%86ия 4. Программное обеспечение.ppt
- Количество слайдов: 56



Программное обеспечение. Молекулярные базы данных Д. б. н. , профессор Мельнов Сергей Борисович

310 Data Collection Software • Контролирует условия выполнения; • Переводит свет на CCD камеру в электроферограмме (исходные данные); • Создает листы образцов и списки инъекций.

• • • Список инъекций в Data Collection Software: Списки образцов для анализа (повторы могут быть легко выполнены); Установка виртуального фильтра на CCDкамеру; Установка времени электрофореза и напряжения; Установка времени инъекции и напряжения; Установка запуска температуры; При необходимости анализ образцов может быть настроен для автоматического разделения цветов и размеров матрицы по внутренним стандартам, с использованием заданных параметров анализа.

DNA Sequencing Analysis Software DNA Sequencing Analysis software может выполнять несколько этапов анализа данных. Эти этапы могут быть выполнены вручную в типовом диспетчере файлов или как часть полностью автоматизированной операции. Автоматизированный анализ начинается с начала сбора данных и заканчивается, когда данные проанализированы Sequencing Analysis software.

Дополнительная автоматическая обработка может осуществляться с использованием идентификации Factura Feature Identification software. Кроме того, в рамках автоматического режима, результаты могут быть распечатаны. Для образцов, с которыми работают на ABI 373 и ABI PRISM 377, программа Sequencing Analysis: • Находит стартовое положение каждой линии в геле.

• Создает файл с данными по образцу для каждой линии, отмеченной как использующейся, а затем передает основную информацию (имя, дата запуска и т. п. ). • Отслеживает линии и передает исходные данные для каждой линии к соответствующему файлу с данными по образцу или отчету базы данных Bio. LIMS.

Поскольку c каждым образцом на ABI PRISM 310 работают индивидуально, программа сбора данных, создает файлы с данными по образцу автоматически. Как только доступны файлы с данными по образцу, программа Sequencing Analysis может: • Создавать проанализированные данные (основанные на исходных данных), в которых идентифицированы основания в последовательности.

• Передавать проанализированные файлы с данными по образцу на ABI PRISM® Factura Feature Identification Software для последующей обработки. Пакет Factura software включен с каждой копией Sequencing Analysis software. Например, Factura используется для выявления и удаления векторной последовательности и неопределенных участков последовательности. • Печатать данные электрофореграммы для каждого файла с данными по образцу после того, как вся требуемая обработка будет закончена.

Seq. Scape Программа, которая разрабатывается для обнаружения и анализа мутации, открытия SNP и проверки допустимости, патогенного выделения подтипов, идентификации аллеломорфа, и подтверждение последовательности. Этот программа также интегрируется для использования с Variant. SEQr ™.

Особенности программы: - Раскладывает данные быстрее и более точно с матобеспечением, оптимизированным для Applied Biosystems. - Получает достоверные значения для каждой пары оснований, консенсусной последовательности и мутации, используя устойчивые алгоритмы, улучшенные возможности дисплея, и детализированные отчеты результатов. - Автоматически обрабатывает необработанные данные генетических анализаторов и генерирует отчеты с мутациями.

Формат FASTA — один из наиболее популярных форматов для представления последовательностей нуклеотидов или аминокислот. Файл в формате FASTA — простой текстовый файл. Первая строка должна начинаться с символа > или ; . Она содержит имя последовательности и некоторую дополнительную информацию, предназначенную для идентификации. Другие строки, начинающиеся с ; , являются комментариями и игнорируются.

После первой строки начинается, собственно, описание последовательности. При кодировании последовательности нуклеотидов, буквы A, C, G, T и U кодируют, соответственно, аденин, цитозин, гуанин, тимин и урацил. Также некоторые буквы кодируют позиции, в которых находится один нуклеотид из некоторого множества (это используется, если неизвестно, какой именно нуклеотид там находится). Символ - (дефис) кодирует неизвестную последовательность произвольной длины.
")
HLA-типирование HLA-диагностика или тканевое типирование применяется сегодня для решения вопросов трансплантации костного мозга (ТКМ) и органов, для диагностики наследственных заболеваний, ассоциированных с генами главного комплекса гистосовместимости (болезни Бехтерева, сахарного диабета, цилиакии, рассеянного склероза и др. ), а также в диагностике некоторых форм бесплодия, связанных с особенностями HLA супругов.

