b705b98c455303dcbe04bc185eb38b8c.ppt
- Количество слайдов: 69



Protein Function Myoglobin and Hemoglobin Chapter 7

O 2 Binding and Allosteric Properties of Hemoglobin • • • Hemoglobin binds and transports H+, O 2 and CO 2 in an allosteric manner Allosteric interaction – of, relating to, undergoing, or being a change in the shape and activity of a protein (as an enzyme) that results from combination with another substance at a point other than the chemically active site a regulatory mechanism where a small molecule (effector) binds and alters an enzymes activity

Protein Function O 2 does not easily diffuse in muscle and O 2 is toxic to biological systems, so living systems have developed a way around this. Physiological roles of: – Myoglobin Transports O 2 in rapidly respiring muscle Monomer - single unit Store of O 2 in muscle high affinity for O 2 Diving animals have large concentration of myoglobin to keep O 2 supplied to muscles – Hemoglobin Found in red blood cells Carries O 2 from lungs to tissues and removes CO 2 and H+ from blood to lungs Lower affinity for O 2 than myoglobin Tetrameter - two sets of similar units ( 2 2)

– Small protein found in muscle – Made up of 153 residues grouped into 8 helix A to H (proline near end) – very small due to the folding 44 x 25 Å – hydrophobic residues oriented towards the interior of the protein – only polar AAs inside are 2 histidines – Red indicates Heme group x-ray crystallography of myoglobin

– Hemoglobin and myoglobin are only slightly related in primary sequence. – Although most amino acids are different between the two sequences, the amino acid changes between the two proteins are generally conservative. – More strikingly, the secondary structures of myoglobin and the subunits of hemoglobin are virtually identical. x-ray crystallography of myoglobin

Myoglobin and Hemoglobin · · Hemoglobin is structurally related to myoglobin very different primary sequence about an 18% homology in the primary sequence 2 alpha subunits and 2 beta subunits in adults there are very small amount of alpha 2 delta 2 hemoglobin - significance of conserved amino acids between myoglobin and hemoglobin • these are the important aas which keep hemes in contact with the protein • Stabilizes helical arraignment • Interacts with heme/iron

3 D structure of hemoglobin and myoglobin · · 1 - 1 units have 35 interactions 1 - 2 units have 19 interaction sites similar units have few polar contacts the two and two subunits face each other through aqueous channels

Synthesis of hemoglobin Hemoglobin synthesis requires the coordinated production of heme and globin. REMEMBER: – Heme is the prosthetic group that mediates reversible binding of oxygen by hemoglobin. – Globin is the protein that surrounds and protects the heme molecule.

Synthesis of heme Heme is synthesized in a complex series of steps involving enzymes in the mitochondrion and in the cytosol of the cell. (1) the condensation of succinyl Co. A and glycine by ALA synthase to form 5 -aminolevulic acid (ALA). – transported to the cytosol (2) A series of reactions produce a ring structure - co-proporphyrinogen III. – returns to the mitochondrion
 an additional reaction produces protoporhyrin IX")
Synthesis of heme Back in the mitochohdrion: (3) an additional reaction produces protoporhyrin IX – This step Primes the center of the ring structure (4) The enzyme ferrochelatase inserts iron into the ring structure of protoporphyrin IX to produce heme!!

Structure of heme prosthetic group Protoporphyrin ring w/ iron = heme Four Pyrrole groups [A to D] linked by methane bridges Fe+2 coordinated by prophyrin N atoms and a N from Histidine (blue) – This is known as His F 8 (8 th residue of the F helix Iron is out of plane due to His 8 bond A molecule of O 2 acts as 6 th ligand

