Понятие о наследственных болезнях человека.ppt
- Количество слайдов: 88



Понятие о наследственных болезнях человека. Медико-генетическое консультирование.

ПЛАН ЛЕКЦИИ Наследственные болезни и их классификация. Хромосомные заболевания человека. Генные болезни. Болезни с наследственным предрасположением. Медико-генетическое консультирование. Пренатальная диагностика наследственных заболеваний.

Наследственные болезни делят на 3 группы: n n n Хромосомные болезни Генные болезни n аутосомно – доминантные n аутосомно – рецессивные Болезни с наследственным предрасположением

n n n Болезни с наследственным предрасположением отличаются от генных болезней тем, что для своего проявления они нуждаются в действии факторов внешней среды. По генетической природе – это две группы болезней: моногенные болезни и полигенные болезни. Моногенные болезни вызываются единичными мутантными генами с сильным эффектом.

n n n Полигенные или их ещё называют мультифакториальными болезнями развиваются в результате совместного действия множества генетических (страдает множество генов) и средовых факторов, каждый из которых в отдельности оказывает лишь слабый эффект в развитии заболевания. Это и определяет различия между моногенными и мультифакториальными болезнями. Мультифакториальные болезни составляют сегодня около 90% всех хронических неинфекционных болезней различных систем и органов человека. К таким болезням относятся: псориаз, сахарный диабет, шизофрения, гипертоническая болезнь, атеросклероз сосудов и др.

С наследственными болезнями имеют дело врачи всех специальностей, т. е. наследственная патология может быть классифицирована и в соответствии с медицинскими специальностями

Классификация наследственных болезней • Наследственные болезни крови • Наследственные сердечно-сосудистые • • заболевания Наследственные болезни внутренних органов Аномалии развития скелета Эндокринные наследственные болезни Наследственные болезни почек Наследственные болезни органов чувств Наследственные болезни нервной системы Психические наследственные болезни

• В среднем до 20% коечного фонда большинства • • • детских клиник приходится на больных с наследственной патологией. Объём груза наследственной патологии представляется в обобщенном виде следующим образом: на 1000 живорождённых 30 -50 имеют тот или иной тип наследственной патологии генные болезни у 5 -14 детей; хромосомные – у 4 -7 детей; врождённые пороки развития – у 19 -22; болезни с выраженным предрасположением – у 7 -10. Следует подчеркнуть, что тяжёлые её формы составляют 1 -2%. Следовательно, на 5 млн. живорождённых приходится 50 -100 тыс. детей с тяжёлой врожденной и наследственной патологией.

В настоящее время существуют трудности в распознавании наследственных заболеваний. n Они обусловлены прежде всего тем, что нозологические формы наследственных болезней очень многообразны (более 2 тыс. ) и каждая характеризуется большим многообразием клинической картины. n Так, в группе нервных болезней известно более 200 наследственных форм. n

n. В связи с таким многообразием врачу трудно овладеть всем запасом знаний, необходимых для диагностики всех наследственных заболеваний, поэтому он должен знать основные принципы, которые помогут ему заподозрить наследственные заболевания и направить пациентов на более квалифицированное медикогенетическое обследование.

ХРОМОСОМНЫЕ ЗАБОЛЕВАНИЯ ЧЕЛОВЕКА n n К хромосомным болезням относятся врождённые пороки развития человеческого организма, обусловленные отклонением от нормального содержания хромосомного материала. Как правило, хромосомные болезни представляют собой спорадические случаи в семье, возникшие в результате мутации в половых клетках одного из родителей. И только 3 -5% являются унаследованными формами, передающимися из поколения в поколение.

n n n Патологические изменения вследствие хромосомных аномалий в организме человека складываются уже в пренатальном периоде его развития. Они обуславливают либо гибель эмбриона или плода, либо создают основную клиническую картину у новорождённого. В среднем около 50% спонтанных абортов и 7% всех мертворождений обусловлены хромосомными нарушениями. На 1000 новорождённых младенцев 7 имеют различные хромосомные болезни.

Классификация хромосомных болезней § Хромосомные болезни, связанные с аномалиями числа хромосом при сохранении их структуры. § Они подразделяются на 3 группы: § Болезни, обусловленные числовыми аномалиями половых Х- и У-хромосом § Болезни, обусловленные числовыми аномалиями аутосом § Болезни, обусловленные увеличением кратности полного гаплоидного набора хромосом – полиплоидии § Хромосомные болезни, обусловленные разрывом хромосом и их перестройкой – хромосомные аберрации: § Транслокации § Делеции § Инверсии § Дупликации

§ Иногда в организме встречаются клетки с различными кариотипами. Такое сочетание кариотипов носит название мозаицизма. § Среди всех хромосомных заболеваний 25% приходится на аутосомные трисомии, 35% на патологию половых хромосом и 40% на сбалансированные и несбалансированные перестройки. § В настоящее время известно более 700 заболеваний, обусловленных структурными нарушениями хромосом.