У человека гены главного комплекса гистосовместимости локализованы на 6 -й хромосоме и кодируют так называемые лейкоцитарные антигены – HLA. Выделяют несколько основных классов системы HLA: I, II и III. Первые антигены MHC были выявлены серологическим методом, их обозначили: HLA-A, HLA-B, HLA-C. Следующий локус обнаружили в результате эксперимента по индукции пролифирации в смешанной культуре лимфоцитов донора и реципиента; его обозначили HLA-D. Затем идентифицировали серологически локус, который был близко сцеплен с HLA-D, он был назван HLA-DR (от D-related). Еще два последующих локуса обозначили соседними с R буквами P и Q – HLA-DP, HLA-DQ.

Позже, выяснили, какие белковые продукты кодируют разные структурные гены из комплекса MHC, гены комплекса разделили на три группы. Каждая группа включает гены, контролирующие синтез полипептидов сходного строения и с одинаковыми функциями одного из трех классов. К I классу отнесли локусы HLA-A, HLA-B, HLA-C, а ко II классу – HLA-D, HLA-DP и HLA-DQ. Гены класса III ответственны за синтез одного из компонентов системы комплемента, фактора некроза опухолей – α и β, ферментов, участвующих в синтезе гормонов.

Среди них также выделяют суперсемейства C (факторы комплемента, которые вовлечены в процессы элиминации чужеродных антигенов) и G (функции окончательно не выяснены, но предполагается, что продукты экспрессии отдельных генов данного семейства участвуют в процессе созревания лейкоцитов). Кроме перечисленных, регион на хромосоме каждого класса содержит и другие гены, правда половина из них не экспрессируются. Расположение генов главного комплекса гистосовместимости человека на 6 -ой хромосоме
. Этот")
Номенклатура HLA аллелей и локусов определяется Номенклатурным Комитетом по факторам системы HLA (WHO). Этот комитет следит за наименованием новых аллелей и регулярно публикует обновления списка вновь открытых аллелей HLA. База данных Комитета IMGT/HLA (www. ebi. ac. uk/imgt/hla) осуществляет централизованное хранение последовательностей аллелей, названных Номенклатурным Комитетом. Список вновь открытых на основании секвенирования HLA аллелей, утверждается Номенклатурным Комитетом по факторам HLA системы. Каждый аллель гена ГКГ имеет уникальное название. Оно включает буквенное обозначение гена, в зависимости от белка, которое он кодирует, а также номер, включающий от 4 до 8 цифр.

Первые две цифры обозначают номер серологического типа белка, третья и четвeртая – подтип. Номера присваивались в таком порядке, в котором произошло открытие этого типа или подтипа антигена. Аллели, чьи номера отличаются в первых четырeх цифрах, могут отличаться на один или несколько нуклеотидов, но при этом меняется аминокислотная последовательность кодируемого белка. Пятая и шестая цифры номера определяют аллели, имеющие замену нуклеотида в кодирующей последовательности, которая не приводит к изменению аминокислотного остатка. А седьмая и восьмая цифры используются для идентификации аллелей, у которых есть замена нуклеотида в интроне или фланкирующих областях.

• • • Также используются дополнительное буквенное обозначение экспрессионного статуса генов: N – неэкспрессирующиеся аллели ( «Null» ); L – низкая экспрессия на клеточную поверхность ( «Low» ); S – ген экспрессирует растворимую молекулу белка, но белок не присутствует на поверхности клетки ( «Secreted» ); С – белок секретируется в цитоплазму, но не присутствует на клеточной мембране ( «Cytoplasm» ); А – аномальная экспрессия, когда есть сомнения, что белок будет синтезироваться ( «Aberrant» ); Q – есть сомнения в том, будет ли экспрессироваться белок, хотя ранние исследования данной мутации показывали, что белок экспрессировался ( «Questionable» ).
")
Определение типа белка ГКГ может проходить в нескольких форматах: – низкое разрешение (два знака) HLA-A*24; – среднее разрешение (четыре знака) HLAA*2402; – высокое разрешение (восемь знаков) HLAA*24020102. Так как полиморфизм не приводящий к замене аминокислоты или в некодирующей области не вызывает изменение структуры белка, приводящее к иммунологической реакции, для точного типирования достаточно определить только первые четыре знака, поэтому типирование среднего разрешения часто называют высоким.

