

первичные иммунодифициты.pptx
- Количество слайдов: 127
 Первичные иммунодифидиты Васильева Д. Д. Дынчева А. А. Петросян С. А. Группа 240 «А»
Первичные иммунодифидиты Васильева Д. Д. Дынчева А. А. Петросян С. А. Группа 240 «А»
 Основные понятия ИММУНОПАТОЛОГИЯ -область иммунологии, изучающая патологические процессы и заболевания иммунной системы. ИММУНОДЕФИЦИТ -заболевание, обусловленное ослаблением функций иммунной системы. АУТОИММУННЫЕ- заболевания, обусловленные избыточным реагированием иммунной системы.
Основные понятия ИММУНОПАТОЛОГИЯ -область иммунологии, изучающая патологические процессы и заболевания иммунной системы. ИММУНОДЕФИЦИТ -заболевание, обусловленное ослаблением функций иммунной системы. АУТОИММУННЫЕ- заболевания, обусловленные избыточным реагированием иммунной системы.
 иммунодифициты ПИД-гетерогенная группа наследственных расстройств, причиной которых являются мутации специфических генов") Первичные (генетически обусловленные) иммунодифициты ПИД-гетерогенная группа наследственных расстройств, причиной которых являются мутации специфических генов с дефектами в одном или нескольких компонентов ИС. Реализация генитического дефекта приводит к появлению многих заболеваний. В основном эти заболевания-результат повышенной чувствительности пациентов к инфекциям, аллергии и предрасположенности к аутоиммунноым, онкологическим заболеваниям.
Первичные (генетически обусловленные) иммунодифициты ПИД-гетерогенная группа наследственных расстройств, причиной которых являются мутации специфических генов с дефектами в одном или нескольких компонентов ИС. Реализация генитического дефекта приводит к появлению многих заболеваний. В основном эти заболевания-результат повышенной чувствительности пациентов к инфекциям, аллергии и предрасположенности к аутоиммунноым, онкологическим заболеваниям.
 Распространненость ПИД В настоящее вемя известно около 150 клинических форм генетически опосредованной иммунной недостаточности. Из них в 130 открыты генные дефекты.
Распространненость ПИД В настоящее вемя известно около 150 клинических форм генетически опосредованной иммунной недостаточности. Из них в 130 открыты генные дефекты.
 Классификация ПИД Все описанные на сегодняшний день генетически опосредованные иммунодифициты разделены на 8 групп, в каждой из которой выделяют подгруппы: Группа 1 -комбинированные Т- и В-клеточные иммунодифициты Группа 2 -преимущественый дефицит Ат Группа 3 -синдромы иммунодифицитов с хлолшо охарактеризованными клиническими признаками Группа 4 - генетические нарушения иммунной регуляции
Классификация ПИД Все описанные на сегодняшний день генетически опосредованные иммунодифициты разделены на 8 групп, в каждой из которой выделяют подгруппы: Группа 1 -комбинированные Т- и В-клеточные иммунодифициты Группа 2 -преимущественый дефицит Ат Группа 3 -синдромы иммунодифицитов с хлолшо охарактеризованными клиническими признаками Группа 4 - генетические нарушения иммунной регуляции
 Группа 6 -дефекты врожденного") Группа 5 -врожденные дефекты фагоцитов(числа, функций и др. ) Группа 6 -дефекты врожденного иммунитета Группа 7 -аутовоспалительные заболевания Группа 8 -дефециты комплемента.
Группа 5 -врожденные дефекты фагоцитов(числа, функций и др. ) Группа 6 -дефекты врожденного иммунитета Группа 7 -аутовоспалительные заболевания Группа 8 -дефециты комплемента.
 Частота встречаемости Пид Преимущественный дефицит АТ Комбинированные Т-и В-коеточные иммунодифициты Другие хорошо охарактеризованные иммунодефицититы Дефициты комплемента Аутовоспалительные нарушения Дефекты врожденного иммунитета: рецепторов и сигнальных компонентов Генитические нарушения иммунной регуляции Врожденные дефекты фагоцитов
Частота встречаемости Пид Преимущественный дефицит АТ Комбинированные Т-и В-коеточные иммунодифициты Другие хорошо охарактеризованные иммунодефицититы Дефициты комплемента Аутовоспалительные нарушения Дефекты врожденного иммунитета: рецепторов и сигнальных компонентов Генитические нарушения иммунной регуляции Врожденные дефекты фагоцитов
 Основные клинические характеристики ПИД Проявление заболевания, как правило с раннего возраста Рецидивирующие инфекционные поражения органов дыхания Рецидивирующие пиогенные(гноеродные) инфекционные заболевания Рецидивирование инфекционных заболеваний, вызванных одним и тем же типом патогена Аутоиммунные или хронические воспалительные заболевания И др.
Основные клинические характеристики ПИД Проявление заболевания, как правило с раннего возраста Рецидивирующие инфекционные поражения органов дыхания Рецидивирующие пиогенные(гноеродные) инфекционные заболевания Рецидивирование инфекционных заболеваний, вызванных одним и тем же типом патогена Аутоиммунные или хронические воспалительные заболевания И др.
 Наиболее частые проявления ПИДрецидивирующие инфекции или инфекции, вызванные оппортунистическими м/о (это м/о которые обычно не приводят к болезни особей с нормальной иммунной системой). При этом природа возбудителя может зависеть от уровня поражения ИС по клеточноопосредованному, гуморальному, фагоцитарному, комплементзависимому или комбинированному типу. Для иммунодифицитов характерны не только инфекционные осложнения, но и поражения различных систем органов.
Наиболее частые проявления ПИДрецидивирующие инфекции или инфекции, вызванные оппортунистическими м/о (это м/о которые обычно не приводят к болезни особей с нормальной иммунной системой). При этом природа возбудителя может зависеть от уровня поражения ИС по клеточноопосредованному, гуморальному, фагоцитарному, комплементзависимому или комбинированному типу. Для иммунодифицитов характерны не только инфекционные осложнения, но и поражения различных систем органов.
 Аутоиммунные проявления У больных с ПИД выявляют разные аутоиммунные нарушения, либо связанные с другими проявлениями иммунодефицита (табл. 1. ), либо носящие определяющий характер (табл. 2. ).
Аутоиммунные проявления У больных с ПИД выявляют разные аутоиммунные нарушения, либо связанные с другими проявлениями иммунодефицита (табл. 1. ), либо носящие определяющий характер (табл. 2. ).
 Табл. 1. Аутоиммунное заболевание Иммунодефициты Идиопатическая тромбоцитопеническая пурпура Ig. А-дефицит, общая вариабельная иммунная недостаточность, хроническая гранулематозная болезнь, синдром Ди Джорджи, синдром Вискотта-Олдрича Аутоиммунная гемолитическая анемия Ig. А-дефицит, общая вариабельная иммунная недостаточность, хроническая гранулематозная болезнь, синдром Ди Джорджи, дефицит HLA-||, синдром Вискотта-Олдрича Системная красная волчанка Дефицит комплемента, хроническая гранулематозная болезнь, Ig. А-дефицит Ювегильный артрит Ig. А-дефицит, Ди Джорджи, синдром Вискотта-Олдрича, Хсцепленная агаммаглбулинемия Склерозирующие холонгиты Дефицит перегруппировки классов иммуноглобулинов Васкулиты Синдром Вискотта-Олдрича, Х-сцепленные лимфопролиферативные заболевания
Табл. 1. Аутоиммунное заболевание Иммунодефициты Идиопатическая тромбоцитопеническая пурпура Ig. А-дефицит, общая вариабельная иммунная недостаточность, хроническая гранулематозная болезнь, синдром Ди Джорджи, синдром Вискотта-Олдрича Аутоиммунная гемолитическая анемия Ig. А-дефицит, общая вариабельная иммунная недостаточность, хроническая гранулематозная болезнь, синдром Ди Джорджи, дефицит HLA-||, синдром Вискотта-Олдрича Системная красная волчанка Дефицит комплемента, хроническая гранулематозная болезнь, Ig. А-дефицит Ювегильный артрит Ig. А-дефицит, Ди Джорджи, синдром Вискотта-Олдрича, Хсцепленная агаммаглбулинемия Склерозирующие холонгиты Дефицит перегруппировки классов иммуноглобулинов Васкулиты Синдром Вискотта-Олдрича, Х-сцепленные лимфопролиферативные заболевания
 Табл. 2. Иммунодефицит Аутоиммунные проявления Аутоиммунный лимфопролиферативный синдром Аутоиммунная гемолитическая анемия, идиопатическая тромбоцитопеническая пурпура, аутоиммунная нейтропения, первичный билиарный цирроз Аутоиммунная полиэндокринопатия с кандиозом и эктодермальной дисплазий Гипоадренализм, сахпрный диабет, аутоиммунный гепатит Х-сцепленная иммунная дисрегуляция, полиэндокронопатия, энтеропатия Аутоиммунная энтеропатия, сахарный диабет, гемолитическая анемия, экзема
Табл. 2. Иммунодефицит Аутоиммунные проявления Аутоиммунный лимфопролиферативный синдром Аутоиммунная гемолитическая анемия, идиопатическая тромбоцитопеническая пурпура, аутоиммунная нейтропения, первичный билиарный цирроз Аутоиммунная полиэндокринопатия с кандиозом и эктодермальной дисплазий Гипоадренализм, сахпрный диабет, аутоиммунный гепатит Х-сцепленная иммунная дисрегуляция, полиэндокронопатия, энтеропатия Аутоиммунная энтеропатия, сахарный диабет, гемолитическая анемия, экзема
 Общая клиническая характеристика ПИД с преимущественным поражением Т-лимфоитов Появление симптомов с 4 -5 мес. Повторные инфекционные заболевания Инфекционные заболевания, вызываемые оппортунистическими м/о Фатальные инфекционные заболевания И д. р.
Общая клиническая характеристика ПИД с преимущественным поражением Т-лимфоитов Появление симптомов с 4 -5 мес. Повторные инфекционные заболевания Инфекционные заболевания, вызываемые оппортунистическими м/о Фатальные инфекционные заболевания И д. р.
 Общая характеристика ПИД с преимущественным поражение Влимфоцитов Появление симптомов в возрасте 7 -9 мес. , после изчезновения материнских Ат Повторные инфекционные заболевания, вызванные инкапсулированными бактериями Хронические очаги инфекции Лимфоидная гиперплазия Повышенная частота аллергических и аутоиммунных заболеваний И др.
Общая характеристика ПИД с преимущественным поражение Влимфоцитов Появление симптомов в возрасте 7 -9 мес. , после изчезновения материнских Ат Повторные инфекционные заболевания, вызванные инкапсулированными бактериями Хронические очаги инфекции Лимфоидная гиперплазия Повышенная частота аллергических и аутоиммунных заболеваний И др.
 Комбинироанный иммуноцифицит") Комбинированные иммунодефициты (ПКИД) Комбинироанный иммуноцифицит
Комбинированные иммунодефициты (ПКИД) Комбинироанный иммуноцифицит
 Общим для комбинированных иммунодефицитов является раннее их проявление, инфицирование множеством микроорганизмов") Комбинированные иммунодефициты (ПКИД) Общим для комбинированных иммунодефицитов является раннее их проявление, инфицирование множеством микроорганизмов (как правило, условнопатогенными), выраженная клиническая симптоматика, тяжелое течение инфекционного процесса. Дети с ПКИД страдают от тяжело протекающих инфекций легких, кожи, слизистой рта и горла, часто наблюдается кандидоз глотки, пищевода, у больных развивается хронический понос, сепсис. Часто ПКИД сочетается с аномалиями скелета; для них характерна лимфоцитопения, снижение количества Т-лимфоцитов и их функциональной активности, гипоплазия тимуса. Для некоторых форм характерна гипогаммаглобулинемия.
Комбинированные иммунодефициты (ПКИД) Общим для комбинированных иммунодефицитов является раннее их проявление, инфицирование множеством микроорганизмов (как правило, условнопатогенными), выраженная клиническая симптоматика, тяжелое течение инфекционного процесса. Дети с ПКИД страдают от тяжело протекающих инфекций легких, кожи, слизистой рта и горла, часто наблюдается кандидоз глотки, пищевода, у больных развивается хронический понос, сепсис. Часто ПКИД сочетается с аномалиями скелета; для них характерна лимфоцитопения, снижение количества Т-лимфоцитов и их функциональной активности, гипоплазия тимуса. Для некоторых форм характерна гипогаммаглобулинемия.
") К группе комбинированных иммунодефицитов относятся: Ретикулярный дисгенез Тяжелый комбинированный иммунодефицит (Х – сцепленный, аутосомно-рецессивный) Иммунодефицит, сцепленный с дефицитом АДА Иммунодефицит, сцепленный с дефицитом ПНФ Синдром голых лимфоцитов Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар) Синдром Вискотта-Олдрича Гипер-Ig. М синдром
К группе комбинированных иммунодефицитов относятся: Ретикулярный дисгенез Тяжелый комбинированный иммунодефицит (Х – сцепленный, аутосомно-рецессивный) Иммунодефицит, сцепленный с дефицитом АДА Иммунодефицит, сцепленный с дефицитом ПНФ Синдром голых лимфоцитов Иммунодефицит с атаксией-телеангиэктазией (синдром Луи-Бар) Синдром Вискотта-Олдрича Гипер-Ig. М синдром
 Ретикулярный дисгенез Заболевание характеризуется дефектностью стволовых кроветворных клеток у новорожденных. В костном мозге практически отсутствуют клетки-предшественники лейкоцитов. В связи с этим наблюдается нарушение кроветворения и лимфопоэза. У больного отмечается глубокая панцитопения (лимфоцитопения). Полностью отсутствуют иммунные реакции гуморального и клеточного типов. Уровень общего иммуноглобулина и отдельных классов близок к нулю. У новорожденных наблюдается гипоплазия вилочковой железы и лимфоидных органов. В селезенке и лимфатических узлах преобладают ретикулярные клетки и макрофаги, отсутствуют фолликулы. Младенцы умирают в течение 1 -го месяца после рождения от септического процесса. Лечение данного заболевания исключительно хирургическое – трансплантация гемопоэтической ткани.