Болезни, связанные с числовыми аномалиями половых хромосом n n Частота этих аномалий составляет 2, 6 на 1000 рождений. У пожилых людей были выявлены колебания числа хромосом в клетках. Отмечена тенденция к утрате Х-хромосом у женщин в возрасте 55 лет и старше и У-хромосом у мужчин в возрасте старше 65 лет. Числовые аномалии половых хромосом чаще всего имеют вид трисомй и моносомий. К ним относятся болезнь Шерешевского – Тернера и болезнь Клайнфельтера.

Болезнь Шерешевского - Тернера n n ü ü ü Болеют только женщины. У них отсутствует одна из Х-хромосом (синдром ХО). Встречается с частотой 1: 3000 новорождённых девочек. Постановка диагноза при этой болезни правомочна лишь в том случае, если у больного имеется три группы отклонений: гипогонадизм и недоразвитие половых признаков врождённые соматические пороки; низкий рост

n n n Отставание в росте уже замечается на 1 году жизни. Но особенно отчётливым становится к 9 -10 годам жизни. Взрослые женщины обычно низкорослые – средний рост 135 см. При этой болезни встречаются различные аномалии развития костного скелета (голова «сфинкса» ). Кардинальный признак заболевания – половой инфантилизм. Вторичные половые признаки отсутствуют или слабо развиты. Больные бесплодны. В 50% случаев такие больные умственно отсталые. Они пассивны, астеничны, склонны к психозам. Продолжительность жизни близка к норме, но наблюдается более раннее старение. Лечение больного в основном симптоматическое и направлено на коррекцию таких дефектов развития, как малый рост и половой инфантилизм (анаболики, половые гормоны).

Синдром Шерешевского-Тернера

Болезнь Клайнфельтера n Болеют только мужчины. Установлено, что больные имеют лишнюю Х-хромосому (кариотип 47, ХХУ). n Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов в их сочетании (мозаицизм). Обнаружено несколько типов полисомий по Х и У- хромосомам – 47, ХХУ; 48, ХХХУ; 47, ХУУ; 48, ХХУУ. n Индивидуумы с полисомией одновременно по Х и У-хромосоме встречаются очень редко (1 на 25 тыс. ). n Синдром Клайнфельтера клинически не диагностируется при рождении и до периода полового созревания ничем не проявляется. Дисбаланс проявляется в период полового созревания. Характерными клиническими признаками этого заболевания являются: высокий рост, длинные конечности, половой инфантилизм, склонность к ожирению. n Одновременно отмечается снижение интеллекта (олигофрения в форме дебильности), нередко у таких больных возникают психические нарушение, антисоциальные поступки и алкоголизм. Для лечения используют андрогены. n

Болезнь Клайнфельтера
 n Синдром плисомии по Х-хромосоме")
Синдром полисомии по Х-хромосоме у женщин ( «сверхженщина» ) n Синдром плисомии по Х-хромосоме включает трисомию (кариотип 47, ХХХ), тетрасомию (48, ХХХХ; 49, ХХХХХ). n Наиболее часто встречается трисомия – 1: 1000 родившихся девочек. n Трисомию заподозрить трудно, так как у её носительницы отклонений может и не быть даже во взрослом состоянии. Плодовитость этих пациенток не страдает. Одним из частых симптомов трисомии Х является та или иная степень умственной отсталости, встречающаяся у 75% больных. n Отмечено повышенное развитие психозов, обнаруживается шизофрения. В общем, число добавочных Х-хромосом увеличивает степень умственной отсталости. Рационального лечения нет.

Синдром полисомии по У-хромосоме у мужчин n Примерно из каждой тысячи мальчиков 1 один имеет ХУУ – хромосомный набор. Клинически этот синдром также ничем не выделяется. n Мужчины имеют высокий рост – около 185 см. Однако у них наблюдается некоторое снижение интеллекта, нередко проявляется агрессивность в поведении, приводящая к совершению антисоциальных поступков. n В настоящее время установлено, что в местах заключения в 10 раз больше, чем нормальных мужчин, попадает мужчин с ХУУ – хромосомным набором. n Рационального лечения нет.
. Любая аутосома")
Болезни, вызванные числовыми аномалиями аутосом Из 46 хромосом человека 22 пары (аутосомы). Любая аутосома может быть вовлечена в патологию. Аномалии 1 -12 пар хромосом обычно приводят к летальному исходу. Наиболее часто среди данных заболеваний встречается болезнь Дауна (1: 700 -800). Ежегодно у нас в стране рождается около 8. 000 детей с болезнью Дауна (трисомия по 21 -й хромосоме). Дети с болезнью Дауна одинаково часто встречается среди обоих полов. Доказана корреляция между рождением таких больных и возрастом матери: чем старее мать, тем больше риска появления ребёнка с болезнью Дауна. Риск возникновения аномального потомства в возрасте 40 -46 лет в 16 раз выше, чем у матерей в 20 -24 года.

