Презентация2.pptx
- Количество слайдов: 20



Презентація на тему: «Спадкові хвороби» Підготувала студентка І курсу Фармації-бакалаврат Телебей Тетяна

Сидром Ангельмана
 -генетична аномалія. Синдром названий по імені британського педіатра Гаррі")
Синдром Ангельмана (Синдром щасливої маріонетки) -генетична аномалія. Синдром названий по імені британського педіатра Гаррі Ангельмана, який вперше описав його в 1965 р. Для нього характерні: 1. Затримка психічного розвитку; 2. Порушення сна; 3. Хаотичні рухи (особливо рук); 4. Частий сміх чи усмішка. При синдромі Ангельмана відсутні деякі гени з 15 -ї хромосоми (у більшості випадків часткова делеція або інша мутація 15 хромосоми). При синдромі Ангельмана страждає материнська хромосома; в разі пошкодження батьківської хромосоми виникає синдром Прадера-Віллі. Каріотип 46 XX або XY, 15 р-. Зазвичай синдром викликається спонтанним хромосомним дефектом, коли відсутня велика суміжна область з 3 -4 мільйонів пар основ (нуклеотидів) ДНК в області q 11 -q 13 15 -ї хромосоми. Частота народження, за різними даними, - 1: 10 000 -20 000 живонароджених немовлят.

Особливості: - У 75 % проблеми з харчуванням , особливо з грудним вигодовуванням , такі немовлята погано набирають вагу ; - затримка в розвитку навичок загальної моторики (вміння сидіти , ходити) ; - Затримка мовного розвитку , нерозвинена мова ( у всіх дітей ) ; - Діти більше розуміють , чим можуть сказати або висловити ; - Дефіцит уваги і гіперактивність ; - Труднощі з навчанням ; - Епілепсія (80% випадків) , порушення виявляються також при електроенцефалографії ; вважається , що у дітей з синдромом Ангельмана спостерігається вторинна (симптоматична ) епілепсія ; - Незвичайні рухи ( невеликий тремор , хаотичні рухи кінцівок) ; - Частий сміх без приводу ; - Ходьба на негнучких ногах - через цю особливість дітей з цим синдромом іноді порівнювали з маріонетками ; - Розмір голови менше середнього , нерідко з сплющеною потилиці ; іноді характерні риси обличчя - широкий рот , рідко розташовані зуби , висунуте вперед підборіддя , висунутий назовні язик; - Порушення сну ; - Страбізм ( косоокість ) в 40 % випадків; - Сколіоз (викривлення хребта ) у 10 % випадків; - Підвищена чутливість до високої температури ; - Почуваються комфортніше у воді.



Джеймс Едгар з Північного Уельсу страждає синдромом Ангельмана. Через нього дитина постійно виглядає радісним. Всьому виною генетична аномалія , ймовірно , пов'язана з геном UBE 3 A. Про це передає The Daily Mail. Крім частого сміху та посмішки , особи, які страждають цим захворюванням , відстають у психічному , інтелектуальному розвитку , погано сплять , у них бувають епілептичні напади і відзначаються хаотичні рухи рук. Буває дефіцит уваги і гіперактивність , проблеми з навчанням , підвищена чутливість до високої температури. За словами матері хлопчика , Рейчел Мартін , дитина дуже ласкавий , радісний, але за ним потрібно постійно стежити , так як він не розуміє , де його може чекати небезпека.
 , рекомендованого для")
Діагностика: 1. Синдром діагностується шляхом генетичного аналізу ( 15 хромосома ) , рекомендованого для новонароджених зі зниженим м'язовим тонусом , відставанням у розвитку загальної моторики і в розвитку мови. 2. Батьки і лікарі повинні звернути увагу на: -випадки дрібного тремору; -хаотичні рухи кінцівок; -ходу з негнучкими ногоми; -специфічний вираз обличчя, занадто частий сміх. 3. Можливі методи аналізу: процес флуоресцентної гібридизації на місці , метилювання ДНК в області 15 q 11 - q 13 , аналіз мутації імпрінтінгового центру , аналіз прямий мутації гена UBE 3 A.

Лікування: Синдром Ангельмана є вродженою генетичною аномалією; в даний час специфічні способи його лікування не розроблені. Однак деякі лікувальні заходи підвищують якість життя людей з даним синдромом: 1. Немовлята з гіпотонусом повинні отримувати масаж та інші види спеціальної терапії (фізіотерапії). 2. Рекомендується використання спеціальних методик розвитку дитини, заняття з логопедом і дефектологом. 3. Порушення сну коригуються призначенням легких снодійних. Д-р Вагстафф (США) вважає, що призначення 0. 3 мг мелатоніну за 30 хвилин-1 годину перед сном покращує сон пацієнтів з синдромом Ангельмана. 4. Напади лікуються так само, як епілепсія. Діти з синдромом Ангельмана часто відчувають більше одного типу нападів. Показана електроенцефалографія. 5. Небажану поведінку. Д -р Чарльз Вільямс ( Гейнсвілл , Флоріда ), що працює в основному з аутичним дітьми , зазначає загальні для аутичних дітей та дітей з синдромом Ангельмана особливості поведінки: помітна аутостимуляція , імпульсивність , нав'язливі , повторювані рухи , інтерес до недоречним предметів , а також складність у спілкуванні з іншими людьми. Лікарі США показують , що для аутичних дітей внутрішньовенні ін'єкції гормону секретину ( знайденого в підшлунковій залозі ) успішно зменшують прояви небажаної поведінки і забезпечують хороший рівень товариськоських та комунікативних навичок.

