поворот участка гена на 180 º нормальная последовательность нуклеотидов инверсия
 нормальная последовательность нуклеотидов вставка нуклеотида")
ИНСЕРЦИЯ (ВСТАВКА) нормальная последовательность нуклеотидов вставка нуклеотида

ЗАМЕНЫ ТРАНЗИЦИЯ замена одного пуринового на пуриновое основание или одного пиримидинового на пиримидиновое основание ТРАНСВЕРСИИ замена пуринового основания на пиримидиновое

Английский врач Гаррод в 1908 году предположил генетическую природу врожденных нарушений обмена веществ ГЕН ФЕРМЕНТ БИОХИМИЧЕСКАЯ РЕАКЦИЯ В это время было известно только 4 заболевания 50 – е гг. XX в. выделяется особая группа болезней – МОЛЕКУЛЯРНЫЕ БОЛЕЗНИ (вызваны дефектом на молекулярном уровне, нарушением в структуре молекулы ДНК)

ГЕННЫЕ БОЛЕЗНИ Моногенные болезни Гетерогенные болезни Факторы внешней среды Множественные Генокопии мутации локуса ( a a 1, a 2 … ) аутосомный тип наследования доминантный Полигенные заболевания Мультифакториальные заболевания сцепленное с полом наследование рецессивный XA Y Xa • гемофилия • талассемия • ФКУ • подагра • СКА • альбинизм • галактоземия • гликогенозы • гипоплазия эмали • рахит, устойчивый к вит. D • дальтонизм • миодистрофия Дюшенна • перламутровая форма ихтиоза

Классификация молекулярных болезней в зависимости от типа белка, подвергающегося изменению обусловлены нарушением синтеза структурных белков обусловлены нарушением синтеза белков – ферментов (ЭНЗИМОПАТИИ) • миодистрофия Дюшенна; • ФКУ; • синдром Марфана; • галактоземия; • нейрофиброматоз; • фруктоземия; • липидозы и др.

Классификация молекулярных болезней по патогенетическому принципу наследственные аномалии комбинированные болезни морфогенеза состояния обмена веществ (врожденные пороки развития) нарушение белкового (аминокислотного) обмена нарушение углеводного обмена нарушение липидного обмена нарушение обмена азотистых оснований

Классификация молекулярных болезней по клиническому принципу (с учетом систем или органов, наиболее вовлеченных в патологический процесс) • • • нервные; нервно – мышечные; кожные; глазные; опорно – двигательного аппарата; сердечно – сосудистой системы; мочеполовой системы; желудочно – кишечного тракта; легких; эндокринные; крови; психические

Общая частота новорожденных с генными болезнями в популяции ~ 1%: 0, 5 % – с аутосомно – доминантным типом наследования; 0, 25 % – с аутосомно – рецессивным типом; 0. 25 % – с Х – сцепленным типом; Y – сцепленные и митохондриальные болезни встречаются крайне редко

Распространенность отдельных форм болезней Распространенность колеблется от 1: 500 до 1: 100000 и ниже 1: 10 000 и чаще – высокая распространенность; 1: 10 000 – 1: 40 000 – средняя распространенность; очень редкие случаи – низкая распространенность В группу распространенных входит ок. 15 генных болезней, но они обусловливают почти 50 % общей частоты больных с наследственной патологией
 Умственная недостаточность")
Распространенность генных болезней Болезнь Первичные гемохроматоз Неполипозный рак толстой кишки (с. Линча) Умственная недостаточность с ломкой Х – хромосомой (с. Мартина - Белла) Муковисцидоз Спинальная мышечная атрофия Миотоническая дистрофия Распространенность Тип наследования 1 : 500 А - Р 1: 200 – 1 : 2 000 А - Д 1 : 1 250 (мальчики) 1: 2 500 (девочки) Х - сцепленный 1 : 1600 – 1; 3000 А - Р 1: 6000 А -Р 1: 8000 – 1: 10 000 А - Д 1: 3000 – 1: 5000 (мальчики) Р - Х - сцепленный 1: 5000 А -Д, А -Р, Р -Х- сцепленнный 1: 10 000 – 1: 15 000 А -Д, А -Р, Р -Х- сцепленнный Фенилкетонурия 1: 10 000 А -Р Ахондроплазия 1: 100 000 А -Д Миопатия Дюшенна -Беккера Синдром Элерса - Данло (все формы) Синдром Марфана