• Болезнь чётко диагностируется с первых лет жизни. • Трисомный вариант болезни Дауна встречается в 98% процентах случаев, в 2% избыточный хромосомный материал транслоцирован на одну из аутосом, чаще группы D и С.

Больные обычно невысокого роста, отличаются слабоумием. Они имеют характерную внешность: небольшая круглая голова со скошенным затылком; косые глазные щели, эпикант; короткий нос с широкой плоской переносицей; маленькие деформированные уши; полуоткрытый рот с высунутым языком и выступающей нижней челюстью; своеобразная походка с неловкими движениями; косноязычность. Бросается в глаза умственная отсталость: около 90% - эмбицильны. Это обусловлено недоразвитием головного мозга. Часто у таких детей встречаются пороки развития сердечно-сосудистой системы, желудочно-кишечного тракта. Больные повышенно восприимчивы к инфекциям и злокачественным заболеваниям (особенно кроветворной системы).

Рождение ребёнка с болезнью Дауна можно рассматривать как явление случайное, вероятность повторения её невелика в случае молодого возраста матери. Лечение при этом страдании мало эффективно. В основном оно симптоматическое. Медико-педагогические и лечебные мероприятия позволяют адаптировать часть больных к посильному труду. Больные могут прожить до 30 лет и более
 – 47, ХХ (ХУ), + 13 Частота этого заболевания колеблется")
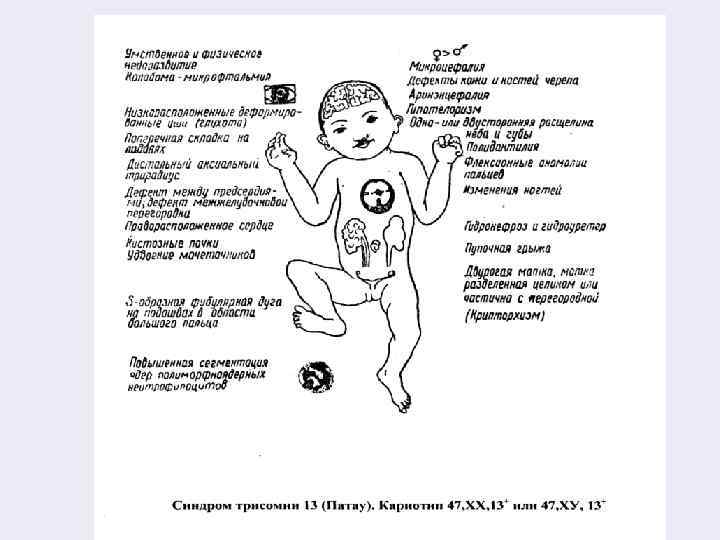
Синдром Патау (трисомия 13) – 47, ХХ (ХУ), + 13 Частота этого заболевания колеблется 1: 3000 – 1: 4000 рождений. Дети (среди больных) преобладают девочки. С этим синдромом рождается у матерей старшего возраста. При этом наблюдается высокая ранняя смертность (в течение первого года жизни погибает около 90% детей). При рождении их вес на 900 г. меньше среднего веса новорождённого.