Ретикулярный дисгенез Заболевание характеризуется дефектностью стволовых кроветворных клеток у новорожденных. В костном мозге практически отсутствуют клетки-предшественники лейкоцитов. В связи с этим наблюдается нарушение кроветворения и лимфопоэза. У больного отмечается глубокая панцитопения (лимфоцитопения). Полностью отсутствуют иммунные реакции гуморального и клеточного типов. Уровень общего иммуноглобулина и отдельных классов близок к нулю. У новорожденных наблюдается гипоплазия вилочковой железы и лимфоидных органов. В селезенке и лимфатических узлах преобладают ретикулярные клетки и макрофаги, отсутствуют фолликулы. Младенцы умирают в течение 1 -го месяца после рождения от септического процесса. Лечение данного заболевания исключительно хирургическое – трансплантация гемопоэтической ткани.
 Х- сцепленный тип ТКИД б) Аутосомно-рецессивный тип") Тяжелый комбинированный иммунодефицит Различают 2 формы: a) Х- сцепленный тип ТКИД б) Аутосомно-рецессивный тип ТКИД
Тяжелый комбинированный иммунодефицит Различают 2 формы: a) Х- сцепленный тип ТКИД б) Аутосомно-рецессивный тип ТКИД
 Х- сцепленный тип ТКИД Болеют только мальчики. Иммунодефицит сцеплен с дефектностью гаммацепи рецептора") a) Х- сцепленный тип ТКИД Болеют только мальчики. Иммунодефицит сцеплен с дефектностью гаммацепи рецептора ИЛ-2 на Т-лимфоцитах, что приводит к нарушению трансдукции сигнала внутрь клетки с рецепторов, воспринимающих ИЛ -2, ИЛ-4, ИЛ-7, ИЛ-9, ИЛ-15. (Эти рецепторы имеют одинаковый тип гамма-цепи). Дефект расположен в хромосоме Xq 13. При этой форме иммунодефицита наблюдается нарушение дифференцировки стволовой лимфоидной клетки в Т- и В-лимфоциты.
a) Х- сцепленный тип ТКИД Болеют только мальчики. Иммунодефицит сцеплен с дефектностью гаммацепи рецептора ИЛ-2 на Т-лимфоцитах, что приводит к нарушению трансдукции сигнала внутрь клетки с рецепторов, воспринимающих ИЛ -2, ИЛ-4, ИЛ-7, ИЛ-9, ИЛ-15. (Эти рецепторы имеют одинаковый тип гамма-цепи). Дефект расположен в хромосоме Xq 13. При этой форме иммунодефицита наблюдается нарушение дифференцировки стволовой лимфоидной клетки в Т- и В-лимфоциты.
 Аутосомно-рецессивный тип ТКИД Болеют в одинаковой степени мальчики и девочки. Этот тип иммунодефицита") б) Аутосомно-рецессивный тип ТКИД Болеют в одинаковой степени мальчики и девочки. Этот тип иммунодефицита может быть сцеплен с одним из следующих факторов: – дефицитом тирозинкиназы ZAP-70, которая участвует в передаче сигнала с Т-клеточного рецептора. Дефект локализован в хромосоме 2 q 12. Для этого вида ТКИД характерно отсутствие CD 8+-клеток в периферической крови. При этом может наблюдаться нормальное содержание CD 4+-клеток и нормальная концентрация иммуноглобулина в крови. Правда, при этом CD 4+-клетки функционально неактивны. – дефицитом JAK-3, участвующем в трансдукции сигнала с рецепторов для ИЛ-2. Дефект локализован в хромосоме 19 p 13. – дефектностью генов, кодирующих RAG-1 и RAG-2, участвующих в реаранжировке генов, кодирующих Т-клеточный рецептор и молекулы иммуноглобулинов. В итоге формируются Т- и В-лимфоциты с дефектными антигенраспознающими рецепторами, неспособные к развитию полноценной иммунной реакции. Дефект расположен в хромосоме 11 р13.
б) Аутосомно-рецессивный тип ТКИД Болеют в одинаковой степени мальчики и девочки. Этот тип иммунодефицита может быть сцеплен с одним из следующих факторов: – дефицитом тирозинкиназы ZAP-70, которая участвует в передаче сигнала с Т-клеточного рецептора. Дефект локализован в хромосоме 2 q 12. Для этого вида ТКИД характерно отсутствие CD 8+-клеток в периферической крови. При этом может наблюдаться нормальное содержание CD 4+-клеток и нормальная концентрация иммуноглобулина в крови. Правда, при этом CD 4+-клетки функционально неактивны. – дефицитом JAK-3, участвующем в трансдукции сигнала с рецепторов для ИЛ-2. Дефект локализован в хромосоме 19 p 13. – дефектностью генов, кодирующих RAG-1 и RAG-2, участвующих в реаранжировке генов, кодирующих Т-клеточный рецептор и молекулы иммуноглобулинов. В итоге формируются Т- и В-лимфоциты с дефектными антигенраспознающими рецепторами, неспособные к развитию полноценной иммунной реакции. Дефект расположен в хромосоме 11 р13.
 Для группы тяжелых комбинированных иммунодефицитов характерны лимфоцитопения, гипоплазия тимуса, снижение количественного содержания Т- и В-лимфоцитов в крови и снижение их функциональной активности. Следует заметить, что содержание гранулоцитов и эритроцитов в периферической крови находится в пределах нормы. У таких детей отсутствуют кожные реакции замедленного типа и подавлена продукция антител на специфические антигены. Содержание Ig. А, М, G в крови снижено.
Для группы тяжелых комбинированных иммунодефицитов характерны лимфоцитопения, гипоплазия тимуса, снижение количественного содержания Т- и В-лимфоцитов в крови и снижение их функциональной активности. Следует заметить, что содержание гранулоцитов и эритроцитов в периферической крови находится в пределах нормы. У таких детей отсутствуют кожные реакции замедленного типа и подавлена продукция антител на специфические антигены. Содержание Ig. А, М, G в крови снижено.
 У младенцев наблюдается задержка развития, снижение массы тела. Они страдают от хронического поноса, пневмонии, грибковых поражений рта, горла, глаз, придаточных пазух носа. Обычно у таких детей наблюдаются вирусные инфекции и микозы или комбинированные инфекции, в результате присоединения условно-патогенной микрофлоры. Дефект гуморального иммунитета проявляется в меньшей степени, чем клеточного, так как они получают антитела с молоком матери. У них иммунная система настолько слаба, что живые ослабленные вакцины (типа полиомиелитной вакцины Сэбина) могут вызвать инфекционное заболевание. Продолжительность жизни таких детей составляет около года. Она может быть продлена путем их изолирования от внешней среды и содержания в стерильных палатах, в которых полностью исключается контакт с микробами. Следует заметить, что такие мероприятия оправданы только в ожидании адекватного лечения.
У младенцев наблюдается задержка развития, снижение массы тела. Они страдают от хронического поноса, пневмонии, грибковых поражений рта, горла, глаз, придаточных пазух носа. Обычно у таких детей наблюдаются вирусные инфекции и микозы или комбинированные инфекции, в результате присоединения условно-патогенной микрофлоры. Дефект гуморального иммунитета проявляется в меньшей степени, чем клеточного, так как они получают антитела с молоком матери. У них иммунная система настолько слаба, что живые ослабленные вакцины (типа полиомиелитной вакцины Сэбина) могут вызвать инфекционное заболевание. Продолжительность жизни таких детей составляет около года. Она может быть продлена путем их изолирования от внешней среды и содержания в стерильных палатах, в которых полностью исключается контакт с микробами. Следует заметить, что такие мероприятия оправданы только в ожидании адекватного лечения.
 Лечение Единственным эффективным способом лечения таких болезней является трансплантация гемопоэтической ткани (костного мозга, клеток эмбриональной печени, стволовых клеток периферической крови) и комплекса – тимус – грудина.
Лечение Единственным эффективным способом лечения таких болезней является трансплантация гемопоэтической ткани (костного мозга, клеток эмбриональной печени, стволовых клеток периферической крови) и комплекса – тимус – грудина.
 Иммунодефицит, сцепленный с АДА и с ПНФ
Иммунодефицит, сцепленный с АДА и с ПНФ
 Дефицит аденозиндезаминазы в лимфоцитах приводит к накоплению в") Иммунодефицит, сцепленный с АДА (аденозиндезаминазой) Дефицит аденозиндезаминазы в лимфоцитах приводит к накоплению в клетках токсических продуктов обмена (дезоксиаденозина и др. ), которые и подавляют функцию лимфоцитов. Заболевание проявляется рецидивирующими инфекциями дыхательных путей, ЛОР-органов. С первых дней жизни у детей наблюдается лимфоцитопения, недостаточность функции Тлимфоцитов. У таких пациентов часто выявляются аномалии развития скелета (деформации, окостенения), отсутствие тимуса.
Иммунодефицит, сцепленный с АДА (аденозиндезаминазой) Дефицит аденозиндезаминазы в лимфоцитах приводит к накоплению в клетках токсических продуктов обмена (дезоксиаденозина и др. ), которые и подавляют функцию лимфоцитов. Заболевание проявляется рецидивирующими инфекциями дыхательных путей, ЛОР-органов. С первых дней жизни у детей наблюдается лимфоцитопения, недостаточность функции Тлимфоцитов. У таких пациентов часто выявляются аномалии развития скелета (деформации, окостенения), отсутствие тимуса.
 Лечение В тяжелых случаях – трансплантация гемопоэтической ткани; переливание эритроцитарной массы как источника АДА; противомикробные препараты. Симптоматическое лечение.
Лечение В тяжелых случаях – трансплантация гемопоэтической ткани; переливание эритроцитарной массы как источника АДА; противомикробные препараты. Симптоматическое лечение.
 Развитие синдрома связано с: недостаточностью ПНФ, подавлением активности") Иммунодефицит, сцепленный с ПНФ (пуриннуклеозидфосфорилазой) Развитие синдрома связано с: недостаточностью ПНФ, подавлением активности рибонуклеотидредуктазы и синтеза в лимфоцитах ДНК. При этом больше угнетается функция Тлимфоцитов, чем В-лимфоцитов.
Иммунодефицит, сцепленный с ПНФ (пуриннуклеозидфосфорилазой) Развитие синдрома связано с: недостаточностью ПНФ, подавлением активности рибонуклеотидредуктазы и синтеза в лимфоцитах ДНК. При этом больше угнетается функция Тлимфоцитов, чем В-лимфоцитов.
 Заболевание проявляется развитием рецидивирующих, трудно поддающихся лечению инфекций. Чаще всего заболевание начинается с поражения дыхательных путей. Затем в процесс вовлекаются другие органы и системы. Часто у больных наблюдаются гиперпластические анемии, спастические тетраплегии, атаксия. В иммунологических исследованиях выявляется снижение функции Тлимфоцитов. Показатели В-системы иммунитета чаще всего в пределах нормы.
Заболевание проявляется развитием рецидивирующих, трудно поддающихся лечению инфекций. Чаще всего заболевание начинается с поражения дыхательных путей. Затем в процесс вовлекаются другие органы и системы. Часто у больных наблюдаются гиперпластические анемии, спастические тетраплегии, атаксия. В иммунологических исследованиях выявляется снижение функции Тлимфоцитов. Показатели В-системы иммунитета чаще всего в пределах нормы.
 Лечение В тяжелых случаях переливание эритроцитарной массы как источника ПНФ противомикробная терапия
Лечение В тяжелых случаях переливание эритроцитарной массы как источника ПНФ противомикробная терапия
 Синдром голых лимфоцитов
Синдром голых лимфоцитов
 Синдром развивается из-за нарушения экспрессии молекул HLA-I и II класса на лимфоцитах и антигенпредставляющих клетках. Заболевание наследуется по аутосомнорецессивному типу, ему подвержены как девочки, так и мальчики.
Синдром развивается из-за нарушения экспрессии молекул HLA-I и II класса на лимфоцитах и антигенпредставляющих клетках. Заболевание наследуется по аутосомнорецессивному типу, ему подвержены как девочки, так и мальчики.
 При этой патологии наблюдается нарушение созревания в тимусе клеток с фенотипом CD 4+, у них также подавлена экспрессия молекул CD 3 и существенно повышено формирование лимфоцитов с фенотипом CD 8+. Отсутствие полноценных Т-лимфоцитов хелперов (CD 4+) и неспособность лимфоцитов экспрессировать HLA –II класса формируют специфическую клиническую картину заболевания.
При этой патологии наблюдается нарушение созревания в тимусе клеток с фенотипом CD 4+, у них также подавлена экспрессия молекул CD 3 и существенно повышено формирование лимфоцитов с фенотипом CD 8+. Отсутствие полноценных Т-лимфоцитов хелперов (CD 4+) и неспособность лимфоцитов экспрессировать HLA –II класса формируют специфическую клиническую картину заболевания.
 Клинически заболевание проявляется на первом году жизни ребенка. Наблюдается задержка роста и развития на фоне частых вирусных инфекций герпетической, аденовирусной и цитомегаловирусной этиологии. Часто у детей происходит поражение придаточных пазух носа, бронхолегочного аппарата, развитие интерстициальной пневмонии. Нередки грибковые инфекции. Наблюдается синдром мальабсорбции, а также трудноизлечимые диареи. Чрезвычайно опасной для таких детей является вакцинация БЦЖ, как и другими живыми вакцинами, поскольку они способны вызвать генерализованные инфекции. Прогноз заболевания в тяжелых случаях неблагоприятный. Дети погибают от септических состояний в течение первых лет жизни. Возможны стертые малосимптомные формы. В этих случаях адекватная симптоматическая терапия позволяет добиться удовлетворительного эффекта.
Клинически заболевание проявляется на первом году жизни ребенка. Наблюдается задержка роста и развития на фоне частых вирусных инфекций герпетической, аденовирусной и цитомегаловирусной этиологии. Часто у детей происходит поражение придаточных пазух носа, бронхолегочного аппарата, развитие интерстициальной пневмонии. Нередки грибковые инфекции. Наблюдается синдром мальабсорбции, а также трудноизлечимые диареи. Чрезвычайно опасной для таких детей является вакцинация БЦЖ, как и другими живыми вакцинами, поскольку они способны вызвать генерализованные инфекции. Прогноз заболевания в тяжелых случаях неблагоприятный. Дети погибают от септических состояний в течение первых лет жизни. Возможны стертые малосимптомные формы. В этих случаях адекватная симптоматическая терапия позволяет добиться удовлетворительного эффекта.