Structure of heme prosthetic group Two hydrophobic sidechains on O 2 binding site of heme elp hold it in place – Valine E 11 and phenylalanine CD 1 oxygenation changes state of Fe – Purple to red color of blood, Fe+3 – brown Oxidation of Fe+2 destroys biological activity of myoglobin Physical barrier of protein is to maintain oxidation state of Fe+2

free vs. bound heme - role of apoprotein Apoprotein - restricts heme dimers keeps iron reduced the protein moiety of a molecule or complex stabilizes transition state (O 2 binding) CO, NO and H 2 S binding greater affinity than O 2 His E 7 decreases affinity of ligands (CO and O 2 ) for Fe+2

Synthesis of globin Alpha gene cluster: Each chromosome 16 has two alpha globin genes that are aligned one after the other on the chromosome. For practical purposes, the two alph globin genes (termed alpha 1 and alpha 2) are identical. The transiently expressed embryonic genes that substitute for alpha very early in development, designated zeta, are also in the alpha globin locus.

Synthesis of globin Beta gene cluster: The genes in the beta globin locus are arranged sequentially from 5' to 3' beginning with the gene expressed in embryonic development (the first 12 weeks after conception; called epislon). The beta globin locus ends with the adult beta globin gene. The sequence of the genes is: epsilon, gamma, delta, and beta.

Synthesis of globin Beta gene cluster: There are two copies of the gamma gene on each chromosome 11. The others are present in single copies. Therefore, each cell has two beta globin genes, one on each of the two chromosomes 11 in the cell. These two beta globin genes express their globin protein in a quantity that precisely matches that of the four alpha globin genes. The mechanism of this balanced expression is still mostly unknown.

What about the other genes? Two distinct globin chains (each with its individual heme molecule) combine to form hemoglobin. One of the chains is designated alpha. The second chain is called "non-alpha". With the exception of the very first weeks of embryogenesis, one of the globin chains is always alpha. A number of variables influence the nature of the non-alpha chain in the hemoglobin molecule. The fetus has a distinct non-alpha chain called gamma. After birth, a different non-alpha globin chain, called beta, pairs with the alpha chain. The combination of two alpha chains and two non-alpha chains produces a complete hemoglobin molecule.

What about the other genes? The combination of two alpha chains and two gamma chains form "fetal" hemoglobin, termed "hemoglobin F". With the exception of the first 10 to 12 weeks after conception, fetal hemoglobin is the primary hemoglobin in the developing fetus. The combination of two alpha chains and two beta chains form "adult" hemoglobin, also called "hemoglobin A". Although hemoglobin A is called "adult", it becomes the predominate hemoglobin within about 18 to 24 weeks of birth.
, epsilon(2) alpha(2), epsilon (2) zeta(2), gamma (2) Fetal")
Developmental gene expression!!!!! Embryonic hemoglobins zeta(2), epsilon(2) alpha(2), epsilon (2) zeta(2), gamma (2) Fetal hemoglobin F- alpha(2), gamma(2) Adult hemoglobins hemoglobin A- alpha(2), beta(2) hemoglobin A 2 - alpha(2), delta(2)

Developmental gene expression!!!!! The globin genes are activated in sequence during development, moving from 5' to 3' on the chromosome. The zeta gene of the alpha globin gene cluster is expressed only during the first few weeks of embryogensis. Thereafter, the alpha globin genes take over. For the beta globin gene cluster, the epsilon gene is expressed initially during embryogensis. The gamma gene is expressed during fetal development. – The combination of two alpha genes and two gamma genes forms fetal hemoglobin, or hemoglobin F.

Developmental gene expression!!!!! Around the time of birth, the production of gamma globin declines in concert with a rise in beta globin synthesis. A significant amount of fetal hemoglobin persists for seven or eight months after birth. Most people have only trace amounts, if any, of fetal hemoglobin after infancy. two alpha genes and two beta genes comprises the normal adult hemoglobin, hemoglobin A. The delta gene, which is located between the gamma and beta genes on chromosome 11 produces a small amount of delta globin in children and adults. The product of the delta globin gene is called hemoglobin A 2, and normally comprises less than 3% of hemoglobin in adults, is composed of two alpha chains and two delta chains.