Внешние признаки больных достаточно типичны и позволяют при рождении заподозрить заболевание или сразу диагностировать его. Окружность черепа уменьшена – микроцефалия, уши неправильной формы, низко расположены. Выявляются аномалии глазных яблок (микрофтальмия или анофтальмия), незаращение верхней губы и нёба, полидактилия, повышенная гибкость суставов. Морфологически определяются множественные пороки развития нервной системы и внутренних органов, в 80% -85% выявляется глухота. Вероятность повторного рождения аномального ребёнка составляет 1 -2%. Лечение неэффективно, прогноз неблагоприятен.
 § Частота синдрома составляет 1: 8000 – 1: 4500 рождений.")
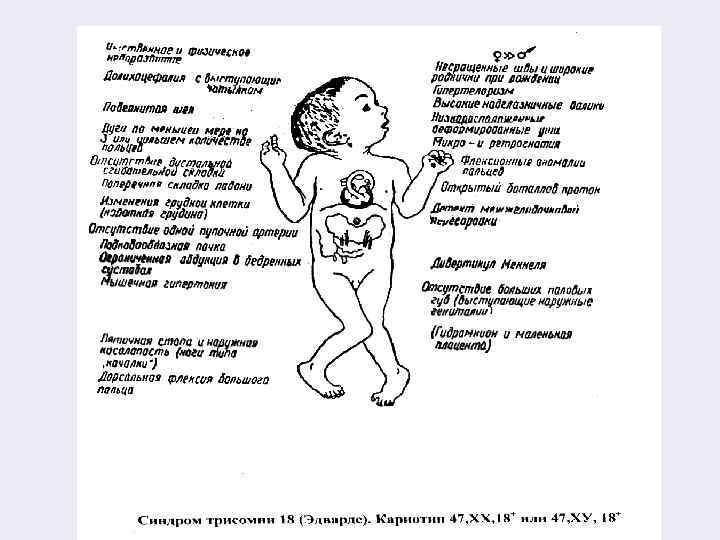
Синдром Эдвардса (трисомия 18) § Частота синдрома составляет 1: 8000 – 1: 4500 рождений. Более 70% всех детей – девочки. § Для этого синдрома характерны резкое пренатальное недоразвитие и множественные пороки развития костной системы: череп долихоцефалитичной формы, нижняя челюсть и ротовое отверстие маленькие, глазные щели узкие, ушные раковины деформированы. § Грудная клетка широкая и короткая, аномальное развитие стопы, имеется флексорное положение кистей. Выявлются выраженные пороки развития внутренних органов. § Дети с синдромом Эдвардса чаще рождаются у пожилых матерей. Специфического лечения нет. § 90% этих детей умирают на первом году жизни.

Болезни, обусловленные увеличением кратности полного гаплоидного набора хромосом – полиплоидия o o У человека обнаружена только трисомия. У мужчин имеется кариотип 69, ХХУ, у женщин – 69, ХХХ. Полиплоидия встречается очень редко. На неё приходится около 22, 5% всех спонтанных абортов с хромосомными аномалиями. Беременность с плодом с триплоидией осложняется токсикозом, повышением хориального гонадотропина. Это уже даёт основание заподозрить трисомию. Отмечается отставание на 6 -7 недель.

o o Основные пороки развития: микрофтальмия, расщелина губы и нёба, низко расположенные и деформированные ушные раковины, синдактилия, гидроцефалия, пороки развития всех внутренних органов. Дети с синдромом триплоидии практически нежизнеспособны. Они погибают в первые часы после рождения. Биопсия из разных участков кожи у таких детей показала, что часть тканей у них триплоидная, часть – диплоидная.

Болезни, связанные со структурными аномалиями хромосом Даже в том случае, если в клетке присутствуют все 46 хромосом, одна или две у них могут быть повреждены. l Эти структурные изменения обычно возникают вследствие разрывов хромосом. l Разрывы хромосом могут возникать самопроизвольно или в результате известных (например, вирусная инфекция, интоксикация, облучение и др. ) или же неустановленных причин, пагубно влияющих на процессы деления соматических и половых клеток. l

l Тенденция к хромосомным разрывам может также передаться ребенку от родителей или прародителя. l Эти структурные хромосомные дефекты встречаются весьма часто: примерно у 1: 400 новорождённых. l Хромосомные разрывы служат причиной разнообразных изменений хромосомной структуры. l К ним следует отнести: транслокации, инверсии, делеции.

Патологии, вызванные транслокациями n n Явление транслокации состоит в переносе участка из одной хромосомы в другую или другое место той же хромосомы. В большинстве случаев наблюдается явление, когда части хромосом меняются лишь местами без потери хромосомного материала. Такие хромосомные транслокации называются сбалансированными (реципрокными). Хромосомные транслокации встречаются у человека довольно часто. Многие из нас, не подозревая об этом, являются носителями различных сбалансированных транслокаций, рискуя произвести на свет дефектное потомство. Этот риск составляет примерно 10 -20% для будущих матерей и около 4% для будущих отцов.

n n n В настоящее время уже идентифицировано более 300 реципрокных транслокаций человека. Существуют также несбалансированные (нереципрокные) транслокации, когда кусок хромосомы полностью теряется за счёт переноса на другую негомологичную хромосому. Описанное явление сопровождается либо ранней гибелью зародыша, либо наличием врождённых дефектов и риском рождения в семье поражённых детей.
")
n n n Бывают также весьма редкие ситуации, когда вследствие хромосомной транслокации (обычно несбалансированной) происходит слияние двух хромосом в одну. При этом возникает клиническая картина заболевания, встречающихся при трисомиях 13, 21 аутосом с развитием наиболее часто синдрома Патау (трисомия 13), синдрома Дауна (трисомия 21). Среди детей с синдромом Дауна около 2% имеют кариотип вместо 47, ХХ (ХУ) - 46, ХХ (ХУ). При это обнаружено, что материал 21 хромосомы был передан на одну из хромосом «группы D» (13 -15).