, на фоне") В иммунограммах больных наблюдается: сниженное содержание в крови Т-хелперов (CD 4+), на фоне либо нормального общего количества лимфоцитов, либо их сниженных значений, отсутствие реакций ГЗТ, снижение или отсутствие бласттрансформирующей способности лимфоцитов на митогены, снижение реактивность клеток в СКЛ, нормальное содержание Влимфоцитов в крови, сниженные концентрации всех классов иммуноглобулинов в сыворотке, сниженная способность лимфоцитов продуцировать специфические антитела на бактериальные и вирусные антигены.
В иммунограммах больных наблюдается: сниженное содержание в крови Т-хелперов (CD 4+), на фоне либо нормального общего количества лимфоцитов, либо их сниженных значений, отсутствие реакций ГЗТ, снижение или отсутствие бласттрансформирующей способности лимфоцитов на митогены, снижение реактивность клеток в СКЛ, нормальное содержание Влимфоцитов в крови, сниженные концентрации всех классов иммуноглобулинов в сыворотке, сниженная способность лимфоцитов продуцировать специфические антитела на бактериальные и вирусные антигены.
 Лечение Антимикробная терапия Пересадка гемопоэтических клеток введение препаратов иммуноглобулинов
Лечение Антимикробная терапия Пересадка гемопоэтических клеток введение препаратов иммуноглобулинов
") Иммунодефицит с атаксиейтелеангиэктазией (синдром Луи-Бар)
Иммунодефицит с атаксиейтелеангиэктазией (синдром Луи-Бар)
 Заболевание связано с дефектностью киназ, участвующих в регуляции клеточного цикла. Заболевание наследуется аутосомно-рецессивно. Дефект локализован в хромосоме 11 q 22.
Заболевание связано с дефектностью киназ, участвующих в регуляции клеточного цикла. Заболевание наследуется аутосомно-рецессивно. Дефект локализован в хромосоме 11 q 22.
 Заболевание начинается в возрасте 3 -5 лет и старше. Проявляется в виде телеангиэктазий, ломкости капилляров, в первую очередь кожи и глаз, атаксии (трудности поддержания равновесия), рецидивирующих инфекций носовых пазух и легких вирусной и бактериальной природы. Часто у пациентов наблюдается витилиго и гиперпигментные пятна на лице после пребывания на солнце. У половины больных наблюдается отставание в умственном развитии, заторможенность, адинамия, ограниченность интересов. Дети очень поздно учатся ходить. Взаимоотношения между развитием иммунодефицита, нервными расстройствами и сосудистыми поражениями до настоящего времени не выяснены.
Заболевание начинается в возрасте 3 -5 лет и старше. Проявляется в виде телеангиэктазий, ломкости капилляров, в первую очередь кожи и глаз, атаксии (трудности поддержания равновесия), рецидивирующих инфекций носовых пазух и легких вирусной и бактериальной природы. Часто у пациентов наблюдается витилиго и гиперпигментные пятна на лице после пребывания на солнце. У половины больных наблюдается отставание в умственном развитии, заторможенность, адинамия, ограниченность интересов. Дети очень поздно учатся ходить. Взаимоотношения между развитием иммунодефицита, нервными расстройствами и сосудистыми поражениями до настоящего времени не выяснены.
 В иммунном статусе больных отмечаются: уменьшение содержания Т-лимфоцитов, сниженный уровень Ig. А, Ig. Е, иногда Ig. G 2, сниженный ответ в РБТ лимфоцитов на митогены (ФГА) и бактериальные антигены. Такие больные способны доживать до 20 -40 лет.
В иммунном статусе больных отмечаются: уменьшение содержания Т-лимфоцитов, сниженный уровень Ig. А, Ig. Е, иногда Ig. G 2, сниженный ответ в РБТ лимфоцитов на митогены (ФГА) и бактериальные антигены. Такие больные способны доживать до 20 -40 лет.
 Лечение Пересадка гемопоэтической ткани Препараты гормонов тимуса Препараты иммуноглобулинов для внутривенного введения Симптоматические средства
Лечение Пересадка гемопоэтической ткани Препараты гормонов тимуса Препараты иммуноглобулинов для внутривенного введения Симптоматические средства
 Синдром Вискотта-Олдрича
Синдром Вискотта-Олдрича
. Для него характерно нарушение активации") Заболевание развивается только у мальчиков (Хсцепленный тип заболевания). Для него характерно нарушение активации CD 4+ и CD 8+- клеток, продукции Ig. М к капсулярным бактериям (у больных не вырабатываются антитела к полисахаридам). При этом у больных наблюдается нормальный уровень Т- и В-лимфоцитов в периферической крови, нормальная концентрация в сыворотке Ig. G и гиперглобулинемия А и Е.
Заболевание развивается только у мальчиков (Хсцепленный тип заболевания). Для него характерно нарушение активации CD 4+ и CD 8+- клеток, продукции Ig. М к капсулярным бактериям (у больных не вырабатываются антитела к полисахаридам). При этом у больных наблюдается нормальный уровень Т- и В-лимфоцитов в периферической крови, нормальная концентрация в сыворотке Ig. G и гиперглобулинемия А и Е.
 Для синдрома характерна триада симптомов: 1. тромбоцитопения 2. экзема 3. частые пиогенные инфекции, которые манифестируют, как правило, с 6 -месячного возраста.
Для синдрома характерна триада симптомов: 1. тромбоцитопения 2. экзема 3. частые пиогенные инфекции, которые манифестируют, как правило, с 6 -месячного возраста.
 Заболевание вначале проявляется геморрагическими кровотечениями, которые локализуются преимущественно в кишечном тракте и проявляются примесью крови в кале (диарея с кровью). Дети также имеют петехиальные кровоизлияния в кожу. С 1 года у больных появляется прогрессирующая экзема, имеющая склонность к переходу в нейродермит. Часто у больных отмечается сухая, покрытая чешуйками кожа. Такие дети характеризуются почти полной утратой сопротивляемости к инфекции. У них обычно развиваются отиты, ангины, назофарингиты, гнойный ринит, конъюктивит, которые сопровождаются высокой температурой и протекают с клиникой септицемии. В итоге заболевание, как правило, заканчивается развитием фатальной инфекции или лимфопролиферативным процессом.
Заболевание вначале проявляется геморрагическими кровотечениями, которые локализуются преимущественно в кишечном тракте и проявляются примесью крови в кале (диарея с кровью). Дети также имеют петехиальные кровоизлияния в кожу. С 1 года у больных появляется прогрессирующая экзема, имеющая склонность к переходу в нейродермит. Часто у больных отмечается сухая, покрытая чешуйками кожа. Такие дети характеризуются почти полной утратой сопротивляемости к инфекции. У них обычно развиваются отиты, ангины, назофарингиты, гнойный ринит, конъюктивит, которые сопровождаются высокой температурой и протекают с клиникой септицемии. В итоге заболевание, как правило, заканчивается развитием фатальной инфекции или лимфопролиферативным процессом.
 Лечение Симптоматическая терапия Пересадка гемопоэтической ткани Введение гаммаглобулина В лечении инфекционных осложнений применяются противомикробные препараты Преимущество отдается внутривенному и энтеральному путям введения препаратов В качестве антибактериальной терапии используются: цефалоспорины (цефтазидим, цефотаксим, цефтриаксон, цефаклор в возрастных дозах – 2 -4 недели); антибиотики пенициллинового ряда (ампиокс, амоксициллин, потенцированный клавуланатом); имипенем+циластатин в возрастных дозах – 2 недели; сульфаниламиды (септрин, бактрим, котримоксазол – более 1 мес. ) В качестве противогрибковых средств назначают: интраконазол, флуконазол в возрастных дозах – 2 -3 недели
Лечение Симптоматическая терапия Пересадка гемопоэтической ткани Введение гаммаглобулина В лечении инфекционных осложнений применяются противомикробные препараты Преимущество отдается внутривенному и энтеральному путям введения препаратов В качестве антибактериальной терапии используются: цефалоспорины (цефтазидим, цефотаксим, цефтриаксон, цефаклор в возрастных дозах – 2 -4 недели); антибиотики пенициллинового ряда (ампиокс, амоксициллин, потенцированный клавуланатом); имипенем+циластатин в возрастных дозах – 2 недели; сульфаниламиды (септрин, бактрим, котримоксазол – более 1 мес. ) В качестве противогрибковых средств назначают: интраконазол, флуконазол в возрастных дозах – 2 -3 недели
 антигистаминные") По показаниям в комплекс лечебных мероприятий могут быть включены: муколитики (амброксол, бромгексин, ацетилцистеин) антигистаминные препараты (клемастин, супрастин, пипольфен, кларитин, зиртек, кестин) местные антисептики (бриллиантовый зеленый, хлоргексидин)
По показаниям в комплекс лечебных мероприятий могут быть включены: муколитики (амброксол, бромгексин, ацетилцистеин) антигистаминные препараты (клемастин, супрастин, пипольфен, кларитин, зиртек, кестин) местные антисептики (бриллиантовый зеленый, хлоргексидин)
 При снижении Hb<50 мг% проводят заместительную терапию эритроцитарной массой. При массивных кровотечениях показана интенсивная кровезаместительная терапия. Состояние детей с синдромом Вискотта-Олдрича характеризуется нестабильностью, поэтому необходимо выбрать оптимальный режим стационарного и амбулаторного лечения
При снижении Hb<50 мг% проводят заместительную терапию эритроцитарной массой. При массивных кровотечениях показана интенсивная кровезаместительная терапия. Состояние детей с синдромом Вискотта-Олдрича характеризуется нестабильностью, поэтому необходимо выбрать оптимальный режим стационарного и амбулаторного лечения
 Гипер-Ig. М синдром
Гипер-Ig. М синдром
 По клинической картине синдром похож на врожденную гипогаммаглобулинемию. Отличие заключается в повышенном содержании у таких больных Ig. М на фоне низких концентраций Ig. G и Ig. А или полного их отсутствия. Синдром сцеплен с Х-хромосомой. Его развитие связано с мутацией в гене, кодирующем продукцию специфического белка CD 40 L на активированных Т-лимфоцитах. Ген CD 40 L расположен на Х-хромосоме в позиции Xq 26. Белок CD 40 L участвует в кооперативном взаимодействии Т - и В-лимфоцитов, Т-лимфоцитов и АПК, которые играют важную роль в развитии иммунных реакций. Его экспрессия на Т-клетках также необходима для формирования В-клеток «памяти» и вторичного иммунного ответа на Т-зависимые антигены.
По клинической картине синдром похож на врожденную гипогаммаглобулинемию. Отличие заключается в повышенном содержании у таких больных Ig. М на фоне низких концентраций Ig. G и Ig. А или полного их отсутствия. Синдром сцеплен с Х-хромосомой. Его развитие связано с мутацией в гене, кодирующем продукцию специфического белка CD 40 L на активированных Т-лимфоцитах. Ген CD 40 L расположен на Х-хромосоме в позиции Xq 26. Белок CD 40 L участвует в кооперативном взаимодействии Т - и В-лимфоцитов, Т-лимфоцитов и АПК, которые играют важную роль в развитии иммунных реакций. Его экспрессия на Т-клетках также необходима для формирования В-клеток «памяти» и вторичного иммунного ответа на Т-зависимые антигены.
 Клинически заболевание проявляется рецидивирующими пиогенными инфекциями. У больных наблюдается повышенная чувствительность к оппортунистическим инфекциям, особенно к пневмоцистным пневмониям и энтеритам, вызываемым Criptosporidium. Пациенты склонны к аутоиммунным заболеваниям: гемолитической анемии, тромбоцитопенической пурпуре, нейтропении. Характерным признаком гипер – Ig. М синдрома является повышенное содержание Ig. М в сыворотке крови. Его содержание может достигать 10, 0 г/л. При этом Ig. А и Ig. Е выявляются в следовых количествах, а концентрация Ig. G составляет менее 2 г/л. У больных определяется нормальное количество В-клеток, экспрессирующих Ig. М, и дефицит В-клеток с поверхностными Ig. G, Ig. А и Ig. Е. Такие больные содержат в лимфоидной ткани высокий уровень Ig. М-секретирующих плазматических клеток. Эти клетки способны инфильтрировать разные ткани, чаще такому процессу подвергаются кишечник, желчный пузырь, печень. В лимфатических узлах наблюдается нарушение структуры В-клеточной зоны. В ряде случаев больные секретируют мономерный Ig. М.
Клинически заболевание проявляется рецидивирующими пиогенными инфекциями. У больных наблюдается повышенная чувствительность к оппортунистическим инфекциям, особенно к пневмоцистным пневмониям и энтеритам, вызываемым Criptosporidium. Пациенты склонны к аутоиммунным заболеваниям: гемолитической анемии, тромбоцитопенической пурпуре, нейтропении. Характерным признаком гипер – Ig. М синдрома является повышенное содержание Ig. М в сыворотке крови. Его содержание может достигать 10, 0 г/л. При этом Ig. А и Ig. Е выявляются в следовых количествах, а концентрация Ig. G составляет менее 2 г/л. У больных определяется нормальное количество В-клеток, экспрессирующих Ig. М, и дефицит В-клеток с поверхностными Ig. G, Ig. А и Ig. Е. Такие больные содержат в лимфоидной ткани высокий уровень Ig. М-секретирующих плазматических клеток. Эти клетки способны инфильтрировать разные ткани, чаще такому процессу подвергаются кишечник, желчный пузырь, печень. В лимфатических узлах наблюдается нарушение структуры В-клеточной зоны. В ряде случаев больные секретируют мономерный Ig. М.
 Механизм формирования иммунологических нарушений при гипер-Ig. M-синдроме.
Механизм формирования иммунологических нарушений при гипер-Ig. M-синдроме.
 Лечение Симптоматическое Антимикробная терапия Применение препаратов иммуноглобулинов для внутривенного введения Больные с гипер-Ig. М синдромом нуждаются в пожизненной заместительной терапии антителосодержащими препаратами. Адекватным препаратом являются иммуноглобулины для внутривенного введения. Может быть использована также нативная плазма от хорошо проверенных доноров.
Лечение Симптоматическое Антимикробная терапия Применение препаратов иммуноглобулинов для внутривенного введения Больные с гипер-Ig. М синдромом нуждаются в пожизненной заместительной терапии антителосодержащими препаратами. Адекватным препаратом являются иммуноглобулины для внутривенного введения. Может быть использована также нативная плазма от хорошо проверенных доноров.