Oxygenation Oxygen binding to hemoglobin is due to the effect of the ligand-binding state of one heme group on the ligand-binding affinity of another. Too far apart to interact! (25 to 37 Å apart) Mechanically transmitted between heme groups by motions of the proteins This means the molecule changes shape!

The two states • There are two general structural states - the deoxy or T form and the oxy or R form. One type of interactions shift is the polar bonds between the alpha 1 and the beta 2 subunits.

– The T form finds the terminals in several important H bonds and salt bridges. – In the T form the C terminus of each subunit are "locked" into position through several hydrogen and ionic bonds. – Shifts into the R state break these and allow an increased movement throughout the molecule. Note that binding of one or more oxygen can have a dramatic affect on the other subunits that have not yet bound an O 2.

Binding of oxygen dramatically alters the interactions and brings about a twisting of the two halves (alpha beta pairs) Much of the quaternary changes takes place in the salt bonds between the C terminals of all four chains

oxy and deoxy quaternary structures are different – change takes place between 1 - 2 and 2 - 1 – amino acids between 1 and 2 help to stabilize each forms

Oxygen binding shifts quaternary structure at long distances –binding of O 2 ligand at 6 th coordinate position pulls Fe into heme –moves proximal histadine (F 8) and the alpha helix it is attached to. –shift in the helix is transmitted throughout of molecule

T and R State of Hemoglobin Below are the two major conformations of hemoglobin as predicted by the models for allosteric activation. Oxygen will bind to hemoglobin in either state; however, it has a signficantly higher affinity for hemoglobin in the R state.

T and R State of Hemoglobin In the absence of oxygen, hemoglobin is more stable in the T state, and is therefore the predominant form of deoxyhemoglobin. R stands for relaxed, while T stands for tense, since this is stabilized by a greater number of ion pairs. Upon a conformational change from the T state to the R state, ion pairs are broken mainly between the a 1 b 2 subunits.

– The T form finds the terminals in several important H bonds and salt bridges. – In the T form the C terminus of each subunit are "locked" into position through several hydrogen and ionic bonds. – Shifts into the R state break these and allow an increased movement throughout the molecule. Note that binding of one or more oxygen can have a dramatic affect on the other subunits that have not yet bound an O 2.
 Bind to Oxygen Move down upon O 2 binding His")
How Do Myoglobin (Hemoglobin) Bind to Oxygen Move down upon O 2 binding His E 7
 Bind to Oxygen Move down upon O 2 binding His")
How Do Myoglobin (Hemoglobin) Bind to Oxygen Move down upon O 2 binding His E 7

So what also allows this to change shape?

Functional Structure of Hemoglobin allosteric regulations Allosteric interaction - the binding of one ligand at one site in a protein that affects the binding of other ligands at other sites in the protein. This can affect binding and can be cooperative (pos or neg). Allosterism is typically seen when sigmoidal binding / activity curves.

diphosphoglycerate BPG present in human red blood cells at approximately 5 mmol/L. It binds with greater affinity to deoxygenated hemoglobin – In bonding to partially deoxygenated hemoglobin it allosterically upregulates the release of the remaining oxygen molecules bound to the hemoglobin, thus enhancing the ability of RBCs to release oxygen near tissues that need it most

So why is this important. Look at the Hill plot and the plot of the of binding vs p. O 2 (figs 7 -8 and 7 -7 respectively). Think of when hemoglobin should be mostly saturated and when it would be best if it had a low saturation / affinity and thus "give up" its oxygen. Use the table below to help your thoughts. Oxygen pressure in various fluids Region or fluid p. O 2 (Torr) Inspired Air 158 Aveolar Air 100 Arterial Blood 90 Capilary 40 Insterstitual Fluid 30 Cell Cytosol 10
 – – Purified Hb has a different O 2 affinity")
2, 3 bisphoglycerate (BPG) – – Purified Hb has a different O 2 affinity than it does in blood 26 fold decrease change in affinity is due to 2, -3 diphosphoglycerate BPG – (BPG replaced by nucleotides IHP and ATP in fish and birds) – - 1 BPG per Hb - binds in central cavity of Hb – - binds preferentially to deoxy Hb – - hydrophobic bonds with Lys and salt bridge with His – - O 2 binding changes conformation and “kicks out” BPG – change in altitude increases concentration of BPG Fetal F Hb has replaced His 143 with Ser - What might the consequences be?