Определение антигенов системы HLA проводят несколькими способами. Изначально антигены ГКГ определяли серологическим методом: из периферической крови выделяли лимфоциты, затем наблюдали микролимфоцитотоксическую реакцию с гистотипируюшими сыворотками. Позже, с развитием молекулярно-генетических методов для точной идентификации МНС антигенов стали использовать полимеразную цепную реакцию (ПЦР). Генетические методы позволили значительно расширитьспектр выявляемых антигенов.

Одним из первых молекулярно-генетических методов определения антигенов МНС является анализ полиморфизма длины рестрикционных фрагментов (RFLP – Restriction Fragment Length Polymorphism). Он основан на наличии специфических сайтов рестикции на каждом гене. При расщеплении данного гена определeнной эндонуклеазой рестрикции при электрофоретическом разделении фрагментов цепи ДНК должны быть идентифицированы фрагменты определeнной длины. При отсутствии рестрикции в полиморфном сайте на электрофореграммах будет выявляться один крупный фрагмент, соответствующий по длине последовательности ДНК между двумя соседними константными сайтами рестрикции для той же эндонуклеазы. При наличии рестрикции в полиморфном локусе на электрофореграмме будет присутствовать меньший по размерам фрагмент, равный расстоянию между полиморфным сайтом рестрикции и одним из ближайших константных сайтов рестрикции. Затем осуществляется гибридизация данных фрагментов по Саузерну с меченными олигонуклеотидами. В настоящий момент, описанный метод применяется редко из-за сложной процедуры, которая не подходит для рутинных исследования диагностических лабораторий.

Более совершенными методами являются гибридизация продуктов амплификации со специфическими олигонуклеотидами, мобилизованными на мембране (Sequence-Specific Oligonucleotide (SSO) probe) и амплификация геномной ДНК со специфическими праймерами и последующим разделением продуктов амплификации в геле (Sequence. Specific Priming (SSP)).

При определении МНСантигенов SSO-методом первоначально проводят амплификацию одного целого гена HLA. Затем продукты ПЦР гибридизуют с меченными специфическими олигонуклеотидами, находящимися на нейлоновой или целлюлозной мембране (стрипе). После того как отмоют все не связавшиеся фрагменты ДНК, происходит окраска меченных олигонуклеотидов. Таким образом выявляются отличия генов вплоть до одного нуклеотида. Недостатком этого метода является то, что очень сложно разделить гетерозиготные аллели на хромосоме человека.

SSP-метод позволяет разделить аллели, так как амплификация гена происходит в лунках с разными праймерами специфичными к полиморфным последовательностям. Идентификация гена происходит по наличию продукта ПЦР. Визуализацию продуктов ПЦР производят электрофоретическим разделением в агарозном геле. Описанные выше методы позволяют определить МНС-антиген только на низком разрешении и с определeнной вероятностью.

Единственным методом, осуществляющим типирование высокого разрешения, является секвенирование последовательности генов. Данная довольно сложная процедура в последние несколько лет стала гораздо проще благодаря созданию полностью автоматических систем ДНКсеквенирования. Первым этапом для точного определения последовательности является выявления аллелей, которые затем по отдельности проходят этап амплификации с флуоресцентными терминирующими дидезоксинуклеотидами. Последней стадией является разделение продуктов реакции с помощью капиллярного электрофореза и детекция флуоресцентных меток лазером.

Современные технологии позволяют в компьютерной программе кроме точного определения последовательности нуклеотидов гена идентифицировать его номер по международной номенклатуре за минимальное время. К сожалению ДНКсеквенирование до сих пор самая дорогостоящая процедура и не может быть использована повсеместно.