Перспективи розвитку: 1. Діти з синдромом Ангельмана розуміють набагато більше, ніж можуть сказати. У деяких випадках у них взагалі немає мови; описані діти зі словниковим запасом близько 5 -10 слів. При цьому діти / люди з синдромом Ангельмана люблять спілкуватися з людьми, грати, як правило, вони дружелюбні і милі. 2. Рекомендується навчати таких дітей мови жестів. Заняття з раннього віку за спеціальними програмами, спрямовані на розвиток навичок дрібної і загальної моторики, у низці випадків дають хороші результати. 3. Перспективи розвитку залежать від ступеня враженості хромосоми. Деякі люди з синдромом Ангельмана здатні освоїти навички самообслуговування і мову на примітивному рівні (зазвичай причиною синдрому в цьому випадку стала мутація), деякі ніколи не зможуть ходити і говорити (це зазвичай відбувається у разі делеції частини хромосоми). 4. З віком, як правило, симптоми гіперактивності та порушення сну пом'якшуються. У дівчаток з синдромом Ангельмана в період статевого дозрівання можуть почастішати напади. 5. Більшість людей з синдромом Ангельмана здатні контролювати екскреторні функції (сечовипускання і дефекацію) днем, деякі - і вночі.

6. Деякі люди з синдромом Ангельмана здатні їсти за допомогою ножа і виделки, одягатися самостійно в разі відсутності на одязі гудзиків, «блискавок» . 7. У дорослому віці може з'явитися ожиріння і погіршитися ситуація зі сколіозом. 8. Менструації, статеве дозрівання індивідів з синдромом Ангельмана відбувається в звичайні терміни. Описаний один випадок вагітності жінки з синдромом Ангельмана: вона народила дівчинку з таким же діагнозом.

Синдром Блума
, (англ. Bloom syndrome), також відомий як синдром Блума-Торре-Махачека - це рідкісний")
Синдром Блума (СБ), (англ. Bloom syndrome), також відомий як синдром Блума-Торре-Махачека - це рідкісний аутосомнорецесивний хромосомний розлад, який характеризується високою частотою розривів і перебудов в хромосомах ураженої людини. Цей синдром був вперше описаний лікарем дерматологом Девідом Блумом (Dr. David Bloom) у 1954 році.

Ознаки і симптоми: 1. Особи, які хворі на СБ, як правило не високого росту, з характерними висипами на шкірі, які виникаютьмайже одразу після першого впливу сонячних променів. Ці висипи, як правило виникають на щоках (викликаючи почервоніння) та мають форму метелика. Але висипи можуть виникати не лише на обличчі, але й на інших частинах тіла, на яких потрапляло сонячне проміння (спина, руки, тощо). 2. Інші клінічні ознаки: високий голос; специфічні риси обличчя, такі як – довге та вузьке обличчя; мікрогнатія нижньої щелепи; великий ніс і вуха; зміни пігментації шкіри, включаючи появу гіпо-і гіпер-пігментних плям та родимих плям; телеангіектазія (розширені кровоносні судини), які можуть проявлятися на шкірі, переважно на верхній частині грудей та спини; помірний імунодефіцитом, який характеризується дефіцитом деяких класів імуноглобулінів, який, як вважають дослідники, призводить до рецидивів пневмонії та інфекцій вух; гіпогонадизм – це патологічний стан, зумовлений недостатньою секрецією андрогенів, тобто організм чоловіків, не може виробляти сперму, а отже, уражені цим захворюванням особи чоловічої статі – безплідні. У жінок ця хвороба проявляється передчасним припиненням менструацій (передчасна менопауза), тобто вони мають знижену здатність до народження дітей. Проте, деякі жінки, які хворіють на синдром Блума можуть мати дітей.

Ускладнення, які виникають у результаті дії захворювання: 1. Хронічні хвороби легень; 2. Цукровий діабет; 3. Виникнення труднощів під час навчання; 4. У деяких осіб, виникає розумова відсталість; 5. Найбільш небезпечним ускладненням можна назвати підвищення ймовірності виникнення пухлинних захворювань. Зв'язок із виникненням раку Внаслідок великої кількості мутацій, які відбуваються при захворюванні на синдром Блума, в уражених осібсуттєво підвищується ризик захворіти на рак. Схильність до онкологічних захворювань характеризується: 1) широким спектром прояву хвороб, який включає лейкози, лімфоми і карциноми; 2) раннім віком виникнення пухлинних захворювань, порівно до решти населення; 3) складністю та важкістю перебігу.