Cooperative interactions between subunits Both models do not fully account for the effects of allosteric effectors – sequential model (D Koshland) binding of one O 2 induces T-R conformation change 1 st change is most difficult due to influence by 3 other subunits binding of next three subunits happens sequentially, with higher affinity (easier T-R changes) kinetics increase to the fully oxy Hb state as more O 2 is bound
 All R or all T no in between as in")
Concerted model (J Monod) All R or all T no in between as in the Koshland model concerted model means as more O 2 binds, the R conformation is favored until all units are in the R conformation regardless of the total units bound to O 2 Affinities do NOT change until conformation changes 1 O 2 - all T; 2 O 2 - nearly even equilibrium; 3 O 2 mostly R; 4 O 2 mostly R form energy from O 2 binding causes the change in equilibrium this model best fits O 2 dissociation curve but with limits.

Simple definition of the concept of cooperativity. In its simplest meaning, cooperativity is a measure of communication between binding sites. Positive cooperativity describes a relationship in which the binding to one site facilitates the binding to the second, third, ect. No cooperativity puts all sites on equal footing and indicates no site to site influence. Negative cooperativity implies binding to one site hinders binding to subsequent sites.

How is this cooperation actually forced on hemoglobin? The structures in one subunit effects the others • Structural differences of bound and unbound Hb – Two structures - T and R form depending on the oxygenation of Hb – T is the deoxy form, R is oxy form – allosteric modifiers shift or stabilize one particular conformation over the other – Monomer interactions, most chain interaction occurs between and chains » through mostly hydrophobic residues » and interactions are few and polar » and contact as a switch

CO 2 effects - In the red blood cells the picture is even more complicated. CO 2 is removed by converting CO 2 to bicarbonate CO 2 + H 2 O <-> HCO 3 - + H+
 formed by the enzyme carbonic anhydrase –H+ produced by this")
bicarbonate (HCO 3 -) formed by the enzyme carbonic anhydrase –H+ produced by this enzyme is removed by the Hb as described above –This allows more CO 2 to be removed in the form of bicarbonate

CO 2 binding aids in reducing O 2 affinity by changing conformation by the production of more H+ (R to T change)

In the lungs the O 2 binds Hb and forces the R conformation
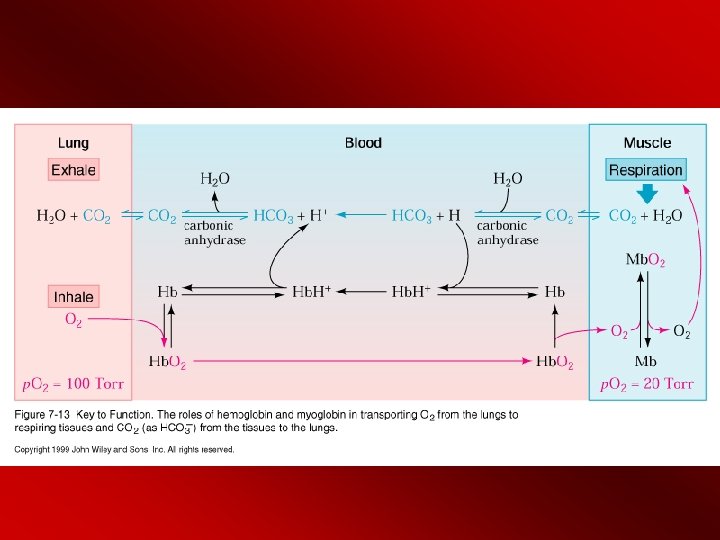