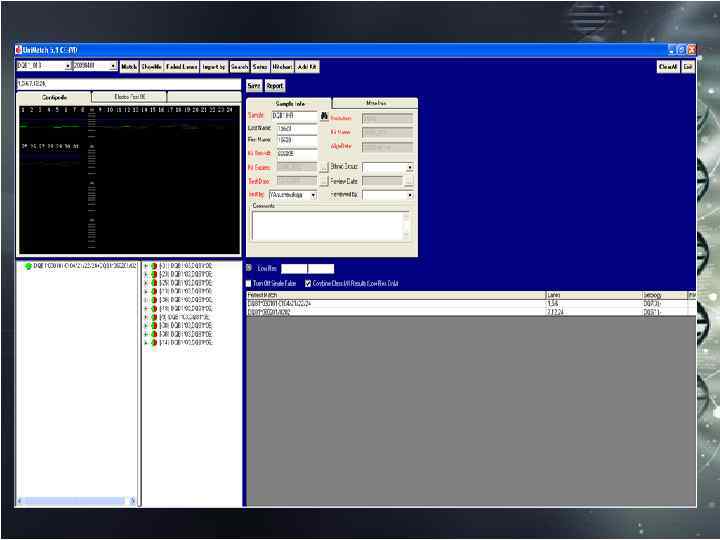
Программа Uni. Match 5. 1 для HLAтипирования
 После проведения электрофореза, позитивные продукты ПЦР-реакции в геле считаются, и заносятся в")
(метод SSP) После проведения электрофореза, позитивные продукты ПЦР-реакции в геле считаются, и заносятся в специальную программу интерпретации результатов. Выбирается лот соответствующего набора, и программа автоматически осуществляет поиск всех возможных вариантов и выдает ответ.

В специальном поле с количеством лунок в наборе отмечаются позитивы. Последняя лунка отрицательный контроль. После интерпретации программа предлагает вариант ответа (слева), и возможные FP и FN. Справа в специальном окне заполняется вся необходимая информация: локус, № образца, ФИО пациента, дата и т. д. После интерпретации генерируестя report и выдается ответ.

Молекулярная база данных Gene. Bank Gen. Bank – база данных генетических последовательностей, поддерживается NIH (Национальный Институт Здоровья США), аннотированная база известных последовательностей ДНК, РНК и белков, с литературными ссылками на первоисточники и информацией биологического характера. Обновляется каждые два месяца. Является частью International Nucleotide Sequence Database Collaboration, которая объединяет три крупнейшие коллекции нуклеотидных последовательностей: DDBJ (NIG), EMBL (EBI) и Gen. Bank (NCBI).

• • • Объем Gen. Bank’а: 1982: 680 338 букв в 606 последовательностях 1992: 101 008 486 букв в 78 608 последовательностях 2002: 28 507 990 166 букв в 22 318 883 последовательностях 2004: 44 575 745 176 букв в 40 604 319 последовательностях 2005: 56 037 734 462 букв в 52 016 762 последовательностях (из ~165 000 организмов) Размер файлов — 196 Gb

Структура документа Gen. Bank’а Описание Последовательность

Описательная часть в документе Gen. Bank’а нужна: • чтобы пользователь банка мог найти интересующую его последовательность; • для хранения дополнительной информации (откуда ДНК, кто проводил эксперимент по секвенированию, биологическая роль данной последовательности и т. д. )
.")
Для удобства пользования описательная часть документа Gen. Bank разбита на так называемые поля (“fields”). Для того, чтобы найти интересующую последовательность в Gen. Bank’е: • Существуют специальные компьютерные программы (например, SRS или Entrez), предназначенные для поиска по ключевым словам в банках последовательностей. • Пользователь указывает в программе, по каким полям нужно искать и какое слово (или слова). Программа выдаёт список записей банка, в которых указанные слова встретились в указанных полях. Чтобы посмотреть, есть ли в соответствующем поле заданное слово необходимо заранее создать индексную таблицу каждого из полей и при каждом запросе обращаться к ней.

Молекулярная база данных EMBL База данных нуклеотидных последовательностей Европейской Молекулярно-Биологической Лаборатории пополняется большей частью непосредственно авторами, определившими первичную структуру фрагмента ДНК или РНК и, кроме последовательности нуклеотидов, содержит разнообразную информацию о каждом фрагменте, включая литературные ссылки, перекрестные ссылки на документы других баз данных, таблицы особенностей и др.
, Gen. Bank (США)")
Существует с 1982 года. База данных – продукт сотрудничества EMBL (ФРГ), Gen. Bank (США) и DDJP (Япония), каждая из этих трех групп собирает свою порцию информации из всех возможных мировых источников, ежедневно обмениваясь новыми и обновленными документами друг с другом.

Сегодня EMBL состоит из 18 разделов. Большая часть разделов соответствует отражает таксономию. EMBL представляет собой плоскую базу данных, состоящую из одной таблицы с множеством строк (= записям). Одна запись = последовательный участок ДНК или РНК. Одна запись состоит из двух частей: 1) аннотации 2) собственно последовательности.