У осіб з синдромом Блума рак може виникнути у будь-якому віці. Середній вік хворих – 25 років. Виникає в сім'ях з високою частотою інбридингу. Найчастіше вражаються особи чоловічої статі. Культура лімфоцитів і фібробластів хворих відрізняється високою частотою хромосомних аберацій і підвищеною чутливістю до УФопромінення , відзначена недостатність ферменту ДНК - лігази I. Клітини при синдромі Блума проявляють хромосомну нестабільність , що приводить до високого рівня соматичних мутацій як внутрішньоутробно , так і в постнатальної життя. У хворих з синдромом Блума відзначається порушення з боку В- (зниження рівня Ig. A і Ig. M ) і Т -системи імунітету. Зокрема, підвищення онкологічної захворюваності при синдромі Блума пов'язують з придушенням проліферативної реакції лімфоцитів на мітогени. У той же час зв'язок дефекту імунної функції з недостатністю ензиму , який бере участь у репарації ДНК лігази I , залишається неясною. Ген синдрому Блума поки не клонований. Співвідношення частоти різних типів раку при синдромі Блума , що відображає їх співвідношення в загальній популяції , підтверджує , що синдром Блума є мутує фенотипом , при якому кожна клітина тіла більш схильна до соматичної мутації. При цьому супутній імунодефіцит неясного походження може також відігравати роль у пухлинної прогресії.

При синдромі Блума відзначається схильність до злоякісних новоутворень внутрішніх органів , крові , кісток , вони розвиваються у віці від 2 до 46 років. Найбільш частими є злоякісні захворювання крові (гострий мієлолейкоз , гострий лімфолейкоз , лімфоми ), рак стравоходу , аденокарцинома кишечника , але описані і рідкісні пухлини: остеогенна саркома , медуллобластома , пухлина Вільмса. Висока ймовірність розвитку раку мови , гортані , легені та молочної залози. У 9 з 165 хворих з синдромом Блума , зареєстрованих у США , були немеланотіческіе раки шкіри , але не меланома. Середній вік цих хворих складав 28 років.
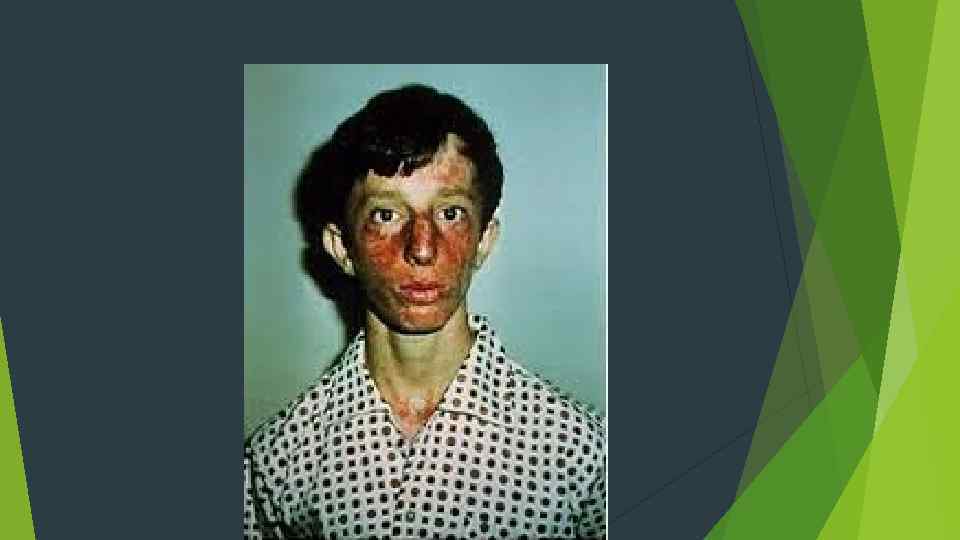


Діагноз синдрому Блума встановлюється на підставі клінічної картини, виявлення патології репарації ДНК, високої частоти сестринських хроматидних обмінів, а також змін у лімфоцитах. В якості методу пренатальної діагностики захворювання рекомендується використовувати оцінку сестринських хроматидних обмінів. Диференціальний діагноз синдрому Блума проводиться з червоним вовчаком, синдромом Коккейна, порфірією, синдромом Ротмунда. Томсона При лікуванні синдрому Блума застосовуються фотозахисні засоби, каротиноїди, вітамін Е, імунокоректори. Хворі повинні спостерігатися дерматоонколога з метою ранньої діагностики немеланотіческіх раків шкіри. В якості профілактики захворювання слід уникати близькоспоріднених шлюбів.
Презентация2.pptx