Bohr Effect The Bohr effect is the reversible shift in Hb affinity for O 2 with changes in p. H. H+ Transport (effect) - O 2 binding to Hb releases H+ due to conformational changes in Hb - deoxyform (T form) brings Asp 94 close to His 146 (fig 7 -11 (b)) - the proximity of an acidic amino acid increases the p. K of histidine (p. Ka is now above the p. H) and results in H+ “binding” to deoxy. Hb in other words the His becomes protonated where it normally would be ionized - increasing p. H stimulates Hb to bind to O 2 - Bottom line - when O 2 binds Hb, H+ is released from several amino acid's functional groups. When O 2 is released, the amino acids become protonized and then "picks" up a H+. So when the H+ is high (acidic conditions) the H+ is driven onto the terminal amino acids driving it into the T conformation
 results")
This all aids the function of Hb. In active tissues respiration, (glycolysis) results in lactic acid formation. These tissues need more O 2. Without the H+ effect Hb would hold on to more of the O 2. The H+ induces Hb to dump 10% more of it's O 2. – CO 2 reversibly binds to N term (carbamate) to remove remaining CO 2 R - NH 2 + CO 2 <-> R - NH - COO- + H+ R is the Hb N term amide The carbamide increases the T formation - deoxy form. The reverse occurs in the lungs. This results in 1/2 of CO 2 removal from tissues.

A quick look at what happens to blood when it all goes wrong

Sickle-Cell Anemia, a Molecular Disease One of the first “molecular” diseases found - sickle cell anemia – sickle cell - blood cell is elongated , mis-shaped (sickle) occurs at low O 2 concentration caused by hemoglobin aggregates inflammation in capillaries and pain red blood cells break down - anemia – between 10% of American blacks and 25% of African blacks are heterozygous for sickle cell anemia – homozygous usually do not survive into adult hood – heterozygous individuals usually have no problem except when in severe oxygen deprivation
 Hb. S vs. Hb. A changes structure – sickle")
Single amino acid (point mutation) Hb. S vs. Hb. A changes structure – sickle cell b chains have a valine in place of glutamate – leads to more Hb S (sickle cell) has 2 more + charges than normal hemoglobin Glu -Val occurs on exterior of protein - does not change O 2 dissociation/allosteric properties of protein
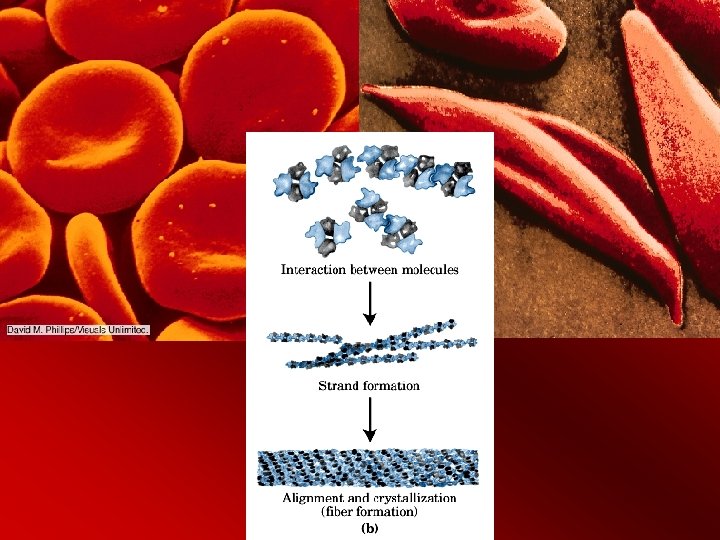

Deoxy Hb. S precipitates – oxy. Hb phenylalanine b 85 and leucine b 88 interior – phe and leu shift to exterior – create a sticky patch with valine (hydrophobic bonding) – nucleation (cluster of aggregate) occurs logarithmically – homozygous - 1000 times faster than heterozygous – that means mixed genes can re-oxygenate faster than polymerization can occur

Malaria – an agent of natural Selection New traits are produced by mutation and are then subject to natural selection. The traits that survive are adaptations. Malaria causes 110 million cases of illness each year – Close to 2 million deaths each year. Rare before the invention of agriculture – Did much to change the selective pressure on human populations

A small change in a gene can have many phenotypic Figure 7. 10 consequences.