Молекулярная база данных Swiss. Prot Swiss-Prot – одна из первых баз данных белковых последовательностей, “gold standard” белковой аннотации. Аннотация выполнена вручную группой профессиональных экспертов на основе экспериментальной информации, описанной в научных статьях. Организована в 1986 году – SIB+EBI+PIR+GU = prof. Amos Bairoch На сегодняшний день – Release 57. 8 509019 последовательностей
")
Uni. Prot. KB Uni. Prot = Swiss-Prot + Tr. EMBL (Translated EMBL sequence database) Tr. EMBL – Release 40. 8 - 9325547 sequences
")
Поиск белка в Uni. Prot. KB (по названию)

Download…

Сохранить в FASTA формате

Стандартная запись Swiss-Prot

Стандартные поля: names and origin, entry information, protein attributes
")
Nice. Zyme (ферменты)

Молекулярная база данных PIR База данных NBRF, с 1984 года коллекционирует данные о белках. Эта база возникла на месте NBRF Protein Sequence Database, созданной M. O. Dayhoff и опубликованной впервые еще в 1965 году в виде атласа белковых последовательностей и структур (Atlas of Protein Sequence and Structure). Сейчас база PIR - продукт сотрудничества NBRF (США), MIPS (Германия) и JIPID (Japan International Protein Information Database).

База данных PIR делится на 4 раздела: PIR 1: содержит полностью классифицированные и аннотированные записи; PIR 2: включает в себя предварительные записи, которые не были полностью рассмотрены и могут содержать избыточность; PIR 3: содержит непроверенные записи; PIR 4: записи - Концептуальное создание искусственных фактических последовательностей нуклеотидов.

Молекулярные базы данных Protein Data Bank, PDB — банк данных 3 -D структур белков и нуклеиновых кислот. База данных Protein Data Bank обновляется еженедельно.

Преобладающая часть структур получена при помощи метода диффракции рентгеновских лучей, около 15 % — при помощи ЯМР белков, и лишь малая часть — при помощи крио-электронной микроскопии. Каждая структура, опубликованная в PDB получает четырёхзначный идентификатор (комбинация цифр и букв латинского алфавита). Данный шифр не может служить идентификатором биомолекул, так как часто разные структуры одной и той же молекулы, например, в различной среде, могут иметь различные PDB ID.

Создание геномных библиотек Рестрикция геномной ДНК на фрагменты и клонирование фрагментов с помощью различных векторов создали основу формирования геномных библиотек. Для этого геномная ДНК разрезается или, как говорят, переваривается определенной рестриктазой, а образующиеся фрагменты клонируются с помощью различных векторов, для чего используют методы рекомбинантной ДНК. Геномная библиотека должна содержать не только гены, но и всю некодирующую ДНК, расположенную между генами.

Поскольку переваривание рестриктазы производят неполное так, что одни сайты, специфические для рестриктазы, разрезаются, а другие нет, то образуются фрагменты ДНК с частично перекрывающимися последовательностяминуклеотидов. Это облегчает последующее восстановление картины расположения фрагментов в нативной ДНК. Кроме геномных библиотек, существуют библиотеки к. ДНК.

Библиотеки к. ДНК содержат только экзоны генов, поскольку получаются на основе зрелой м. РНК с помощью фермента, называемого обратной транскриптазой. Обратная транскриптаза на матрице м. РНК создает комплементарную нить ДНК, которая затем превращается в обычную двунитевую ДНК с помощью ДНК-полимеразы. Затем такие молекулы к. ДНК клонируются в бактериальных клетках точно так же, как и геномная ДНК. Еще один тип ДНК-библиотек — хромоспецифические библиотеки.

Для их создания хромосомы разделяют с помощью проточной цитометрии, которая позволяет выделить отдельные хромосомы. ДНК, полученная после такой сортировки, будет преимущественно представлять определенную хромосому. Затем получают фрагменты ДНК отдельной хромосомы перевариванием с той или иной рестриктазой и клонируют их обычным путем. Различные библиотеки ДНК широко используют при реализации программы «Геном человека» , а также для других целей, например при поиске полиморфных ДНК-маркеров.

Спасибо за внимание!
%86ия 4. Программное обеспечение.ppt