 Т-клеточные иммунодефициты
Т-клеточные иммунодефициты
 К группе Т-клеточных иммунодефицитов относятся: 1. Синдром Ди-Джорджи 2. Синдром Незелофа 3. Хронический кожно-слизистый кандидоз
К группе Т-клеточных иммунодефицитов относятся: 1. Синдром Ди-Джорджи 2. Синдром Незелофа 3. Хронический кожно-слизистый кандидоз
 Клинически Т-лимфоцитарные иммунодефициты проявляются, главным образом, развитием вирусных и грибковых инфекций кожи, ногтей, волосистой части головы, слизистых оболочек бронхолегочного аппарата, кишечника, гениталий. Часто первыми признаками иммунодефицита является молочница, осложнения после вакцинации БЦЖ, тяжелые формы инфекций, обусловленных вирусом простого герпеса 1 -го и 2 го серотипов (ВПГ-1, ВПГ-2) и вирусом ветряной оспы – опоясывающего лишая (ВПГ-3).
Клинически Т-лимфоцитарные иммунодефициты проявляются, главным образом, развитием вирусных и грибковых инфекций кожи, ногтей, волосистой части головы, слизистых оболочек бронхолегочного аппарата, кишечника, гениталий. Часто первыми признаками иммунодефицита является молочница, осложнения после вакцинации БЦЖ, тяжелые формы инфекций, обусловленных вирусом простого герпеса 1 -го и 2 го серотипов (ВПГ-1, ВПГ-2) и вирусом ветряной оспы – опоясывающего лишая (ВПГ-3).
 Специфические дефекты и характер иммунных расстройств у детей с первичными Т-лимфоцитарными иммунодефицитами Иммунодефицит Специфический дефект Характер нарушения Характер иммунных расстройств 1. Синдром Ди. Джорджи Гипоплазия тимуса и паращитовидных желез вследствие недоразвития 3 -го и 4 го глоточных карманов Нарушение созревания пре–Т- клеток в Т-клетки Лимфоцитопения Снижение количества и функции Т-клеток, реакций ГЗТ. 2. Синдром Незелофа Гипоплазия или атрофия тимуса Нарушение созревания пре–Т- клеток в Т-клетки Лимфоцитопения Снижение количества и функции Т-клеток, реакций ГЗТ. 3. Хрониче-ский кожно -слизистый кандидоз Не выявлен Специфический дефект Т-клеток реагировать на Candida –АГ Селективный дефицит развития иммунной реакции на Candida – АГ
Специфические дефекты и характер иммунных расстройств у детей с первичными Т-лимфоцитарными иммунодефицитами Иммунодефицит Специфический дефект Характер нарушения Характер иммунных расстройств 1. Синдром Ди. Джорджи Гипоплазия тимуса и паращитовидных желез вследствие недоразвития 3 -го и 4 го глоточных карманов Нарушение созревания пре–Т- клеток в Т-клетки Лимфоцитопения Снижение количества и функции Т-клеток, реакций ГЗТ. 2. Синдром Незелофа Гипоплазия или атрофия тимуса Нарушение созревания пре–Т- клеток в Т-клетки Лимфоцитопения Снижение количества и функции Т-клеток, реакций ГЗТ. 3. Хрониче-ский кожно -слизистый кандидоз Не выявлен Специфический дефект Т-клеток реагировать на Candida –АГ Селективный дефицит развития иммунной реакции на Candida – АГ
 Синдром Ди-Джорджи
Синдром Ди-Джорджи
 Заболевание развивается в результате нарушения эмбрионального развития структур 3 -4 глоточных карманов, что проявляется в недоразвитии или отсутствии тимуса и паращитовидных желез. Болеют дети обоего пола. Для детей характерна триада аномалий: 1. гипоплазия тимуса 2. гипоплазия паращитовидных желез 3. аномалия дуги аорты.
Заболевание развивается в результате нарушения эмбрионального развития структур 3 -4 глоточных карманов, что проявляется в недоразвитии или отсутствии тимуса и паращитовидных желез. Болеют дети обоего пола. Для детей характерна триада аномалий: 1. гипоплазия тимуса 2. гипоплазия паращитовидных желез 3. аномалия дуги аорты.
 Ребенок рождается с симптомами сердечных нарушений, цианозом, одышкой в покое, шумовой симптоматикой при аускультации. Часто заболевание сочетается с такими аномалиями, как: волчья пасть, аномалия дуги аорты в комбинации с аномалиями крупных сосудов, аномалия грудины, катаракта; из-за недоразвития паращитовидных желез у детей наблюдаются неонатальные тетании, которые развиваются через 1 -2 суток после рождения. Для детей характерны: гипертелоризм – широкая переносица, увеличение расстояния между зрачками, низко посаженные уши. Дети с этим синдромом подвержены рецидивирующим вирусным и бактериальным инфекциям. Характер течения инфекции зависит от степени недоразвития тимуса (его полной аплазии или гипоплазии). При отсутствии тимуса развиваются плохо контролируемые генерализованные инфекции, способные привести к гибели. Большую роль в исходе заболевания играет сопутствующая патология. При серьезных дефектах в сердечно-сосудистой системе прогноз заболевания, как правило, неблагоприятный.
Ребенок рождается с симптомами сердечных нарушений, цианозом, одышкой в покое, шумовой симптоматикой при аускультации. Часто заболевание сочетается с такими аномалиями, как: волчья пасть, аномалия дуги аорты в комбинации с аномалиями крупных сосудов, аномалия грудины, катаракта; из-за недоразвития паращитовидных желез у детей наблюдаются неонатальные тетании, которые развиваются через 1 -2 суток после рождения. Для детей характерны: гипертелоризм – широкая переносица, увеличение расстояния между зрачками, низко посаженные уши. Дети с этим синдромом подвержены рецидивирующим вирусным и бактериальным инфекциям. Характер течения инфекции зависит от степени недоразвития тимуса (его полной аплазии или гипоплазии). При отсутствии тимуса развиваются плохо контролируемые генерализованные инфекции, способные привести к гибели. Большую роль в исходе заболевания играет сопутствующая патология. При серьезных дефектах в сердечно-сосудистой системе прогноз заболевания, как правило, неблагоприятный.
 Иммунный статус таких детей характеризуется снижением содержания в периферической крови Тклеток (CD 3+, CD 4+, CD 8+) и их функциональной активности, отсутствием кожных реакций ГЗТ. У таких детей не отторгаются кожные трансплантаты. Изменения в гуморальном звене иммунитета не всегда развиваются. Чаще определяется нормальное или повышенное содержание В-лимфоцитов в крови. Концентрация иммуноглобулинов в сыворотке в пределах нормы. Это свидетельствует в пользу того, что такие больные могут быть иммунизированы против ряда бактериальных и вирусных инфекций (дифтерии, столбняка, полиомиелита, кори, краснухи и др. ).
Иммунный статус таких детей характеризуется снижением содержания в периферической крови Тклеток (CD 3+, CD 4+, CD 8+) и их функциональной активности, отсутствием кожных реакций ГЗТ. У таких детей не отторгаются кожные трансплантаты. Изменения в гуморальном звене иммунитета не всегда развиваются. Чаще определяется нормальное или повышенное содержание В-лимфоцитов в крови. Концентрация иммуноглобулинов в сыворотке в пределах нормы. Это свидетельствует в пользу того, что такие больные могут быть иммунизированы против ряда бактериальных и вирусных инфекций (дифтерии, столбняка, полиомиелита, кори, краснухи и др. ).
 Лечение При полной аплазии тимуса-трансплантация железы. При гипоплазии железы – назначают препараты тимуса. Лечение пороков сердца ведется по стандартам, принятым в кардиологии, а недостаточности паращитовидных желез – по эндокринологическим стандартам. Иммунная недостаточность, за редким исключением, не определяет прогноз и спектр клинических проявлений заболевания. В большинстве случаев, если пациент переживает 6 -месячный возраст, наблюдается постепенное спонтанное восстановление Т-клеточного иммунитета.
Лечение При полной аплазии тимуса-трансплантация железы. При гипоплазии железы – назначают препараты тимуса. Лечение пороков сердца ведется по стандартам, принятым в кардиологии, а недостаточности паращитовидных желез – по эндокринологическим стандартам. Иммунная недостаточность, за редким исключением, не определяет прогноз и спектр клинических проявлений заболевания. В большинстве случаев, если пациент переживает 6 -месячный возраст, наблюдается постепенное спонтанное восстановление Т-клеточного иммунитета.
 Синдром Незелофа
Синдром Незелофа
 Заболевание наследуется по аутосомно-рецессивному типу. У детей с этим синдромом наблюдается недоразвитый эмбриоподобный тимус, который не способен поддерживать Т-клеточную дифференцировку. Заболевание проявляется в раннем детском возрасте. Характеризуется задержкой развития ребенка, его роста и массы тела. У таких детей наблюдаются вирусные, бактериальные и протозойные затяжные инфекции. Часто развивается сепсис с гнойными очагами в коже, легких и других органах, гемолитическая анемия с положительной реакцией Кумбса, аутоиммунная тромбоцитопения. Нередко бактериальный сепсис сочетается с грибковым. Больные очень чувствительны к вирусам ВПГ-1, ВПГ-2, ВПГ-3.
Заболевание наследуется по аутосомно-рецессивному типу. У детей с этим синдромом наблюдается недоразвитый эмбриоподобный тимус, который не способен поддерживать Т-клеточную дифференцировку. Заболевание проявляется в раннем детском возрасте. Характеризуется задержкой развития ребенка, его роста и массы тела. У таких детей наблюдаются вирусные, бактериальные и протозойные затяжные инфекции. Часто развивается сепсис с гнойными очагами в коже, легких и других органах, гемолитическая анемия с положительной реакцией Кумбса, аутоиммунная тромбоцитопения. Нередко бактериальный сепсис сочетается с грибковым. Больные очень чувствительны к вирусам ВПГ-1, ВПГ-2, ВПГ-3.
 Иммунный статус таких больных характеризуется лимфоцитопенией, сниженным содержанием в периферической крови Т-лимфоцитов (CD 3+, CD 4+, CD 8+) и низкой их функциональной активностью (РБТЛ, СКЛ), резким угнетением кожных реакций ГЗТ. Уровень В-лимфоцитов и Ig – в норме или несколько снижен. Продукция специфических антител чаще подавлена. Фагоцитарная функция нейтрофилов при этой патологии не изменена или даже несколько повышена. Гибель таких детей наступает в первые недели жизни. Изучение структуры тимуса показало отсутствие кортикальных лимфоцитов и телец Гассаля. В лимфатических узлах наблюдается гипоплазия Т-зон.
Иммунный статус таких больных характеризуется лимфоцитопенией, сниженным содержанием в периферической крови Т-лимфоцитов (CD 3+, CD 4+, CD 8+) и низкой их функциональной активностью (РБТЛ, СКЛ), резким угнетением кожных реакций ГЗТ. Уровень В-лимфоцитов и Ig – в норме или несколько снижен. Продукция специфических антител чаще подавлена. Фагоцитарная функция нейтрофилов при этой патологии не изменена или даже несколько повышена. Гибель таких детей наступает в первые недели жизни. Изучение структуры тимуса показало отсутствие кортикальных лимфоцитов и телец Гассаля. В лимфатических узлах наблюдается гипоплазия Т-зон.
 Лечение Рансплантация тимуса Введение препаратов тимуса, иммуноглобулинов Трансплантация костного мозга
Лечение Рансплантация тимуса Введение препаратов тимуса, иммуноглобулинов Трансплантация костного мозга
 Хронический кожно-слизистый кандидоз
Хронический кожно-слизистый кандидоз
 Хронический кожно-слизистый кандидоз развивается вследствие селективного дефицита ответа Тлимфоцитов на Candida- антиген. При этом гуморальный ответ в пределах физиологической нормы. Клинически дефект проявляется в поражении кожи, ногтей, волосистой части головы и слизистых оболочек грибами рода Candida albicans. При этом исследования свидетельствуют о нормальном содержании в периферической крови Т-лимфоцитов (CD 3+), нормальной их бласттрансформирующей способности на ФГА и пролиферативной активности в СКЛ. РБТЛ на Candida- антиген и кожная реакция ГЗТ на этот антиген отрицательная. Гуморальный иммунный ответ у таких больных не нарушен.
Хронический кожно-слизистый кандидоз развивается вследствие селективного дефицита ответа Тлимфоцитов на Candida- антиген. При этом гуморальный ответ в пределах физиологической нормы. Клинически дефект проявляется в поражении кожи, ногтей, волосистой части головы и слизистых оболочек грибами рода Candida albicans. При этом исследования свидетельствуют о нормальном содержании в периферической крови Т-лимфоцитов (CD 3+), нормальной их бласттрансформирующей способности на ФГА и пролиферативной активности в СКЛ. РБТЛ на Candida- антиген и кожная реакция ГЗТ на этот антиген отрицательная. Гуморальный иммунный ответ у таких больных не нарушен.
 Лечение Назначается противомикозная терапия А так же препараты, стимулирующие активность клеточного иммунитета
Лечение Назначается противомикозная терапия А так же препараты, стимулирующие активность клеточного иммунитета
 Гуморальные иммунодефициты
Гуморальные иммунодефициты
 Дефекты в В-звене иммунитета могут проявляться в нарушении созревания В-клеток, в нарушении формирования плазматических клеток, продукции всех классов иммуноглобулинов или отдельных их классов. Пациенты с этими нарушениями обычно демонстрируют нормальное развитие клеточных иммунных реакций. Больные проявляют высокую устойчивость к вирусным инфекциям. Корь, паротит, ветряная оспа, краснуха у таких лиц протекают обычно. У них часто иммунодефицитные состояния клинически проявляются в средней и легкой форме тяжести. Селективный дефицит Ig. А в большинстве случаев протекает бессимптомно. Клиническая манифестация гуморального иммунодефицита обычно происходит на 1 -2 году жизни ребенка, а отдельных его форм – в подростковом возрасте или даже во взрослом периоде.
Дефекты в В-звене иммунитета могут проявляться в нарушении созревания В-клеток, в нарушении формирования плазматических клеток, продукции всех классов иммуноглобулинов или отдельных их классов. Пациенты с этими нарушениями обычно демонстрируют нормальное развитие клеточных иммунных реакций. Больные проявляют высокую устойчивость к вирусным инфекциям. Корь, паротит, ветряная оспа, краснуха у таких лиц протекают обычно. У них часто иммунодефицитные состояния клинически проявляются в средней и легкой форме тяжести. Селективный дефицит Ig. А в большинстве случаев протекает бессимптомно. Клиническая манифестация гуморального иммунодефицита обычно происходит на 1 -2 году жизни ребенка, а отдельных его форм – в подростковом возрасте или даже во взрослом периоде.