РАЗДЕЛ II ЭНЗИМОПАТИИ – ПРИЧИНА НАРУШЕНИЙ МЕТАБОЛИЧЕСКИХ ПРОЦЕССОВ
 БЛОК 1) ТОКСИЧЕСКОЕ ДЕЙСТВИЕ НАКАПЛИВАЮЩИХСЯ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ ОБМЕНА; 2) ОТСУТСТВИЕ ПРОДУКТОВ РЕАКЦИИ")
МЕТАБОЛИЧЕСКИЙ (ЭНЗИМАТИЧЕСКИЙ) БЛОК 1) ТОКСИЧЕСКОЕ ДЕЙСТВИЕ НАКАПЛИВАЮЩИХСЯ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ ОБМЕНА; 2) ОТСУТСТВИЕ ПРОДУКТОВ РЕАКЦИИ

НАРУШЕНИЕ АМИНОКИСЛОТНОГО ОБМЕНА • ФЕНИЛКЕТОНУРИЯ • АЛКАПТОНУРИЯ • АЛЬБИНИЗМ • БОЛЕЗНЬ КЛЕНОВОГО СИРОПА • ЦИСТИНОЗ и др.
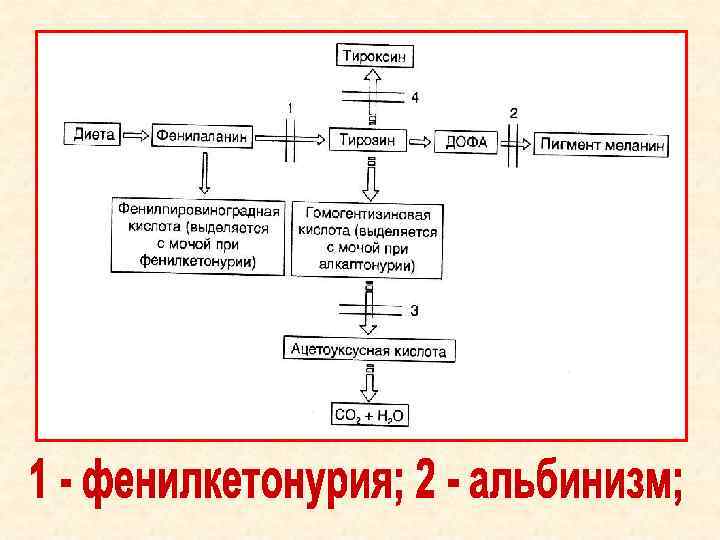
 Причина: невозможность превращения фенилаланина в тирозин Наследуется по аутосомно")
ФЕНИЛКЕТОНУРИЯ (фенилпировиноградная олигофрения/ б. Фёллинга) Причина: невозможность превращения фенилаланина в тирозин Наследуется по аутосомно - рецессивному типу Частота встречаемости: 1: 5000 – 1: 10000 Поражаются лица обоего пола, но девочки несколько чаще