n n Клиническая картина транслокационного варианта и простой формы трисомии аутосомы 21 идентичны. Распознаются они только с помощью цитогенетических методов. При появлении транслокационного варианта кариотипа 46, ХХ (ХУ) у женщины или мужчины вызывали 100% риск с болезнью Дауна.

Патологии, вызванные инверсией § Хромосома может разорваться в двух местах. При этом освобождённый участок поворачивается на 1800 и вновь встаёт на прежнее место. Этот процесс называется инверсией. § Поскольку функция генов в некоторой степени определяется их положением, то это явление может сопровождаться неприятными последствиями.

§ У некоторых носителей таких инверсий выявляются аномалии типа умственной отсталости или пороков развития. § Фенотип других, не обнаруживая какихлибо изменений, но в браках с ними регистрировались спонтанные аборты. § У представителей третьей группы не обнаруживалось вообще никаких аномалий.

Патологии, вызванные делециями хромосом § Иногда часть хромосомы может просто оторваться и исчезнуть. Это явление носит название делеция. § За счёт делеций возникает большая группа хромосомных заболеваний. И снова, в зависимости от выпавшей части или от того, какая хромосома вовлечена в процесс, могут возникать различные врождённые дефекты. § Природа этих дефектов варьирует – от незначительных до высокой степени серьёзных.
 и короткое (р)")
§ Как известно, из цитогенетики, у каждой хромосомы выявляется длинное (q) и короткое (р) плечо. § В настоящее время описана клиническая картина многих аутосомных аномалий: 4 р-, 5 р-, 9 р-, 11 q-, 13 q-, 18 р-, 18 q-, 21 q-, 22 q-.

n n n Остановимся на клинике более известного синдрома 5 р-, или синдрома Лежена. Его ещё называют синдромом «кошачьего крика» . Встречается с частотой 1: 40. 000 – 1: 50. 000 рождений. Наиболее характерным для синдрома является специфический плач, напоминающий «кошачий крик» . Кроме этого выявляются умственное и физическое недоразвитие, микроцефалия, низко расположенные деформированные ушные раковины, моноголидный разрез глаз, мышечная гипотония. «Кошачий крик» обусловлен изменением гортани, однако, с возрастом этот крик исчезает. Врождённые пороки развития встречаются редко. Большинство детей умирает в раннем возрасте. Специфического лечения нет.

Ребёнок с выраженными признаками синдрома кошачьего крика: микроцефалия, лунообразное лицо, эпикант, широкая плоская спинка носа, низко расположенные ушные раковины.

Генные болезни делятся на 2 группы: n Моногенные болезни, обусловленные дефектом одного гена. n Полигенные (мультифакториальные) болезни, связанные с нарушением взаимодействия нескольких генов и факторов окружающей среды.

По типу наследования моногенные болезни делятся на: n Аутосомно-доминантные. n Аутосомно-рецессивные. n Сцепленные с полом.

К болезням с выявленным биохимическим дефектом относятся наследственные энзимопатии. Классифицируются они на болезни аминокислотного, углеводного, липидного, стероидного, пуринового и пиримидинового обменов.
. n n Болезнь впервые описал в 1934 г.")
Среди множества аминоацидопатий рассмотрим фенилкетонурию (ФКУ). n n Болезнь впервые описал в 1934 г. норвежский врач Феллинг у умственно отсталых больных со специфическим «мышиным» запахом мочи. Среди умственно отсталых детей, которые содержатся в больницах около 1% страдает фенилкетонурией. У больных с данным заболеванием нарушено превращение фенилаланина в тирозин из-за резкого снижения активности фермента – фенилаланингидроксилазы. Ген, контролирующий все эти реакции локализуется в 12 хромосоме. Это приводит к повышению концентрации фенилаланина в крови на 4 день жизни ребёнка.

n n n Частота встречаемости фенилкетонурией в среднем 1: 10. 000. Частота гетерозиготности по ФКУ составляет 1, 25%. Наследование аутосомно-рецессивное. Ребёнок с ФКУ рождается здоровым, заболевание проявляется через 3 -6 месяцев после рождения, тогда как его биохимический маркёр – гиперфенилаланинемия – через 3 -4 дня после начала кормления. По этому маркёру заболевание может быть выявлено ещё в роддоме.