 Клинически дефицит гуморального иммунитета проявляется развитием бактериальных инфекций, вызванных стафилококками, стрептококками, пневмококками, гемофильной палочкой. Чувствительность таких детей к энтерококкам и грамотрицательным бактериям выражена несколько меньше. У детей с недостаточностью гуморального иммунитета (антителообразования) наблюдаются: часто повторяющиеся бронхиты, хронические инфекции ЛОР-органов, гнойные синуситы, отиты, лимфоадениты, бронхоплевропневмонии, бактериальные инфекции кожи и подкожной клетчатки (пиодермии, фурункулез, абсцессы, флегмоны), септические гранулемы, урогенитальные инфекции (гнойные вульвиты, пиелонефриты с частыми обострениями), рецидивирующий гнойный конъюктивит, гастроэнтеропатии с хронической диареей неясной этиологии, дисбактериозы.
Клинически дефицит гуморального иммунитета проявляется развитием бактериальных инфекций, вызванных стафилококками, стрептококками, пневмококками, гемофильной палочкой. Чувствительность таких детей к энтерококкам и грамотрицательным бактериям выражена несколько меньше. У детей с недостаточностью гуморального иммунитета (антителообразования) наблюдаются: часто повторяющиеся бронхиты, хронические инфекции ЛОР-органов, гнойные синуситы, отиты, лимфоадениты, бронхоплевропневмонии, бактериальные инфекции кожи и подкожной клетчатки (пиодермии, фурункулез, абсцессы, флегмоны), септические гранулемы, урогенитальные инфекции (гнойные вульвиты, пиелонефриты с частыми обострениями), рецидивирующий гнойный конъюктивит, гастроэнтеропатии с хронической диареей неясной этиологии, дисбактериозы.
 Общий вариабельный иммунодефицит") Различают следующие формы гуморального иммунодефицита: Гипогаммаглобулинемия, сцепленная с Ххромосомой (болезнь Брутона) Общий вариабельный иммунодефицит (общая вариабельная гипогаммаглобулинемия) Транзиторная гипогаммаглобулинемия детей (медленный иммунологический старт) Дисгаммаглобулинемия (избирательный дефицит иммуноглобулинов)
Различают следующие формы гуморального иммунодефицита: Гипогаммаглобулинемия, сцепленная с Ххромосомой (болезнь Брутона) Общий вариабельный иммунодефицит (общая вариабельная гипогаммаглобулинемия) Транзиторная гипогаммаглобулинемия детей (медленный иммунологический старт) Дисгаммаглобулинемия (избирательный дефицит иммуноглобулинов)
 Иммуно-дефицит Специфический дефект Характер нарушения Характер иммунных расстройств 1. Болезнь Брутона Дефект гена тирозинкиназы. Нарушение трансдукции активационных сигналов в В-клетках, важных для их созревания Нарушение созревания пре. В-клеток в Влимфоциты Отсутствие или низкое содержание В -клеток и всех классов иммуноглобулинов 2. Общий вариабельный иммунодефицит не известен Нарушение способности В-клеток дифференцироваться в плазматические клетки Нарушена способность продуцировать АТ Нарушение механизмов контроля и переключения синтеза иммуноглобулинов Селективные дефициты Ig. А, Ig. М, Ig. G, Ig. Е и дефицит образования антител 3. Дис-гамма-глобулинемия -
Иммуно-дефицит Специфический дефект Характер нарушения Характер иммунных расстройств 1. Болезнь Брутона Дефект гена тирозинкиназы. Нарушение трансдукции активационных сигналов в В-клетках, важных для их созревания Нарушение созревания пре. В-клеток в Влимфоциты Отсутствие или низкое содержание В -клеток и всех классов иммуноглобулинов 2. Общий вариабельный иммунодефицит не известен Нарушение способности В-клеток дифференцироваться в плазматические клетки Нарушена способность продуцировать АТ Нарушение механизмов контроля и переключения синтеза иммуноглобулинов Селективные дефициты Ig. А, Ig. М, Ig. G, Ig. Е и дефицит образования антител 3. Дис-гамма-глобулинемия -
 Болезнь Брутона
Болезнь Брутона
 Заболевание сцеплено с Х-хромосомой, имеет рецессивный тип наследования. Дефектный ген локализован в хромосоме Xq 21. Болеют только мальчики.
Заболевание сцеплено с Х-хромосомой, имеет рецессивный тип наследования. Дефектный ген локализован в хромосоме Xq 21. Болеют только мальчики.
 Заболевание развивается в результате нарушения созревания пре-В-клеток в В-лимфоциты. В свою очередь, этот процесс связан с дефектом тирозинкиназы, которая участвует в трансдукции активирующих сигналов созревания В-лимфоцитов. Заболевание, как правило, проявляется со второго полугодия жизни ребенка развитием хронических и рецидивирующих инфекций бронхо-легочного аппарата, придаточных пазух носа, среднего уха, бактериальных инфекций кожи и подкожной клетчатки, диареи и дисбактериоза. У больных не наблюдается реакций со стороны лимфатических узлов, селезенки в острый воспалительный период. У таких лиц не бывает гиперплазии миндалин, аденоидов. Часто наблюдается развитие аллергических реакций (на лекарственные средства, продукты и др. ).
Заболевание развивается в результате нарушения созревания пре-В-клеток в В-лимфоциты. В свою очередь, этот процесс связан с дефектом тирозинкиназы, которая участвует в трансдукции активирующих сигналов созревания В-лимфоцитов. Заболевание, как правило, проявляется со второго полугодия жизни ребенка развитием хронических и рецидивирующих инфекций бронхо-легочного аппарата, придаточных пазух носа, среднего уха, бактериальных инфекций кожи и подкожной клетчатки, диареи и дисбактериоза. У больных не наблюдается реакций со стороны лимфатических узлов, селезенки в острый воспалительный период. У таких лиц не бывает гиперплазии миндалин, аденоидов. Часто наблюдается развитие аллергических реакций (на лекарственные средства, продукты и др. ).
 Иммунный статус таких больных характеризуется отсутствием или низким содержанием В-лимфоцитов в периферической крови (<1% CD 19+, CD 20+, CD 22+) и низким содержанием в сыворотке всех классов иммуноглобулинов, особенно Ig. G (<200 мг%) и Ig. А (<20 мг%). В лимфатических узлах и лимфоидной ткани слизистых оболочек отсутствуют плазматические клетки. Количество Т-лимфоцитов в норме. Реакции ГЗТ имеют обычный вид. Устойчивость к вирусным инфекциям в целом сохранена. Гистологические исследования свидетельствуют о том, что в периферических лимфоидных органах отсутствуют зародышевые центры. Больные с наследственной гипогаммаглобулинемией нуждаются в пожизненной заместительной терапии антителосодержащими препаратами. Адекватными препаратами являются иммуноглобулины для внутривенного введения. В лечении этой категории больных может быть также использована нативная плазма от хорошо проверенных доноров. По показаниям больным могут быть рекомендованы муколитики, антигистаминные препараты, местные антисептики. Больным с врожденной ГГГ, страдающим хроническими бронхолегочными инфекциями, нужны ежедневные вибрационный массаж и постуральный дренаж, при обострениях требуются санационные бронхоскопии.
Иммунный статус таких больных характеризуется отсутствием или низким содержанием В-лимфоцитов в периферической крови (<1% CD 19+, CD 20+, CD 22+) и низким содержанием в сыворотке всех классов иммуноглобулинов, особенно Ig. G (<200 мг%) и Ig. А (<20 мг%). В лимфатических узлах и лимфоидной ткани слизистых оболочек отсутствуют плазматические клетки. Количество Т-лимфоцитов в норме. Реакции ГЗТ имеют обычный вид. Устойчивость к вирусным инфекциям в целом сохранена. Гистологические исследования свидетельствуют о том, что в периферических лимфоидных органах отсутствуют зародышевые центры. Больные с наследственной гипогаммаглобулинемией нуждаются в пожизненной заместительной терапии антителосодержащими препаратами. Адекватными препаратами являются иммуноглобулины для внутривенного введения. В лечении этой категории больных может быть также использована нативная плазма от хорошо проверенных доноров. По показаниям больным могут быть рекомендованы муколитики, антигистаминные препараты, местные антисептики. Больным с врожденной ГГГ, страдающим хроническими бронхолегочными инфекциями, нужны ежедневные вибрационный массаж и постуральный дренаж, при обострениях требуются санационные бронхоскопии.
") Общий вариабельный иммунодефицит (общая вариабельная гипогаммаглобулинемия)
Общий вариабельный иммунодефицит (общая вариабельная гипогаммаглобулинемия)
 Заболевание может возникнуть в любом возрасте, часто проявляется на 14 -36 годах жизни, манифистирует развитием рецидивирующих и хронических инфекций бактериальной природы бронхолегочного тракта, придаточных пазух носа, уха, бактериальными поражениями кожи и подкожной клетчатки, развитием энтероколита и дисбактериоза. Иммунодефицит развивается в результате нарушения способности В-лимфоцитов дифференцироваться в плазматические клетки и продуцировать антитела. У больных наблюдаются низкие концентрации в сыворотке крови Ig. А, Ig. М, Ig. G (суммарная ниже 300 мг%), сниженная продукция специфических антител на патоген ( в ответ на иммунизацию). При этом у таких лиц в крови наблюдается нормальный или несколько сниженный уровень В-лимфоцитов. Показатели Т-звена иммунитета в пределах нормы.
Заболевание может возникнуть в любом возрасте, часто проявляется на 14 -36 годах жизни, манифистирует развитием рецидивирующих и хронических инфекций бактериальной природы бронхолегочного тракта, придаточных пазух носа, уха, бактериальными поражениями кожи и подкожной клетчатки, развитием энтероколита и дисбактериоза. Иммунодефицит развивается в результате нарушения способности В-лимфоцитов дифференцироваться в плазматические клетки и продуцировать антитела. У больных наблюдаются низкие концентрации в сыворотке крови Ig. А, Ig. М, Ig. G (суммарная ниже 300 мг%), сниженная продукция специфических антител на патоген ( в ответ на иммунизацию). При этом у таких лиц в крови наблюдается нормальный или несколько сниженный уровень В-лимфоцитов. Показатели Т-звена иммунитета в пределах нормы.
 Лечение таких больных симптоматическое. Обязательным является назначение антибактериальных препаратов. Больные с общим вариабельным иммунодефицитом нуждаются в пожизненной заместительной терапии антителосодержащими препаратами. Заместительная терапия впервые выявленных больных проводится в режиме насыщения.
Лечение таких больных симптоматическое. Обязательным является назначение антибактериальных препаратов. Больные с общим вариабельным иммунодефицитом нуждаются в пожизненной заместительной терапии антителосодержащими препаратами. Заместительная терапия впервые выявленных больных проводится в режиме насыщения.
 (медленный иммунологический старт)") Транзиторная гипогаммаглобулинемия детей (ТГД) (медленный иммунологический старт)
Транзиторная гипогаммаглобулинемия детей (ТГД) (медленный иммунологический старт)
 Естественное иммунодефицитное состояние, встречающееся у 5 -8% детей. Отмечается в возрасте 7 месяцев – 4 лет. Развивается в силу запаздывания продукции собственных иммуноглобулинов (Ig. G). Обычно с 5 -6 месячного возраста у таких детей наблюдается развитие рецидивирующих бактериальных инфекций верхних дыхательных путей, кожи и подкожной клетчатки, урогенитального тракта, частые диареи. Диагноз выставляется детям при снижении сывороточной концентрации одного или нескольких изотипов иммуноглобулинов: Ig. G<500 мг%, Ig. А< 20 мг%, Ig. М<40 мг%, при исключении других иммунодефицитных состояний.
Естественное иммунодефицитное состояние, встречающееся у 5 -8% детей. Отмечается в возрасте 7 месяцев – 4 лет. Развивается в силу запаздывания продукции собственных иммуноглобулинов (Ig. G). Обычно с 5 -6 месячного возраста у таких детей наблюдается развитие рецидивирующих бактериальных инфекций верхних дыхательных путей, кожи и подкожной клетчатки, урогенитального тракта, частые диареи. Диагноз выставляется детям при снижении сывороточной концентрации одного или нескольких изотипов иммуноглобулинов: Ig. G<500 мг%, Ig. А< 20 мг%, Ig. М<40 мг%, при исключении других иммунодефицитных состояний.
 ТГД имеет доброкачественное течение и по существу представляет собой затяжной вариант физиологического состояния гипогаммаглобулинемии, свойственный детям в возрасте 3 -6 месяцев, когда запасы полученных внутриутробно материнских Ig. G истощаются, а собственный синтез еще недостаточен. ТГД может также обнаруживаться у практически здоровых детей как случайная находка. ТГД, как правило, заканчивается самопроизвольным выздоровлением и не требует патогенетической иммунокоррекции.
ТГД имеет доброкачественное течение и по существу представляет собой затяжной вариант физиологического состояния гипогаммаглобулинемии, свойственный детям в возрасте 3 -6 месяцев, когда запасы полученных внутриутробно материнских Ig. G истощаются, а собственный синтез еще недостаточен. ТГД может также обнаруживаться у практически здоровых детей как случайная находка. ТГД, как правило, заканчивается самопроизвольным выздоровлением и не требует патогенетической иммунокоррекции.
 Лечение симптоматическое, направленное на купирование инфекций. В тяжелых случаях показана заместительная терапия препаратами иммуноглобулинов.
Лечение симптоматическое, направленное на купирование инфекций. В тяжелых случаях показана заместительная терапия препаратами иммуноглобулинов.
 При этих состояниях наблюдается снижение в сыворотке крови одного -двух") Дисгаммаглобулинемия (избирательный дефицит иммуноглобулинов) При этих состояниях наблюдается снижение в сыворотке крови одного -двух классов иммуноглобулинов при нормальном или повышенном содержании других. Развитие данной патологии связывают с нарушением механизмов контроля за синтезом иммуноглобулинов и переключения их продукции с одного класса на другой.