Обмен фенилаланина в норме

Схема нарушения обмена при тяжелой форме фенилкетонурии

Схема нарушений обмена при классической фенилкетонурии
 2) ребенок при рождении выглядит здоровым симптомы болезни появляются ко второму")
Клиническая картина: 1) 2) ребенок при рождении выглядит здоровым симптомы болезни появляются ко второму полугодию жизни: • • • беспокойство; рвота; экзематозные изменения кожи; судороги; отставание в умственном развитии; задержка моторного развития (поздно начинают сидеть и ходить) 3) поза больного и походка своеобразны: (мышечная гипертония) • 4) сидят в “положении портного”, поджав ноги (мышечная при ходьбе делают маленькие шаги, покачиваются пот больных имеет неприятный (“заплесневелый”, “мышиный”, “волчий” запах; обусловлен метаболитами фенилпировиноградной кислоты) 5) у большинства волосы светлые, глаза голубые, кожа почти полностью лишена пигментации
 светлая кожа и волосы; 2) через лишенную пигментации радужную оболочку")
АЛЬБИНИЗМ Клинические проявления: 1) светлая кожа и волосы; 2) через лишенную пигментации радужную оболочку просвечивает красноватая сосудистая сеть
 Клинические признаки: 1) моча при стоянии приобретает темно – коричневый")
АЛКАПТОНУРИЯ (б. темной мочи) Клинические признаки: 1) моча при стоянии приобретает темно – коричневый цвет; 2) потемнение кончика носа, кончиков ушей, склер глаз 3) в более позднем возрасте – артропатия

НАРУШЕНИЕ УГЛЕВОДНОГО ОБМЕНА • ГЛИКОГЕНОЗЫ; • ГАЛАКТОЗЕМИЯ; • МУКОПОЛИСАХАРИДОЗЫ и др.

ГЛИКОГЕНОЗЫ - группа наследственных нарушений углеводного обмена; - наследуются по аутосомно – рецессивному типу; - частота встречаемости : 1: 68 000; - встречается одинаково часто как среди мальчиков, так и среди девочек; - обусловлены дефицитом ферментов, участвующих в процессах синтеза или распада гликогена; - характеризуются избыточным накоплением гликогена в органах и тканях; - выделяют 13 типов гликогенозов ( в зависимости от энзимного дефекта)

Схема мобилизации гликогена
 накопление гликогена")
Недостаточность глюкоза – 6 – фосфатазы ( I тип – б. Гирке) накопление гликогена в печени и почках гепатомегалия, увеличение размеров почек Гипогликемия, ацидоз вялость, апатия, повышение аппетита вторичное нарушение жирового обмена – гиперлипидемия отложение жира в подкожной жировой клетчатке и внутренних органах ВНЕШНИЙ ВИД БОЛЬНОГО круглое «кукольное» лицо, маленький рост, короткие конечности и шея, значительно увеличенный в размерах живот
 •")
Недостаточность амило – 1, 6 – глюкозидазы (III тип – б. Форбеса _Кори) • • накопление гликогена в печени, мышцах, сердце; гепатомегалия, кардиомегалия, мышечная слабость Недостаточность фосфорилазы в мышцах (V - б. Мак – Ардля) • • накопление гликогена в мышцах; мышечная слабость, боли в мышцах и судороги при физической нагрузке; Недостаточность фосфорилазы в печени (VI - б. Херса) • • • избыток гликогена накапливается в печени, скелетных мышцах; выраженная гепатомегалия; гипогликемия и гиперлипидемия развиваются редко;
 наиболее злокачественная форма")
Недостаточность кислой α – глюкозидазы (II тип – генерализованный гликогеноз/б. Помпе) наиболее злокачественная форма гликогенозов; составляет почти 10 % всех гликогенозов единственная лизосомная болезнь из глиогенозов, другие варианты гликогенозов связаны с дефектом ферментов, локализованных в цитоплазме α – глюкозидаза – гликогенрасщепляющий фермент лизосом

Лизосомальный механизм

Препарат печени при дефиците лизосомальной кислой α – глюкозидазы: повсюду видны “аномальные лизосомы” (плотные, заполненные гликогеном темные частицы), отсутствует цитоплазматический гликоген