n Классическая клиническая картина ФКУ складывается из тетрады признаков: умственная отсталость, судорожный синдром, нарушение пигментного обмена, склонность к дерматитам. n n n Ведущим клиническим симптомом болезни является олигофрения, которая достигает различной степени выраженности. Очень рано отмечаются судорожные припадки. При физическом обследовании обращает на себя внимание то, что ребёнок выглядит более белокурым, у него светла кожа и голубые глаза. Это обусловлено недостаточностью образования пигмента меланина. От детей исходит заплесневелый мышиный запах.

n Основным биохимическим маркёром всех форм ФКУ является увеличение плазменной фенилаланина. концентрации Обычно используют пробу Феллинга – в 2 -5 мл свежей мочи добавляют 10 капель 5% раствора трёххлористого железа и уксусной кислоты. n Проба считается положительной при появлении через несколько секунд сине -зелёного окрашивания мочи. n

n n n Массовый скрининг на ФКУ можно осуществлять имунноферментным методом на аппарате «Флюороскан» . Лечение. Как только диагноз ФКУ подтверждён, больной ребёнок госпитализируется в больницу. Лечение заключается в назначении больному диеты с низким содержанием фенилаланина. Кроме того, назначается патентованные препараты, содержащие гидролизаты белков. Лечение назначают обычно в 2 -3 месяца, так как более поздно начатое лечение даёт меньший эффект. Лечение гидролизатом обычно продолжается 5 -8 лет без перерыва. В настоящее время разработаны методы дородовой диагностики ФКУ.

Болезни с наследственным предрасположением характеризуются следующими закономерностями: n Частота заболевания среди населения обычно выше, чем при моногенном заболевании. Полигенные болезни наблюдаются примерно у 20% населения. Так, шизофренией болеют около 1% населения, диабетом 5%, аллергическими заболеваниями страдают более 10%, гипертония – около 30%. n Для каждого типа брака родителей ожидаемый риск поражения очередного ребёнка тем выше, чем больше больных детей уже имеется в семье и наоборот.

n n Широкий клинический полиморфизм заболевания. Проявление заболевания зависит от возраста больного, эндокринных влияний, нерационального питания и других неблагоприятных факторов. Менделевские законы передачи заболевания не соответствуют ожидаемым при рецессивном или доминантном типе наследования. Количество поражённых обычно ниже, чем теоретически ожидаемое. Большое значение имеет выявление так называемого маркёра наследственной предрасположенности к определяемому заболеванию. Например, предрасположенность к сахарному диабету связана со снижением толерантности к глюкозе и др.

Медикогенетическое консультирование

n n n В настоящее время основной метод предупреждения возникновения и распространения наследственных болезней заключается в медико-генетическом консультировании. Медико-генетическое консультирование – это обмен информацией между врачом и будущими родителями, а также людьми, поражёнными болезнью, или их родственниками по вопросу о возможности проявления или повторения в семье наследственного заболевания. Главная его цель заключается в предупреждении рождения больного ребёнка.

Суть консультирования состоит в следующем: n n n Установление точного диагноза. Определения типа наследственного заболевания в данной семье. Составление прогноза рождения ребёнка с наследственной болезнью. Расчёт величины риска повторения заболевания в семье. Помощь семье в принятии правильного решения. Пропаганда медико-генетических знания среди врачей и населения.

Поводом для направления на консультацию являются следующие ситуации: § Наличие у женщины более двух предшествующих спонтанных абортов. § Мертворождения при исключении тяжелой гинекологической и соматической патологии у женщины. § Рождение ребенка с пороками развития, умственной отсталостью, судорогами, слепотой и глухотой. § Наличие у женщины рецидивирующих хронических заболеваний легких.

§ Упорная диарея у ребенка при исключении инфекционной природы заболевания. § Наличие у ребенка тяжелых поражений кожи (нейродермиты, экзема и др. ), не поддающихся терапии. § Наличие любого наследственного заболевания у родственников. § Несовместимость супругов по резусфактору крови, если в анамнезе имеются указания на резус-конфликт.

§ Бездетный брак, если исключены заболевания половой сферы у супругов. § Первичная и вторичная аменорея. § Работа супругов на вредном производстве. § Сведения о принятии супругами перед зачатием или женщиной до 16 недель беременности больших доз лекарственных или токсических препаратов.

§ Сведения о проведении рентгенологического обследования малого таза перед или во время беременности. § Вирусные инфекции, перенесенные женщиной во время беременности. § Непереносимость лекарственных препаратов или пищевых продуктов. § Лица, состоящие в кровном родстве. § Наличие у ребенка резкого отставания в физическом и половом развитии.