Дисгаммаглобулинемия (избирательный дефицит иммуноглобулинов) При этих состояниях наблюдается снижение в сыворотке крови одного -двух классов иммуноглобулинов при нормальном или повышенном содержании других. Развитие данной патологии связывают с нарушением механизмов контроля за синтезом иммуноглобулинов и переключения их продукции с одного класса на другой.
 Наблюдаются следующие варианты селективного дефицита иммуноглобулинов: Селективный дефицит Ig. А Селективный дефицит Ig. М Селективный дефицит Ig. G (Ig. G 2 и Ig. G 4) Селективный дефицит Ig. Е Дефицит антител при нормо- или гипергаммаглобулинемии. Среди перечисленных селективных дефицитов иммуноглобулинов наиболее часто встречается дефицит Ig. А (1/500 – 1/700 человек). Имеются данные о семейных случаях.
Наблюдаются следующие варианты селективного дефицита иммуноглобулинов: Селективный дефицит Ig. А Селективный дефицит Ig. М Селективный дефицит Ig. G (Ig. G 2 и Ig. G 4) Селективный дефицит Ig. Е Дефицит антител при нормо- или гипергаммаглобулинемии. Среди перечисленных селективных дефицитов иммуноглобулинов наиболее часто встречается дефицит Ig. А (1/500 – 1/700 человек). Имеются данные о семейных случаях.
 Селективный дефицит Ig. А Состояние развивается из-за неспособности В-лимфоцитов дифференцироваться в Ig. А-секретирующие клетки. Диагноз ставится детям старше 1 года, если концентрация Ig. А в сыворотке ниже 5 мг% при достаточном уровне других классов иммуноглобулинов и отсутствии признаков других ИДС (например, атаксии-телеангиэктазии). Дефицит Ig. А клинически может проявляться самыми различными симптомами; в отдельных случаях может протекать бессимптомно. Частыми проявлениями бывают рецидивирующие инфекции слизистых оболочек дыхательных путей, урогенитального тракта, кишечника, инфекции ЛОР-органов. Низкий уровень Ig. А предрасполагает к аллергии, сопутствует ряду аутоиммунных заболеваний. Лечебные мероприятия при дефиците Ig. А, главным образом, сводятся к лечению вторичных инфекционных осложнений и активации сохранных звеньев иммунитета в целях компенсации недостатка продукции Ig. А. Иммуностимуляция проводится по клиническим показаниям. Таким больным показано назначение препаратов: бронхомунал, рибомунил, биостим, ликопид, натрия нуклеонат, полиоксидоний. Лечение проводится амбулаторно.
Селективный дефицит Ig. А Состояние развивается из-за неспособности В-лимфоцитов дифференцироваться в Ig. А-секретирующие клетки. Диагноз ставится детям старше 1 года, если концентрация Ig. А в сыворотке ниже 5 мг% при достаточном уровне других классов иммуноглобулинов и отсутствии признаков других ИДС (например, атаксии-телеангиэктазии). Дефицит Ig. А клинически может проявляться самыми различными симптомами; в отдельных случаях может протекать бессимптомно. Частыми проявлениями бывают рецидивирующие инфекции слизистых оболочек дыхательных путей, урогенитального тракта, кишечника, инфекции ЛОР-органов. Низкий уровень Ig. А предрасполагает к аллергии, сопутствует ряду аутоиммунных заболеваний. Лечебные мероприятия при дефиците Ig. А, главным образом, сводятся к лечению вторичных инфекционных осложнений и активации сохранных звеньев иммунитета в целях компенсации недостатка продукции Ig. А. Иммуностимуляция проводится по клиническим показаниям. Таким больным показано назначение препаратов: бронхомунал, рибомунил, биостим, ликопид, натрия нуклеонат, полиоксидоний. Лечение проводится амбулаторно.
 Селективный дефицит Ig. М При этой форме дефицита наблюдаются с раннего возраста рецидивирующие инфекции разной локализации (стафилококковая пиодермия, менингококковая септицемия, язвенный колит с длительной диареей). Менингококковая инфекция для лиц с дефицитом Ig. М часто является фатальной.
Селективный дефицит Ig. М При этой форме дефицита наблюдаются с раннего возраста рецидивирующие инфекции разной локализации (стафилококковая пиодермия, менингококковая септицемия, язвенный колит с длительной диареей). Менингококковая инфекция для лиц с дефицитом Ig. М часто является фатальной.
 Селективный дефицит Ig. G Может наблюдаться дефицит одного или одновременно нескольких субклассов Ig. G. При этом концентрация Ig. А и Ig. М в пределах возрастной нормы или могут быть несколько повышены. При дефиците Ig. G наблюдаются респираторные, бронхолегочные инфекции, часто вызванные пневмококками, палочкой инфлюенцы. При дефиците Ig. G 2 развивающиеся инфекции протекают наиболее тяжело. Дефицит Ig. G 4 может протекать бессимптомно.
Селективный дефицит Ig. G Может наблюдаться дефицит одного или одновременно нескольких субклассов Ig. G. При этом концентрация Ig. А и Ig. М в пределах возрастной нормы или могут быть несколько повышены. При дефиците Ig. G наблюдаются респираторные, бронхолегочные инфекции, часто вызванные пневмококками, палочкой инфлюенцы. При дефиците Ig. G 2 развивающиеся инфекции протекают наиболее тяжело. Дефицит Ig. G 4 может протекать бессимптомно.
 Лечение Антибиотики Заместительную терапию препаратами иммуноглобулинов для внутривенного введения.
Лечение Антибиотики Заместительную терапию препаратами иммуноглобулинов для внутривенного введения.
 Дефициты системы фагоцитов
Дефициты системы фагоцитов
 Дефицит в системе фагоцитов может быть связан с нарушением любого из этапов процесса фагоцитоза: хемотаксиса, адгезии, поглощения, дегрануляции, киллинга и разрушения объекта фагоцитоза. Клинически дефекты фагоцитоза проявляются развитием гнойно-воспалительных процессов в коже, слизистых покровах и паренхиматозных органах. Основными возбудителями инфекционного процесса при этом выступают грамотрицательные кишечные и пиогенные бактерии (E. coli, бактерии родов Pseudomonas, Klebsiella, Staphylococcus), грибы (Candida, Aspergillus, Mucor).
Дефицит в системе фагоцитов может быть связан с нарушением любого из этапов процесса фагоцитоза: хемотаксиса, адгезии, поглощения, дегрануляции, киллинга и разрушения объекта фагоцитоза. Клинически дефекты фагоцитоза проявляются развитием гнойно-воспалительных процессов в коже, слизистых покровах и паренхиматозных органах. Основными возбудителями инфекционного процесса при этом выступают грамотрицательные кишечные и пиогенные бактерии (E. coli, бактерии родов Pseudomonas, Klebsiella, Staphylococcus), грибы (Candida, Aspergillus, Mucor).
.") Дефицит системы фагоцитов наблюдается при: Хроническом гранулематозе. Синдроме Чедиака-Хигаси. Синдроме гипериммуноглобулинемии Е (Синдром Джоба). Дефиците экспрессии молекул адгезии (Дефект адгезии лейкоцитов) и других.
Дефицит системы фагоцитов наблюдается при: Хроническом гранулематозе. Синдроме Чедиака-Хигаси. Синдроме гипериммуноглобулинемии Е (Синдром Джоба). Дефиците экспрессии молекул адгезии (Дефект адгезии лейкоцитов) и других.
 Хронический гранулематоз Различают Х-сцепленную форму, которая встречается у 70% больных, и аутосомнорецессивную форму, наблюдающуюся у 30 % больных. Дефект фагоцитоза обусловлен нарушением в клетках кислородзависимого метаболизма, неспособностью их генерировать активные формы кислорода. В клетках больных наблюдается снижение активности НАДФ-оксидазы, отсутствие или дефектный цитохром b 558. Наряду с этим снижена способность мононуклеарных клеток выступать в качестве антигенпрезентирующих единиц за счет нарушения процессинга и презентации антигена. Заболевание может впервые проявиться как в детском возрасте, так и у взрослых.
Хронический гранулематоз Различают Х-сцепленную форму, которая встречается у 70% больных, и аутосомнорецессивную форму, наблюдающуюся у 30 % больных. Дефект фагоцитоза обусловлен нарушением в клетках кислородзависимого метаболизма, неспособностью их генерировать активные формы кислорода. В клетках больных наблюдается снижение активности НАДФ-оксидазы, отсутствие или дефектный цитохром b 558. Наряду с этим снижена способность мононуклеарных клеток выступать в качестве антигенпрезентирующих единиц за счет нарушения процессинга и презентации антигена. Заболевание может впервые проявиться как в детском возрасте, так и у взрослых.
 Одними из первых клинических симптомов заболевания является появление гнойничковых инфильтратов в коже и экзематозный дерматит вокруг рта, носа и ушей. В последующем воспалительные гранулемы и абсцессы возникают в различных органах (чаще всего в легких), при этом развивается гепато- и спленомегалия, увеличиваются лимфатические узлы. Появление гранулем связано с неспособностью фагоцитов (ПМЯЛ и макрофагов) к киллингу и перевариванию поглощенных микроорганизмов (St. aureus, Serratia, Esherichia, Pseudomonas), вырабатывающих перекись водорода (каталазоположительных). В НСТ-тесте нейтрофилы проявляют низкую кислородзависимую метаболическую активность. Количественное содержание и функциональная активность Т- и В-лимфоцитов, а также уровень комплемента у больных в пределах нормы.
Одними из первых клинических симптомов заболевания является появление гнойничковых инфильтратов в коже и экзематозный дерматит вокруг рта, носа и ушей. В последующем воспалительные гранулемы и абсцессы возникают в различных органах (чаще всего в легких), при этом развивается гепато- и спленомегалия, увеличиваются лимфатические узлы. Появление гранулем связано с неспособностью фагоцитов (ПМЯЛ и макрофагов) к киллингу и перевариванию поглощенных микроорганизмов (St. aureus, Serratia, Esherichia, Pseudomonas), вырабатывающих перекись водорода (каталазоположительных). В НСТ-тесте нейтрофилы проявляют низкую кислородзависимую метаболическую активность. Количественное содержание и функциональная активность Т- и В-лимфоцитов, а также уровень комплемента у больных в пределах нормы.
 нуждаются в постоянной (пожизненной) антибактериальной терапии, требующейся") Лечение симптоматическое. Больные с хроническим гранулематозом (ХГ) нуждаются в постоянной (пожизненной) антибактериальной терапии, требующейся даже в период ремиссии инфекционных проявлений. В зависимости от степени восприимчивости к инфекциям, устанавливаемой индивидуально, больные либо получают постоянно триметоприм – сульфаметоксазол, либо чередование пероральных антибиотиков широкого спектра действия (цефалоспоринов, полусинтетических пенициллинов, оксихинолонов и др. ) в сочетании с антимикотическими препаратами в возрастной дозировке. Интенсивность противомикробной терапии может достигать многомесячного применения одновременно 2 -3 препаратов (при абсцессах легких и внутренних органов). Нередким инфекционным осложнением при ХГ являются грибковые поражения легких, внутренних органов, кожи, слизистых покровов. Попытки радикальной коррекции иммунного дефекта при ХГ имеют ограниченный успех. Показано переливание лейкоцитарной массы, пересадка костного мозга.
Лечение симптоматическое. Больные с хроническим гранулематозом (ХГ) нуждаются в постоянной (пожизненной) антибактериальной терапии, требующейся даже в период ремиссии инфекционных проявлений. В зависимости от степени восприимчивости к инфекциям, устанавливаемой индивидуально, больные либо получают постоянно триметоприм – сульфаметоксазол, либо чередование пероральных антибиотиков широкого спектра действия (цефалоспоринов, полусинтетических пенициллинов, оксихинолонов и др. ) в сочетании с антимикотическими препаратами в возрастной дозировке. Интенсивность противомикробной терапии может достигать многомесячного применения одновременно 2 -3 препаратов (при абсцессах легких и внутренних органов). Нередким инфекционным осложнением при ХГ являются грибковые поражения легких, внутренних органов, кожи, слизистых покровов. Попытки радикальной коррекции иммунного дефекта при ХГ имеют ограниченный успех. Показано переливание лейкоцитарной массы, пересадка костного мозга.
 Синдром Чедиака-Хигаси Аутосомно-рецессивное заболевание, характеризующееся развитием тяжелых рецидивирующих пиогенных инфекций кожи (абсцессы кожи и подкожной клетчатки), органов дыхания (бронхиты, пневмонии, лимфоаденопатии), вызванных чаще всего стрептококками и стафилококками, сопровождающееся инфильтрацией ткани лимфоидными клетками.
Синдром Чедиака-Хигаси Аутосомно-рецессивное заболевание, характеризующееся развитием тяжелых рецидивирующих пиогенных инфекций кожи (абсцессы кожи и подкожной клетчатки), органов дыхания (бронхиты, пневмонии, лимфоаденопатии), вызванных чаще всего стрептококками и стафилококками, сопровождающееся инфильтрацией ткани лимфоидными клетками.
 Развитие инфекций связано с потерей переваривающей способности нейтрофилов в результате нарушения механизма высвобождения лизосомных ферментов в фагосомы (фаголизосомы). Нейтрофилы таких больных характеризуются наличием гигантских гранул (лизосом). Для больных также характерен частичный альбинизм глаз и кожи. В лабораторных исследованиях выявляется нарушение хемотаксиса и переваривающей способности нейтрофилов на фоне нормального содержания и функции Т- и В-лимфоцитов, а также уровня комплемента.
Развитие инфекций связано с потерей переваривающей способности нейтрофилов в результате нарушения механизма высвобождения лизосомных ферментов в фагосомы (фаголизосомы). Нейтрофилы таких больных характеризуются наличием гигантских гранул (лизосом). Для больных также характерен частичный альбинизм глаз и кожи. В лабораторных исследованиях выявляется нарушение хемотаксиса и переваривающей способности нейтрофилов на фоне нормального содержания и функции Т- и В-лимфоцитов, а также уровня комплемента.
. Прогноз неблагоприятный.") Лечение симптоматическое с использованием антибиотиков. Полезны высокие дозы витамина С (500 мг/сутки). Прогноз неблагоприятный.
Лечение симптоматическое с использованием антибиотиков. Полезны высокие дозы витамина С (500 мг/сутки). Прогноз неблагоприятный.