Клинические проявления: распространенное отложение гликогена в печени, почках, сердечной мышце, скелетной мускулатуре и др. отложение гликогена в печени Гепатомегалия в сердечной мышце кардиомегалия, сердечная недостаточность в межреберных мышцах и диафрагме пневмонии, дыхательная недостаточность ВНЕШНИЙ ВИД круглое пастозное лицо, увеличенный язык, мышечная гипотония, задержка роста; высокая летальность

ГАЛАКТОЗЕМИЯ - наследственная энзимопатия, передается по аутосомно - рецессивному типу; - частота заболевания 1: 50000 - в основе лежит нарушение превращения галактозы в глюкозу
 начинается сразу после рождения или позднее; 2) больные дети плохо переносят")
Клиническая картина: 1) начинается сразу после рождения или позднее; 2) больные дети плохо переносят молоко и рано отказываются от груди; 3) после кормления молоком – обильная рвота, иногда поносы; 4) гипогликемия; гипергалактоземия 5) увеличение печени, желтуха; 6) расширение поверхностных вен живота, асцит; 7) токсическое действие галактозы на мозг, отек мозга 8) катаракта; 9) умственная отсталость; задержка роста 10) у нелеченых больных прогноз неблагоприятный

Швейцарский вариант - отсутствие поражения печени; - патологические изменения связаны с высокой концентрацией галактозы в крови и тканях
 Основные ферментативные дефекты при МПС - нарушение метаболизма ГАГов")
МУКОПОЛИСАХАРИДОЗЫ (> 14 видов) Основные ферментативные дефекты при МПС - нарушение метаболизма ГАГов

Клинические проявления МПС

Гаргоилизм: • большая голова; • внутренние углы глаз далеко отстоят друг от друга; • нос широкий, основание носа погружено; • шея короткая; • увеличение печени и живота; • изуродованный, деформированный скелет

Типичный вид больного с с. Гурлера Рентгенограмма кисти больного с с. Гурлера

Больной с с. Гурлера – Шейе: интеллект сохранен, скованность в суставах рук и ног
 энзимопатии, наследуемые по аутосомно – рецессивному типу;")
НАРУШЕНИЕ ЛИПИДНОГО ОБМЕНА ЛИПИДОЗЫ (болезни накопления липидов) энзимопатии, наследуемые по аутосомно – рецессивному типу; связаны с накоплением липидов в различных органах и тканях (состав накапливаемых липидов при отдельных заболеваниях различен); • примеры : болезнь Гоше болезнь Тея – Сакса – Шаффера болезнь Нимана – Пика и др.

Болезнь Гоше – в селезенке, печени, костях откладывается цереброзид (болезненность костей, гепатоспленомегалия, повреждение костного мозга, изменения ЦНС, вплоть до исчезновения нейронов в спинном мозге) Болезнь Тея - Сакса - Шаффера – Шаффера накопление ганглиозидов в нервной системе (нарушение пихического и умственного развития, судороги, слепота) Болезнь Нимана – Пика – накопление сфингомиелина в печени, селезенке, костном мозге, лимфатических узлах, коже. В начале проявления схожи с б. Тея – Сакса – Шаффера, Мальчик 11 лет. затем полная слепота, глухота, увеличение Грубые черты лица, макулопапулезные Гигантские клетки в костном мозге, печени, живота, высыпания на коже, содержащие липоид желто – коричневая окраска кожи. ). (сфингомиелин) гепатомегалия
 пуриновые")
НАРУШЕНИЕ МЕТАБОЛИЗМА ПУРИНОВЫХ ОСНОВАНИЙ ПОДАГРА (аутосомно – доминантный тип наследования с неполной пенетрантностью) пуриновые основания нуклеопротеидов ксантиноксидаза мочевая кислота выпадение кристаллов и отложение их в суставах деформация сустава и нарушение функций
 РАХИТ, РЕЗИСТЕНТНЫЙ К ВИТАМИНУ D • наследуется по –")
НАРУШЕНИЕ ВИТАМИННОГО ОБМЕНА ГЕНУИННЫЙ (ПЕРВИЧНЫЙ) РАХИТ, РЕЗИСТЕНТНЫЙ К ВИТАМИНУ D • наследуется по – Х – сцепленному доминантному типу; • вызван дефектом фермента, осуществляющего синтез активной формы витамина D; • клинически характеризуется грубыми дефектами костей; ребенок резко отстает в росте и развитии костей