§ Лица, у которых в первом браке родился больной ребенок, и они выясняют степень риска рождения больного ребенка во втором браке. § Возраст женщины старше 35 лет, а мужчины – 40 лет (самый благоприятный возраст для родов – от 18 до 30 лет). § Наличие у беременной пристрастия к курению (особенно опасно курение более 15 сигарет в день).

§ Спортсменки высших разрядов, актрисы, балерины. § Тяжелые травмы во время беременности. § Категория женщин, окончивших специальную школу для умственно отсталых. § Венерические заболевания во время беременности.

В первую очередь медико-генетическое консультирование должны получать молодые супружеские пары, прежде чем иметь ребёнка. n n В этом случае не нужно появление на свет ребёнка с врождёнными дефектами для того, чтобы задним числом молодые осознали, что могли бы избежать несчастья или, хотя бы уменьшить опасность, прибегнув к пренатальной диагностике. Однако, в настоящее время только незначительная часть семей (1 -10% в разных странах мира), которым требуется совет генетика, обращаются в консультацию. Это слишком мало.

В г. Ростове-на-Дону медико-генетические консультации существуют в НИИАПе, Рост. ГМУ, где работают врачи-генетики высокой квалификации.


Этапы медико-генетического консультирования 1. Уточнение диагноза наследственного заболевания с помощью генетических методик исследования: генеалогических цитогенетических биохимических иммунологических и др.

2. Определение прогноза потомства. l На этом этапе задача врача-генетика заключается в определении риска рождения больного ребёнка. l В соответствии с основными законами наследственности передача наследственных заболеваний возможна несколькими путями.

l В тех случаях, когда у ребёнка появляется заболевание, как у одного из родителей, говорят о доминантном типе наследования. В настоящее время таких заболеваний около 950. l Когда у здоровых родителей появляется больной ребёнок, можно говорить о рецессивном типе наследования. Риск иметь больного ребёнка у родителей с рецессивным заболеванием равен 25%. Всего известно 790 рецессивно-наследуемых заболеваний.

Когда передача наследственного заболевания зависит от пола, то говорят о наследовании, сцепленном с полом (Х-сцепленном типе наследования). l Риск заболевания у мальчиков и носительство у девочек составляет 50%. l Заболеваний, передающихся по сцепленному с полом типу, в настоящее время насчитывается около 150. l

Большая группа наследственных заболеваний возникает в результате взаимодействия многих генов с другими генами и факторами внешней среды – так называемые мультифакториальные заболевания. l При этом из-за множественности одновременно действующих факторов вероятность их повторного совпадения, а значит и вероятность повторения заболевания низка. l

Расчёт риска проводится с использованием специально разработанных таблиц эмпирического риска при мультифакториальных заболеваниях.

Эмпирический риск Заболевание или порок n развития Болезнь Дауна: – Простая трисомия – Транслокационная форма n n n n Синдромы Клайнфельтера и Шерешевского- Тернера Расщепление губы (заячья губа) Расщепление нёба (волчья пасть) Врождённый порок сердца Сахарный диабет Аллергические заболевания Эпилепсия Шизофрения Риск в % 0, 5 – 4 0 – 25 1 5 – 10 3– 4 1– 2 5 – 20 10 – 15 15 – 20

n n n Генетический риск до 5% расценивается как низкий, не являющийся противопоказанием к продолжению деторождения в семье. Риск от 6 -20% принято считать средним, в этом случае рекомендации относительно планирования дальнейших беременностей зависят не только от величины риска, но и от тяжести медицинских и социальных последствий данного заболевания, а также от возможностей пренатальной диагностики. Генетический риск свыше 20% относится к категории высокого риска. При отсутствии пренатальной диагностики соответствующей патологии дальнейшее деторождение данной семье не рекомендуется.

ПРЕНАТАЛЬНАЯ ДИАГНОСТИКА НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ Во всех случаях предпочтительней предсказать появление врождённого дефекта, чем установить его уже после рождения. Для этих целей разработаны различные методы пренатальной диагностики, позволяющие установить точный диагноз тяжёлой или даже смертельной наследственной болезни у плода ещё в начале беременности. Оптимальные сроки консультации беременных женщин 6 -8 недель для определения плана пренатальной диагностики.

Показания для пренатальной диагностики n n n Наличие в семье точно-установленного наследственного заболевания. Носительство матерью рецессивного Хсцепленного заболевания. Хромосомная патология у предыдущего ребёнка. Множественные врождённые пороки развития у предыдущего ребёнка. Возраст будущей матери старше 35 лет.