 Заболевание характеризуется развитием холодных (из-за отсутствия нормальной воспалительной реакции)") Синдром гипериммуноглобулинемии Е (Синдром Джоба) Заболевание характеризуется развитием холодных (из-за отсутствия нормальной воспалительной реакции) абсцессов кожи и подкожной клетчатки, лимфоузлов, повторными гнойными отитами с холодным течением. Часто у больных наблюдается хроническая экзема. Особую опасность представляют тяжелые эпизоды острых пневмоний, в т. ч. деструктивных, абсцессы
Синдром гипериммуноглобулинемии Е (Синдром Джоба) Заболевание характеризуется развитием холодных (из-за отсутствия нормальной воспалительной реакции) абсцессов кожи и подкожной клетчатки, лимфоузлов, повторными гнойными отитами с холодным течением. Часто у больных наблюдается хроническая экзема. Особую опасность представляют тяжелые эпизоды острых пневмоний, в т. ч. деструктивных, абсцессы
 Названные симптомы развиваются из-за нарушения хемотаксиса нейтрофилов, повышенного высвобождения под влиянием гиперпродукции Ig. Е из тучных клеток гистамина, который блокирует развитие воспаления и хемотаксис нейтрофилов. Главный дефект при этой патологии заключен в неспособности Тн 1 -клеток продуцировать гамма-интерферон и связанной с этим повышенной функцией Тн 2 -клеток, а также гиперпродукцией Ig. Е, что и обусловливает атипичное течение воспаления. Для детей с этим синдромом характерными признаками являются диспластические черты лица, спонтанные переломы трубчатых костей. При лабораторном обследовании у больных выявляется нарушение хемотаксиса нейтрофилов, повышенный уровень Ig. Е (1000 МЕ/мл), сниженная продукция Тлимфоцитами гамма-интерферона, повышенная концентрация в крови гистамина (75 -100 мкг/л).
Названные симптомы развиваются из-за нарушения хемотаксиса нейтрофилов, повышенного высвобождения под влиянием гиперпродукции Ig. Е из тучных клеток гистамина, который блокирует развитие воспаления и хемотаксис нейтрофилов. Главный дефект при этой патологии заключен в неспособности Тн 1 -клеток продуцировать гамма-интерферон и связанной с этим повышенной функцией Тн 2 -клеток, а также гиперпродукцией Ig. Е, что и обусловливает атипичное течение воспаления. Для детей с этим синдромом характерными признаками являются диспластические черты лица, спонтанные переломы трубчатых костей. При лабораторном обследовании у больных выявляется нарушение хемотаксиса нейтрофилов, повышенный уровень Ig. Е (1000 МЕ/мл), сниженная продукция Тлимфоцитами гамма-интерферона, повышенная концентрация в крови гистамина (75 -100 мкг/л).
 Лечение Патогенетическая иммунокоррекция до настоящего времени не предложена. Лечение сводится к предупреждению и устранению клинических проявлений заболевания, главным образом инфекционных. Реже требуют лечения дерматит и патологические переломы. Больные с синдромом Джоба нуждаются в пожизненной антимикробной терапии. В качестве антибактериальной терапии наиболее часто назначают цефалоспорины (цефтазидим, цефотаксим, цефтриаксон, цефаклор), аминогликозиды (амикацин, гентамицин, тобрамицин), антибиотики пенициллинового ряда (ампиокс, амоксициллин, потенцированный клавуланатом), имипенем+циластатим, сульфаниламиды (септрин, бактрим, котримоксазол). В противогрибковой терапии используются интраконазол, флуконазол. Также показано применение индукторов интерферона.
Лечение Патогенетическая иммунокоррекция до настоящего времени не предложена. Лечение сводится к предупреждению и устранению клинических проявлений заболевания, главным образом инфекционных. Реже требуют лечения дерматит и патологические переломы. Больные с синдромом Джоба нуждаются в пожизненной антимикробной терапии. В качестве антибактериальной терапии наиболее часто назначают цефалоспорины (цефтазидим, цефотаксим, цефтриаксон, цефаклор), аминогликозиды (амикацин, гентамицин, тобрамицин), антибиотики пенициллинового ряда (ампиокс, амоксициллин, потенцированный клавуланатом), имипенем+циластатим, сульфаниламиды (септрин, бактрим, котримоксазол). В противогрибковой терапии используются интраконазол, флуконазол. Также показано применение индукторов интерферона.
 (LAD-синдром) Дефект адгезии лейкоцитов приводит к нарушению") Дефицит экспрессии молекул адгезии (Дефект адгезии лейкоцитов) (LAD-синдром) Дефект адгезии лейкоцитов приводит к нарушению бактерицидной функции фагоцитов. Развивается в результате нарушения экспрессии на мембране клеток бета-цепи интегринов: LFA-1, Mac-1. Из-за отсутствия на лейкоцитах молекул адгезии нарушается их взаимодействие с другими клетками, нейтрофилы не связываются с клетками эндотелия и не мигрируют в ткани в ответ на хемотаксические стимулы. Клинически дефект адгезии лейкоцитов проявляется в развитии рецидивирующих кожных абсцессов, синуситов, пневмоний, трахеобронхитов, септицемий, бактериальных инфекций. У больных нарушена способность к формированию гноя, наблюдается лейкоцитоз (15 -20 × 109 /л), лихорадка неясной этиологии.
Дефицит экспрессии молекул адгезии (Дефект адгезии лейкоцитов) (LAD-синдром) Дефект адгезии лейкоцитов приводит к нарушению бактерицидной функции фагоцитов. Развивается в результате нарушения экспрессии на мембране клеток бета-цепи интегринов: LFA-1, Mac-1. Из-за отсутствия на лейкоцитах молекул адгезии нарушается их взаимодействие с другими клетками, нейтрофилы не связываются с клетками эндотелия и не мигрируют в ткани в ответ на хемотаксические стимулы. Клинически дефект адгезии лейкоцитов проявляется в развитии рецидивирующих кожных абсцессов, синуситов, пневмоний, трахеобронхитов, септицемий, бактериальных инфекций. У больных нарушена способность к формированию гноя, наблюдается лейкоцитоз (15 -20 × 109 /л), лихорадка неясной этиологии.
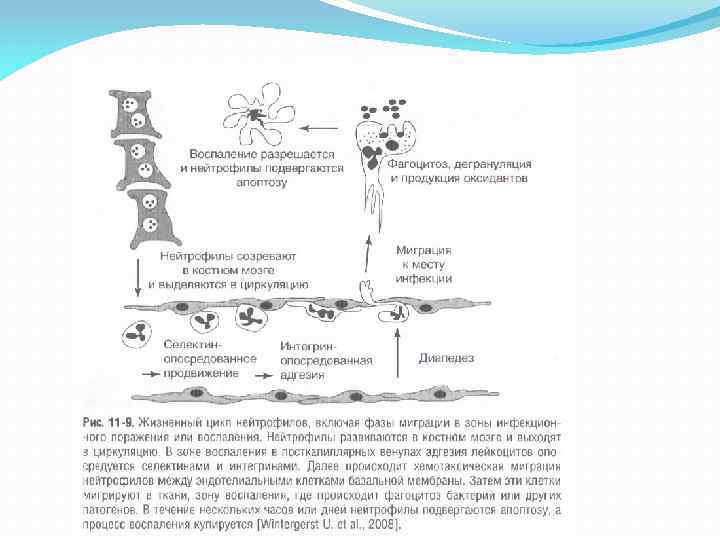
 Лечение антибактериальное назначение стимуляторов хемотаксиса лейкоцитов переливание лейкоцитарной массы.
Лечение антибактериальное назначение стимуляторов хемотаксиса лейкоцитов переливание лейкоцитарной массы.
 Молекулярно-генетические дефекты и характер иммунных расстройств у больных с первичным дефицитом системы фагоцитов. Заболевание, с которым Специфический дефект сцеплен иммнодефицит Характер нарушения 1. Хрониче-ский гранулематоз Нарушение cпособности Снижение киллерной фагоцитов продуцировать способности фагоцитов активные формы кислорода и, как следствие, нарушение их способности к киллингу и перевариванию поглощенных каталазоположительных микробов Снижение активности НАДФ-окси-даз, дефект цитохрома b 558. Cytp 91 phox Cytp 67 phox Cytp 22 phox 2. Синдром Чедиака-Хигаси - Характер иммунных расстройств Нарушение хемотаксиса и способности нейтрофилов высвобождать лизосомные ферменты в фагосомы Неспособность нейтрофилов разрушать бактерии 3. Синдром гипер-иммуноглобулинемии Е (синдром Джоба) Снижение продукции гамма-интерферона, повышенная секреция Ig. Е, чрезмерное высвобождение гистамина Нарушение хемотаксиса нейтрофилов Нарушение функции нейтрофилов, гамма интерферона, Ig. Е, гистамина 4. Дефицит экспрессии молекул адгезии Дефект CD 18 Нарушение адгезии лейкоцитов Снижение бактерицидной активности фагоцитов
Молекулярно-генетические дефекты и характер иммунных расстройств у больных с первичным дефицитом системы фагоцитов. Заболевание, с которым Специфический дефект сцеплен иммнодефицит Характер нарушения 1. Хрониче-ский гранулематоз Нарушение cпособности Снижение киллерной фагоцитов продуцировать способности фагоцитов активные формы кислорода и, как следствие, нарушение их способности к киллингу и перевариванию поглощенных каталазоположительных микробов Снижение активности НАДФ-окси-даз, дефект цитохрома b 558. Cytp 91 phox Cytp 67 phox Cytp 22 phox 2. Синдром Чедиака-Хигаси - Характер иммунных расстройств Нарушение хемотаксиса и способности нейтрофилов высвобождать лизосомные ферменты в фагосомы Неспособность нейтрофилов разрушать бактерии 3. Синдром гипер-иммуноглобулинемии Е (синдром Джоба) Снижение продукции гамма-интерферона, повышенная секреция Ig. Е, чрезмерное высвобождение гистамина Нарушение хемотаксиса нейтрофилов Нарушение функции нейтрофилов, гамма интерферона, Ig. Е, гистамина 4. Дефицит экспрессии молекул адгезии Дефект CD 18 Нарушение адгезии лейкоцитов Снижение бактерицидной активности фагоцитов
 Дефицит системы комплемента
Дефицит системы комплемента
 Генетический дефект в системе комплемента способен затрагивать любой из его компонентов. Дефект может выражаться в полном отсутствии какого-либо компонента комплемента либо в снижении его количества, что указывает на нарушение не в структурном, а в регуляторном гене. Различают следующие виды дефицитов: 1. дефицит С 1, С 2, С 3, С 4, С 5 -С 9, 2. дефицит С 1 – ингибитора, 3. дефицит фактора I.
Генетический дефект в системе комплемента способен затрагивать любой из его компонентов. Дефект может выражаться в полном отсутствии какого-либо компонента комплемента либо в снижении его количества, что указывает на нарушение не в структурном, а в регуляторном гене. Различают следующие виды дефицитов: 1. дефицит С 1, С 2, С 3, С 4, С 5 -С 9, 2. дефицит С 1 – ингибитора, 3. дефицит фактора I.
 Дефицит компонента комплемента Клинические проявления С 1, С 4, С 2 Иммунокомплексная патология (системной красной волчанка, гломерулонефрит) С 3 Рецидивирующие пиогенные инфекции (пневмонии, септицемии, отиты, менингиты) С 5 -С 9 Рецидивирующие нейссериальные инфекции С 1 -ингибитора Врожденный ангионевротический отек С 3 b-ингибитора (фактора I) Рецидивирующие пиогенные инфекции С 3 b Низкий уровень С 3, С 5, фактора В Пневмонии, менингиты, синуситы, отиты, септицемии
Дефицит компонента комплемента Клинические проявления С 1, С 4, С 2 Иммунокомплексная патология (системной красной волчанка, гломерулонефрит) С 3 Рецидивирующие пиогенные инфекции (пневмонии, септицемии, отиты, менингиты) С 5 -С 9 Рецидивирующие нейссериальные инфекции С 1 -ингибитора Врожденный ангионевротический отек С 3 b-ингибитора (фактора I) Рецидивирующие пиогенные инфекции С 3 b Низкий уровень С 3, С 5, фактора В Пневмонии, менингиты, синуситы, отиты, септицемии
 Наследственный ангионевротический отек Врожденное заболевание, наследуемое по аутосомнодоминантному типу. Проявляется в виде рецидивирующих самоорганизующихся отеков кожных покровов и слизистых оболочек.
Наследственный ангионевротический отек Врожденное заболевание, наследуемое по аутосомнодоминантному типу. Проявляется в виде рецидивирующих самоорганизующихся отеков кожных покровов и слизистых оболочек.
 Наиболее часто отеки охватывают слизистые оболочки глотки, гортани, желудка, кишечника, несколько реже лица и конечностей. Отеки плотные по консистенции, как правило, ограничены по площади, имеют беловатую окраску, не сопровождаются зудом и сыпью. Длительность отека 24 -72 часа. Отек слизистой кишечника способен привести к его непроходимости, отек гортани – к асфиксии. Развитие отеков наблюдается с различной периодичностью. Они могут отсутствовать несколько лет подряд или в течение одного года появляться десятки раз. Отеки могут быть спровоцированы случайной травмой, операционным вмешательством, психоэмоциональным и физическим перенапряжением, химическими и лекарственными веществами. В 40% случаев отеки развиваются спонтанно, без видимых причин.
Наиболее часто отеки охватывают слизистые оболочки глотки, гортани, желудка, кишечника, несколько реже лица и конечностей. Отеки плотные по консистенции, как правило, ограничены по площади, имеют беловатую окраску, не сопровождаются зудом и сыпью. Длительность отека 24 -72 часа. Отек слизистой кишечника способен привести к его непроходимости, отек гортани – к асфиксии. Развитие отеков наблюдается с различной периодичностью. Они могут отсутствовать несколько лет подряд или в течение одного года появляться десятки раз. Отеки могут быть спровоцированы случайной травмой, операционным вмешательством, психоэмоциональным и физическим перенапряжением, химическими и лекарственными веществами. В 40% случаев отеки развиваются спонтанно, без видимых причин.
 Существует две патофизиологические формы врожденного ангионевротического отека: истинная, при которой наблюдается фактический дефицит ингибитора – С 1 компонента комплемента вариантная, при которой определяется нормальное содержание ингибитора С 1 в крови, но функциональная активность его подавлена.