Грубые деформации костей при рахите, резистентном к витамину D

РАЗДЕЛ III МОЛЕКУЛЯРНЫЕ БОЛЕЗНИ, ОБУСЛОВЛЕННЫЕ НАРУШЕНИЕМ СИНТЕЗА СТРУКТУРНЫХ БЕЛКОВ
; • МУКОВИСЦИДОЗ; • СИНДРОМ")
• СЕРПОВИДНО – КЛЕТОЧНАЯ АНЕМИЯ; • ТАЛАССЕМИЯ (АНЕМИЯ КУЛИ); • МУКОВИСЦИДОЗ; • СИНДРОМ МАРФАНА; • СИНДРОМ ЭЛЕРСА – ДАНЛОСА; • МИОДИСТРОФИЯ ДЮШЕННА – БЕККЕРА; И ДР.
 • была описана J. Herrick в")
СЕРПОВИДНО – КЛЕТОЧНАЯ АНЕМИЯ (ДРЕПАНОЦИТОЗ / S- ГЕМОГЛОБИНОПАТИЯ) • была описана J. Herrick в 1910 году • распространена у жителей – стран тропической Африки, – бассейна Средиземного моря, особенно на ближнем Востоке, – некоторых районах Индии • на генетическом уровне у мутантного гена, кодирующего одну из цепей глобина, замена одного нуклеотида и в м – РНК происходит замена аденина на уроцил • на молекулярном уровне – замена в β – цепи глутаминовой кислоты на валин

Мутация гена β - глобина Первичные биохимические изменения Аномальный глобин Аномальный Hb S Первичные физиологические эффекты СЕРПОВИДНЫЕ Er уменьшение продолжительности жизни серповидных Er агглютинация серповидных Er и серповидных нарушение кровообращения агглютинация клеток в селезенке Анемии Нарушение притока крови Вторичные физиологические пролиферация к желудку, увеличение к сердцу эффекты к легким клеток сердца костного мозга кишечнику к мышцам, суставам к мозгу к почкам увеличение массы костного мозга почечная паралич ревматизм Внешние нарушение недостаточность фенотипические физических и сердечной аномалии боли пневмонии проявления развития костей психических деятельности в увеличение функций животе селезенки
 эритроциты при СКА")
Форма эритроцитов в норме Дрепаноциты (серповидные) эритроциты при СКА
 нарушение синтеза цепей α, β – цепей глобина в результате наследственного")
ТАЛАССЕМИЯ (АНЕМИЯ КУЛИ) нарушение синтеза цепей α, β – цепей глобина в результате наследственного дефекта т - РНК Мишеневидные эритроциты
 - наследуется по аутосомно – рецессивному типу; - ген муковисцидоза локализован")
МУКОВИСЦИДОЗ (кистозный фиброз) - наследуется по аутосомно – рецессивному типу; - ген муковисцидоза локализован 7 q 31 – 32; - в гене муковисцидоза обнаружено около 1000 мутаций , из них 200 – 300 дают патологический эффект; - частота: в Европе – 1: 2500; в восточных популяциях и у африканского чернокожего населения – 1: 100 000 - гетерозиготы по муковисцидозу устойчивы к туберкулезу

Патогенез муковисцидоза ПАТОЛОГИЧЕСКИЙ ГЕН отсутствие трансмембранного регулятора проводимости нарушение транспорта хлоридов в эпителиальных клетках инфекции

Патогистологическая картина поджелудочной железы: кисты и расширенные протоки

Патогистологическая картина легкого: бронхоэктазы со слизистым воспалительным экссудатом в просвете