На сегодняшний день возможна диагностика практически всех хромосомных болезней и более 100 наследственных болезней, биохимический дефект при которых установлен достоверно.

Методы пренатальной диагностики n n n Амниоцентез. Биохимическое исследование амниотической жидкости. Ультразвуковое исследование. Рентгенография. Фетоскопия. Биопсия хориона.

l l l 3. Третий этап - заключительный В итоге, проведя все генетические исследования в данной семье, врач-генетик даёт заключение об имеющейся болезни или вероятности возникновения заболевания в будущем. Давая своё заключение врач должен помочь семье принять ей правильное решение. Его роль заключается, прежде всего, в том, чтобы объяснить семье с реальных позиций, а именно природу и происхождение заболевания у ребёнка или родственника, величину риска и его значение с практической точки зрения. Получив такую информацию, родители сами должны принять решение по поводу больного ребёнка, продолжении беременности, и рождении будущих детей.

В ходе консультирования приходится сталкиваться с самыми разнообразными вопросами: опасен ли для потомства тот или иной лекарственный препарат, каковы последствия облучения, усыновлять ли ребёнка с отягощённой наследственностью, прерывать ли беременность и т. д. Ø Полезно сразу же внушить родителям мысль, что они не одиноки в своём несчастье. Обычно родители несколько успокаиваются, когда им сообщают, что примерно 1 из 7 беременностей кончается выкидышем, а 1 из 25 новорождённых имеет какие-то серьёзные аномалии. Ø Окончательное решение по интересующему родителей вопросу они принимают самостоятельно. Ø

Планирование семьи • Планирование дальнейшего деторождения в • • семье, в которой зарегистрирован случай наследственного заболевания, зависит от целого ряда факторов. Ведущую роль при этом играет уровень риска рождения больного ребенка. Генетический риск обычно выражается в процентах. Его разделят на низкий, средний и высокий.

n n При низком риске патологические изменения можно ожидать только у 5% потомков. В этом случае обычно нет противопоказаний для дальнейшего деторождения. Средний риск регистрируется, если вероятность рождения больного ребенка достигает 6 -20%. В такой ситуации планирование деторождения определяется предполагаемой тяжестью заболевания и возможностью дородовой диагностики. n Высокий генетический риск (более 20%) обычно сопровождается необходимостью исследования плода при беременности для обнаружения у него патологических изменений. Если дородовая диагностика невозможна, а заболевание является очень тяжелым, то часто супруги отказываются от дальнейшего деторождения.

n n Повышенный генетический риск часто сопровождает кровнородственные браки. Планирование деторождения может сопровождаться отказом от брака двух гетерозиготных носителей одного патологического гена. Возраст супругов также имеет большое значение пли планировании семьи. Показано увеличение рождения детей с наследственной патологий особенно у женщин старше 35 лет и у мужчин 40 лет, поэтому желательно окончить деторождение до этого возраста.

Окончательное решение супругов о количестве детей в семье зависит также от их интеллектуального и социального уровня, степени родства с больным родственником, количеством индивидуумов с наследственной патологией в семье.

При планировании беременности применяется так называемое периконцепционная профилактика, которая включает следующие этапы: n Выбор времени зачатия. Многочисленными исследованиями было показано, что риск рождения ребёнка с ВПР является особенно низким, если зачатие приходится на конец лета – начало осени. Самая высокая вероятность появления больного потомства регистрируется при начале беременности в весенние месяцы. n Обследование супругов до беременности для выявления у них различных инфекционных, эндокринных и других заболеваний. Прием поливитаминных препаратов в течение 2 -3 х месяцев до зачатия. n Эти лекарственные средства должны содержать фолиевую кислоту до 0, 4 – 1 мг в сутки, аскорбиновую кислоту, альфа-токоферол, витамины группы В.

n n Прием женщиной поливитаминов до 10 -12 недель беременности. Наблюдение за беременностью, которое включает методы дородовой диагностики, позволяющие обнаружить заболевание у плода. Широкое распространение таких профилактических мероприятий, серьёзное отношение супругов к рождению ребёнка наряду с общегосударственными программами по охране окружающей среды и совершенствование здравоохранения буду способствовать уменьшению груза наследственной и врождённой патологии.

Вопросы лекционного рейтинга n n n 1. На какие группы делятся наследственные болезни ? 2. Какова классификация хромосомных болезней ? 3. Что такое медико-генетическое консультирование ? 4. Перечислите этапы медико-генетического консультирования. 5. Назовите показания для пренатальной диагностики. 6. Какие этапы включает периконцепционная профилактика?
Понятие о наследственных болезнях человека.ppt