Существует две патофизиологические формы врожденного ангионевротического отека: истинная, при которой наблюдается фактический дефицит ингибитора – С 1 компонента комплемента вариантная, при которой определяется нормальное содержание ингибитора С 1 в крови, но функциональная активность его подавлена.
 Ангионевротический отек развивается, когда его количество или активность снижается более, чем на 70% от нормы. При этом происходит активация системы комплемента и его компонента С 2, который, как полагают, играет ключевую роль в каскаде биохимических реакций. Под влиянием активированных компонентов комплемента усиливается продукция кининов (брадикинина) и высвобождение их в окружающую ткань. В результате их действия происходит расслабление гладкой мускулатуры, вазодилатация, повышается проницаемость сосудистой стенки. Все эти эффекты приводят к пропотеванию жидкости из сосудов в ткань и развитию отека. Следует помнить, что наряду с врожденной формой ангионевротического отека существует приобретенная и аллергическая формы ангионевротического отека.
Ангионевротический отек развивается, когда его количество или активность снижается более, чем на 70% от нормы. При этом происходит активация системы комплемента и его компонента С 2, который, как полагают, играет ключевую роль в каскаде биохимических реакций. Под влиянием активированных компонентов комплемента усиливается продукция кининов (брадикинина) и высвобождение их в окружающую ткань. В результате их действия происходит расслабление гладкой мускулатуры, вазодилатация, повышается проницаемость сосудистой стенки. Все эти эффекты приводят к пропотеванию жидкости из сосудов в ткань и развитию отека. Следует помнить, что наряду с врожденной формой ангионевротического отека существует приобретенная и аллергическая формы ангионевротического отека.
 Лечение врожденного ангионевротического отека в острый период – переливание нативной или свежезамороженной плазмы. Развитие отека гортани требует немедленной госпитализации больного в хирургическую клинику (клинику ЛОР-болезней). При необходимости производят эндотрахеальную интубацию или трахеостомию. Развитие абдоминального синдрома требует консультации хирурга. Профилактическое лечение таких больных включает: назначение даназола (данола), метилтестостерона, εаминокапроновой кислоты, транексамовой кислоты а при достижении клинической ремиссии дозу препарата снижают.
Лечение врожденного ангионевротического отека в острый период – переливание нативной или свежезамороженной плазмы. Развитие отека гортани требует немедленной госпитализации больного в хирургическую клинику (клинику ЛОР-болезней). При необходимости производят эндотрахеальную интубацию или трахеостомию. Развитие абдоминального синдрома требует консультации хирурга. Профилактическое лечение таких больных включает: назначение даназола (данола), метилтестостерона, εаминокапроновой кислоты, транексамовой кислоты а при достижении клинической ремиссии дозу препарата снижают.
 Основные клинические симптомы и тесты лаб. Диагностики ПИД
Основные клинические симптомы и тесты лаб. Диагностики ПИД
 Успешную диагностику врожденных нарушений ИС можно провести на основании оценки разнообразных клинических симптомов ПИД и выявления ряда особенностей и проявлений. Выявление и анализ этих симптомов в сочетании с общеклиническими лаб. исследованиями позволяют врачу с большой долей вероятностью заподозрить у пациента наличие врожденного дефекта ИС.
Успешную диагностику врожденных нарушений ИС можно провести на основании оценки разнообразных клинических симптомов ПИД и выявления ряда особенностей и проявлений. Выявление и анализ этих симптомов в сочетании с общеклиническими лаб. исследованиями позволяют врачу с большой долей вероятностью заподозрить у пациента наличие врожденного дефекта ИС.
 Основные клинические симптомы и лаб. показатели, характерные для первичных иммунодефицитов Клинические симптомы Лабораторные показатели Рецидивирующие, плохо поддающиеся традиционной терапии инфекционные заболевания респираторноного и пищ. тракта Лейкопения, лимфопения, нейтропения, гемолитическая анемия, тромбоцитопения[лимфаденопатия в сочетании с гепатоспленомегалией(при исключении злокачественных новооббразований и специфических инфекционных заболеваний)], снижение уровня Ig Оппортунистические инфекции Упорный кандидоз слизистых оболочек и кожи, особенно у детей старше 1 года Упорная диарея, мальабсорбция Повторные бактериальные инфекционные поражения кожи, ПЖК, лимф. узлов и внутренних органов Отставание в развитии у младенцев на фоне необычных инфекций и диареи Лимфаденопатия в сочетании гепатоспленомегалией(при исключении злокачественных новообразований и специф. инфекционных заболеваний) Артриты, дерматомиозит, склеродермия Гипоплазия лимфоидной ткани
Основные клинические симптомы и лаб. показатели, характерные для первичных иммунодефицитов Клинические симптомы Лабораторные показатели Рецидивирующие, плохо поддающиеся традиционной терапии инфекционные заболевания респираторноного и пищ. тракта Лейкопения, лимфопения, нейтропения, гемолитическая анемия, тромбоцитопения[лимфаденопатия в сочетании с гепатоспленомегалией(при исключении злокачественных новооббразований и специфических инфекционных заболеваний)], снижение уровня Ig Оппортунистические инфекции Упорный кандидоз слизистых оболочек и кожи, особенно у детей старше 1 года Упорная диарея, мальабсорбция Повторные бактериальные инфекционные поражения кожи, ПЖК, лимф. узлов и внутренних органов Отставание в развитии у младенцев на фоне необычных инфекций и диареи Лимфаденопатия в сочетании гепатоспленомегалией(при исключении злокачественных новообразований и специф. инфекционных заболеваний) Артриты, дерматомиозит, склеродермия Гипоплазия лимфоидной ткани
, субпопуляций Т-лимфоцитов(CD") Тесты оценки Т-клеточного звена Скрининговые тесты: 1. Абсолютное число лимфоцитов, Т-лимфоцитов(CD 3), субпопуляций Т-лимфоцитов(CD 4, 8) 2. Ультразвуковое исследование или рентгенография тимуса у младенцев 3. Кожные тесты ГЗТ. Подтверждающие тесты: 1. Лимфопролиферативный ответ на митогены, АГ и аллогенные кл. 2. Образование цитокинов: Th 1 -типа(ИНФгамма, ФНОальфа, ИЛ-2); Th 2 -типа(ИЛ-4, 5, 6, 10, 13) Функциональный ответ Т-кл. на цитокины Выявление активационных молекул на мембране Тлифоцитов(CD 25, HLA-DR) Молекулярный анализ специфических дефектов развития Т-кл. Выявление молекул адгезии(интегрина CD 11 a/CD 18)
Тесты оценки Т-клеточного звена Скрининговые тесты: 1. Абсолютное число лимфоцитов, Т-лимфоцитов(CD 3), субпопуляций Т-лимфоцитов(CD 4, 8) 2. Ультразвуковое исследование или рентгенография тимуса у младенцев 3. Кожные тесты ГЗТ. Подтверждающие тесты: 1. Лимфопролиферативный ответ на митогены, АГ и аллогенные кл. 2. Образование цитокинов: Th 1 -типа(ИНФгамма, ФНОальфа, ИЛ-2); Th 2 -типа(ИЛ-4, 5, 6, 10, 13) Функциональный ответ Т-кл. на цитокины Выявление активационных молекул на мембране Тлифоцитов(CD 25, HLA-DR) Молекулярный анализ специфических дефектов развития Т-кл. Выявление молекул адгезии(интегрина CD 11 a/CD 18)
") Тесты оценки B-клеточного звена Скрининговые тесты: 1. Опледеление содержания В-лимфоцитовв переферической крови(CD 19, 20) 2. Определение концентрации сывороточных иммуноглобулинов Ig A, M, E, G 3. Определение субклассов Ig. G(1 -4) 4. Выявление специфических АТ Подтверждающие тесты: 1. Тест на образование В-кл. иммуноглобулинов in vitro 2. Регуляция синтеза иммуноглобулинов 3. Молекулярный анализ генитических нарушений.
Тесты оценки B-клеточного звена Скрининговые тесты: 1. Опледеление содержания В-лимфоцитовв переферической крови(CD 19, 20) 2. Определение концентрации сывороточных иммуноглобулинов Ig A, M, E, G 3. Определение субклассов Ig. G(1 -4) 4. Выявление специфических АТ Подтверждающие тесты: 1. Тест на образование В-кл. иммуноглобулинов in vitro 2. Регуляция синтеза иммуноглобулинов 3. Молекулярный анализ генитических нарушений.
 Тесты оценки фагоцитарного звена Скрининговые тесты: 1. Определение абсолютного числа нейтрофлов и моноцитов 2. Показатели фагоцитоза 3. Генерация активных форм кислорода(НСТ-тест). Подтверждающие тесты: 1. Оценка хемотаксиса 2. Определение завершенности фагоцитоза 3. Определение экспрессии молекул адгезии на повнрхностной мембране нейтрофилов(CD 11 а, CD 11 b, CD 11 с, CD 18).
Тесты оценки фагоцитарного звена Скрининговые тесты: 1. Определение абсолютного числа нейтрофлов и моноцитов 2. Показатели фагоцитоза 3. Генерация активных форм кислорода(НСТ-тест). Подтверждающие тесты: 1. Оценка хемотаксиса 2. Определение завершенности фагоцитоза 3. Определение экспрессии молекул адгезии на повнрхностной мембране нейтрофилов(CD 11 а, CD 11 b, CD 11 с, CD 18).
 Лечение ПИД Патогенетическая, клиническая и прогностическая вариабельность этой группы заболеваний делает их терапию достаточно сложной задачей. Выбор терапии, как правило, основывается на оценке состояния больного, характере и глубине иммунных расстройств. Терапия иммунодефицитных состояний включает: 1) антимикробную терапию, 2) заместительную терапию, 3) реконструктивную терапию.
Лечение ПИД Патогенетическая, клиническая и прогностическая вариабельность этой группы заболеваний делает их терапию достаточно сложной задачей. Выбор терапии, как правило, основывается на оценке состояния больного, характере и глубине иммунных расстройств. Терапия иммунодефицитных состояний включает: 1) антимикробную терапию, 2) заместительную терапию, 3) реконструктивную терапию.
, противогрибковых и противовирусных средств. При появлении") Антимикробная терапия включает применение антибактериальных препаратов (антибиотиков), противогрибковых и противовирусных средств. При появлении признаков активной инфекции стартовая терапия назначается с учетом характера дефекта иммунной системы. При генерализованной инфекции проводится госпитализация больного и назначается внутривенное введение комбинаций антибиотиков с максимально широким спектром действия до выяснения типа патогена (посевы крови) и/или достижения эффекта. При отсутствии эффекта следует назначать противогрибковые препараты (амфотерицин В). Заместительная терапия предполагает, в первую очередь, регулярные внутривенные инфузии иммуноглобулина. Альтернативный вариант – инфузия свежезамороженной плазмы. Однако, при использовании этого метода очень высокий риск парентеральной инфекции, в связи с чем желательно привлечение постоянных доноров. Реконструктивная терапия включает трансплантацию гемопоэтической ткани и генную терапию. В настоящее время в мире проведены сотни пересадок костного мозга при врожденных дефектах иммунной системы. В 50 -70 % случаев наблюдается выраженный терапевтический эффект. Наиболее серьезными проблемами трансплантации гемопоэтических клеток являются слабая приживаемость донорских клеток и развитие реакции трансплантат против хозяина. Имеются сообщения об успешной пересадке гена, кодирующего аденозиндезаминазу, больным с дефицитом данного фактора.
Антимикробная терапия включает применение антибактериальных препаратов (антибиотиков), противогрибковых и противовирусных средств. При появлении признаков активной инфекции стартовая терапия назначается с учетом характера дефекта иммунной системы. При генерализованной инфекции проводится госпитализация больного и назначается внутривенное введение комбинаций антибиотиков с максимально широким спектром действия до выяснения типа патогена (посевы крови) и/или достижения эффекта. При отсутствии эффекта следует назначать противогрибковые препараты (амфотерицин В). Заместительная терапия предполагает, в первую очередь, регулярные внутривенные инфузии иммуноглобулина. Альтернативный вариант – инфузия свежезамороженной плазмы. Однако, при использовании этого метода очень высокий риск парентеральной инфекции, в связи с чем желательно привлечение постоянных доноров. Реконструктивная терапия включает трансплантацию гемопоэтической ткани и генную терапию. В настоящее время в мире проведены сотни пересадок костного мозга при врожденных дефектах иммунной системы. В 50 -70 % случаев наблюдается выраженный терапевтический эффект. Наиболее серьезными проблемами трансплантации гемопоэтических клеток являются слабая приживаемость донорских клеток и развитие реакции трансплантат против хозяина. Имеются сообщения об успешной пересадке гена, кодирующего аденозиндезаминазу, больным с дефицитом данного фактора.
 Вакцинация больных с первичными иммунодефицитами
Вакцинация больных с первичными иммунодефицитами
 Вакцинация больных с ПИД может быть опасна, неэффективна или очень важна. В случаях, когда сохранена способность организма к какому -либо иммунному ответу, иммунизация не только не запрещена, но и показана, в том числе и в более интенсивных, чем для здорового ребенка, режимах. Возможно использование только убитых вакцин. Вакцинация может иметь и диагностическую ценность, так как ясно показывает способность организма к развитию иммунных реакций и степень иммунных расстройств. Больным первичным иммунодефицитом введение живых вакцин противопоказано в связи с возможностью развития инфекции. После эффективной реконструктивной терапии больные первичным иммунодефицитом нуждаются в иммунизации, как и здоровые дети, однако ее можно проводить в возрасте не ранее 2 лет и не менее чем через 1 год после трансплантации гемопоэтических клеток.
Вакцинация больных с ПИД может быть опасна, неэффективна или очень важна. В случаях, когда сохранена способность организма к какому -либо иммунному ответу, иммунизация не только не запрещена, но и показана, в том числе и в более интенсивных, чем для здорового ребенка, режимах. Возможно использование только убитых вакцин. Вакцинация может иметь и диагностическую ценность, так как ясно показывает способность организма к развитию иммунных реакций и степень иммунных расстройств. Больным первичным иммунодефицитом введение живых вакцин противопоказано в связи с возможностью развития инфекции. После эффективной реконструктивной терапии больные первичным иммунодефицитом нуждаются в иммунизации, как и здоровые дети, однако ее можно проводить в возрасте не ранее 2 лет и не менее чем через 1 год после трансплантации гемопоэтических клеток.
 Конец!
Конец!