СИНДРОМ МАРФАНА (арахнодактилия/ с. “паучьих пальцев” - впервые описан Вильямсом в 1876 г; - в последующие годы наблюдался Марфаном (1896), - наследуется по аутосомно – доминантному типу; - связан с нарушением синтеза фибриллина (кодируется геном фибриллина 1, картированным в локусе 15 q 21. 1); - частота в популяции 1: 10000 -1: 15000; - возможна мутация de novo

Основные клинические проявления С. Марфана Изменение костно - суставной системы Изменения Патология Смешанные дефекты ССС глаз мягких тканей (40 -60%) удлиненные конечности, дефекты длинные перегородок пальцы ("паучьи") скудный слой изменение подкожной жировой хрусталика клетчатки разболтанно дефекты сть суставов клапанов миопия плоскостопи е катаракта килевидная грудная клетка аномалии в строении аорты косоглазие грыжи большие некрасивые уши

Новорожденный с с. Марфана

МИОДИСТРОФИЯ ДЮШЕННА – БЕККЕРА • вызвана мутацией в гене, ответственном за синтез белка дистрофина (участвует в поддержании целостности мембран) Гистохимическая картина мышц в норме и при миопатии Дюшенна

• тип наследования: Х – сцепленнно рецессивный; • ген дистрофина располагается в коротком плече Х - коротком плече хромосомы; • это самый длинный из всех ныне изученных генов; • в нем > 2*106 пар оснований; • в связи с большой длиной в гене часто возникают перестройки, ведущие к мутациям • доля свежих мутаций – 30%

Клиническое разделение: 1. Миодистрофия Дюшенна: – первые симптомы появляются в возрасте 2– 3 лет; – дети поздно начинают ходить; – “утиная походка, ” псевдогипертрофия икроножных мышц; – в более позднем возрасте атрофия мышц распространяется до кисти; – наблюдается псевдогипертрофия внутренних органов

Миодистрофия Дюшенна Лордоз, псевдогипертрофия икроножных мышц, атрофия бедренных мышц

Трудности при распрямлении после наклона
 Миодистрофия Беккера: Ø по клиническим формам, напоминает миодистрофию Дюшенна; Ø симптомы менее")
2) Миодистрофия Беккера: Ø по клиническим формам, напоминает миодистрофию Дюшенна; Ø симптомы менее выраженны; Ø проявляется в 10 – 15 лет
 - наследственное заболевание соединительной ткани - тип наследования -")
СИНДРОМ ЭЛЕРСА – ДАНЛО (ДАНЛОСА) - наследственное заболевание соединительной ткани - тип наследования - аутосомно-доминантный - в основе - мутация гена, отвечающего за синтез различных типов коллагена • • • ген коллагена I типа альфа I (Col I AI); ген коллагена I типа альфа II (Col I A II); ген коллагена III типа альфа I (Col III AI) (2 q 31) ; ген коллагена V типа альфа I (Col. V AI) (9 q 34. 2 -q 34. 3); ген коллагена V типа альфа II (Col. V AII) (2 q 31); - различают 10 типов синдрома.

КЛИНИЧЕСКАЯ КАРТИНА В основе патогенеза - дегенеративные изменения коллагеновой и эластичной ткани. Первые проявления болезни начинаются в детском возрасте: • повышенная эластичность кожи, ее растяжимость (особенно на лице и в области больших суставов. Кожа локтевых, коленных суставов истончена, пергаментообразна, сквозь нее проступает сеть телеангиэктазий • перерастяжение связочного аппарата (чрезмерную подвижность суставов и частые подвывихи) • ломкость и ранимость ее сосудов

В процесс вовлекается слизистая оболочка щек и языка: возникают пузырьковые высыпания Заболевание сопровождается • аневризмой аорты, • дивертикулами пищевода, • дилятацией кишечника, • эвертерацией диафрагмы, • вывихом хрусталика, • аномалиями зубов.
Молекулярные основы болезней обмена веществ.ppt