Malaria – an agent of natural Selection Most victims of malaria are young children Where malaria occurrence is high, so is the HBs allele – Odd, as Sickle Cell Anemia is nearly always fatal before reproductive age – HBs allele confers resistance to malaria So in areas of high occurrence to malaria, the HBs allele may cause a genetic disorder, but increases the overall fitness of a population where malaria occurs.

Malaria – an agent of natural Selection

Rh Factor There are four blood groups but eight blood types. The Rh-factor!! 85% Positive (US population) 15% Negative Genetic factor Can cause Hemolytic Disease and death of infants.

The genetics of the Rh factor Another blood grouping system independent of ABo – the Rhfactor – three genes: located very close together on the same chromosome. First C & c, second D & d, third E & e Unlike the ABo system there is no co-dominance, c, d, and e are recessive to C, D, and E. ccddee is known as Rh-negative. All others Rh-positive.

Hemolytic disease If a child is Rh+, a Rh- Mother can begin to produce antibodies Rh+ red blood cells – Rh factor crosses placenta and mother makes antibodies In subsequent pregnancies these antibodies can cross the placenta and cause hemolysis of a Rh+ Childs red blood cells. – Can lead to mental retardation or death Prevented by giving Rh- women a Rh immunoglobulin injection no later than 72 hours after birth. Attacks any of the babies Abs in mother before her own antibodies are produced
")
Hemolytic disease Figure 7. 5 (1)
")
Hemolytic disease Figure 7. 5 (2)
 • Prevented by giving Rh- women a Rh")
Hemolytic disease Figure 7. 5 (3) • Prevented by giving Rh- women a Rh immunoglobulin injection no later than 72 hours after birth. • Attacks any of the babies Abs in mother before her own antibodies are produced.

O 2 affinity p. H affects the binding of oxygen to Hb In the lungs (p. O 2 = 100 mm Hg), Hb is 98% saturated at both p. H 7. 4 and 7. 2. binding of oxygen to Hb in the lungs is not affected by changing the p. H and oxygen will load normally.

O 2 affinity at the tissues The change in the p. H results is a lower % saturation of Hb even though the p. O 2 in the tissues has remained at 40 mm Hg (for this example). the % saturation at p. H 7. 4 with a p. O 2 of 40 mm Hg is about 70% – meaning 30% of the oxygen coming to the tissue is released.

O 2 affinity at the tissues At a p. H of 7. 2, the value is close to 60% (meaning 40% of the oxygen coming to the tissue is released). Thus more oxygen is delievered to tissues at a lower p. H even when the p. O 2 remains unchanged. Lowering the p. H appears to shift the entire red curve to the right.
 In")
Alteration in oxygen binding by: - H+, CO 2 2, 3 bisphoglycerate (BPG) In deoxyhemoglobin, the N-terminal amino groups of the α-subunits and the C-terminal Histidine of the β-subunits participate in ion pairs. The formation of ion pairs causes them to decrease in acidity. Thus, deoxyhemoglobin binds one proton for every two O 2 released. In oxyhemoglobin, these ion pairings are absent and these groups increase in acidity. Consequentially, a proton is released for every two O 2 bound. Specifically, this reciprocal coupling of protons and oxygen is the Bohr effect.
 Simple")
Alteration in oxygen binding by: - H+, CO 2 2, 3 bisphoglycerate (BPG) Simple rule of thumb- – High H+ conc and higher CO 2 levels decrease O 2 affinity for Hb – High O 2 concentration decrease H+ and CO 2 affinity for Hb acts as a H+ buffer in respiring tissue

The End, at last!
b705b98c455303dcbe04bc185eb38b8c.ppt