ПИДС диагностика и терапия.ppt
- Количество слайдов: 100



ПЕРВИЧНЫЕ ИММУНОДЕФИЦИТЫ В ПРАКТИКЕ ГЕМАТОЛОГА Щербина А. Ю. ФНКЦ Детской Гематологии, Онкологии и Иммунологии имени Дмитрия Рогачева, Москва

Иммунная система Клетки Органы Растворимые вещества

ИММУНОДЕФИЦИТЫ – значимые, длительно существующие дефекты звеньев иммунитета, сопровождающиеся клиническими проявлениями

Опухоли Аутоиммунные состояния ПОВРЕЖДЕННЫЕ КЛЕТКИ Деструкция тканей, шок, ЧУЖ. хр. воспаление ЧУЖ. ОРГАНИЗМЫ ВЕЩЕСТВА Частые Аллергия инфекции

ИММУНОДЕФИЦИТЫ ПЕРВИЧНЫЕ ВТОРИЧНЫЕ q Повышенная чувствительность к инфекциям q Аутоиммунные заболевания q Опухоли q Аллергия

ВТОРИЧНЫЕ ИММУНОДЕФИЦИТЫ Ø Дефицит и потери белка Ø Спленэктомия ØОпухоли Ø Иммуносупрессивная химиотерапия Ø Иррадиация Ø Инфекция (в т. ч. ВИЧ)

ВТОРИЧНЫЕ ИММУНОДЕФИЦИТЫ Необходимо найти причину! Нет причины – ПИДС? Инфицирование ЦМВ и ЭБВ не является причиной иммунодефицита!

ИССЛЕДОВАНИЕ в США 2007 г. • Время хапаздывания диагноза – 12. 4 г. • 49% - стойкие изменения органов вследствие ИДС

КОМПОНЕНТЫ ИММУНИТЕТА Врожденный • Микробицидные вещества • Фагоциты • Комплемент Приобретенный • Клетки • Иммуноглобулины • Цитокины

ВТОРИЧНЫЕ ИММУНОДЕФЦИТЫ Как правило комбинированные

ПИДС – ГЕНЕТИЧЕСКИ ОБУСЛОВЛЕННЫЕ ЗАБОЛЕВАНИЯ

")
ПЕРВИЧНЫЕ ИДС • Гуморальные дефекты • Комбинированные дефекты ( клеточного и гуморального звена) • Количественные и качественные дефекты фагоцитов • Дефекты врожденного иммунитета • Аутовоспалительные заболевания • Дефекты системы комплемента • Синдромальные формы ПИДС (в т. ч дефекты репарации ДНК и др) • ПИДС с иммунной дисрегуляцией • Соматические фенокопии

ОБЩИЕ ЗАКОНОМЕРНОСТИ • ГИПОДИАГНОСТИКА

Пол и возраст живущих пациентов Муж Жен Частота Возраст, лет

ОБЩИЕ ЗАКОНОМЕРНОСТИ • ГИПОДИАГНОСТИКА • РАЗНООБРАЗИЕ ПРОЯВЛЕНИЙ

Вариабельность ПИДС обусловлена: • Множественными функциями иммунной системы • Поражением других органов и систем (синдромальная патология) • Различной степенью выраженности симптомов при одном заболевании

Общие закономерности • ГИПОДИАГНОСТИКА • РАЗНООБРАЗИЕ ПРОЯВЛЕНИЙ • РАЗЛИЧНЫЕ «МАСКИ»

ИНФЕКЦИОННЫЕ МАСКИ – атипичное течение или возбудители ТКИН

ИНФЕКЦИОННЫЕ МАСКИ – атипичное течение или возбудители ХГБ

ИНФЕКЦИОННЫЕ МАСКИ – атипичное течение или возбудители Гипер-Ig. E синдром

ИНФЕКЦИОННЫЕ МАСКИ – атипичное течение или возбудители Деф. STAT 1

Аллергические «маски» ПИДС Гипер Ig. E синдром

Аллергические «маски» ПИДС Синдром Омен

Аллергические «маски» ПИДС Дефицит С 1 ингибитора

FHL CAPS
 • Эндокринопатия смешанного генеза")
ЭНДОКРИНОЛОГИЧЕСКИЕ МАСКИ • Иммунная эндокринопатия: гипопаратиреоз, гипокортицизм (APECED, IPEX) • Эндокринопатия смешанного генеза (гипопаратиреоз при синдроме Ди. Джорджи)

ГАСТРОЭНТЕРОЛОГИЧЕСКИЕ МАСКИ • Диаррея, с-м мальабсорбции: - ТКИН - IPEX - XLP - гипер-Ig. D синдром • Тяжелое ВЗК: - деф. ИЛ-10 - IPEX - ОВИН
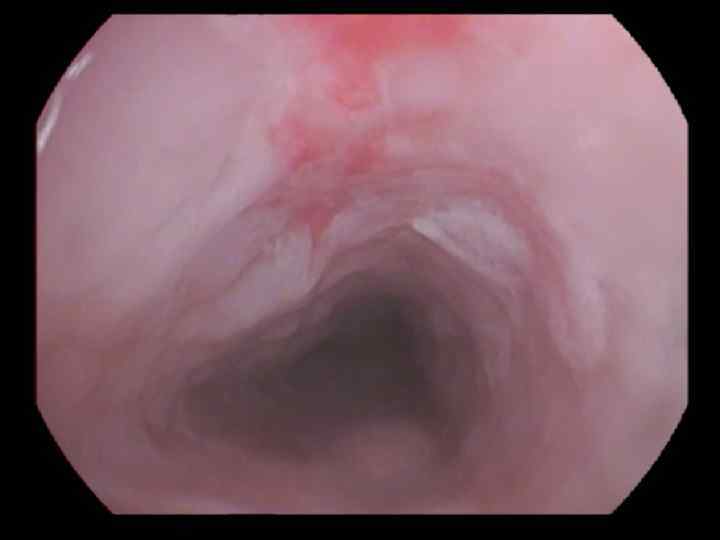

АУТОИММУННЫЕ МАСКИ 1. Нефрит 2. Артрит 3. Васкулит 4. Дерматомиозит 5. СКВ и др.

ПСЕВДО-АУТОИММУННЫЕ МАСКИ – Аутовоспалительные заболевания • Лихорадка • Сыпи • Поражение суставов, костей • Поражение внутренних органов • Выраженная воспалительная активность

ГЕПАТОСПЛЕНОМЕГАЛИЯ

ЛИМФАДЕНОПАТИЯ

ПОРАЖЕНИЕ КОСТЕЙ с-м СRMO
 • Цитопении вследствие нарушения структуры,")
ГЕМАТОЛОГИЧЕСКИЕ МАСКИ • Аутоиммунные цитопении (анемия, тромбоцитопения, нейтропения) • Цитопении вследствие нарушения структуры, дифференцировки/пролиферации, либо метаболизма клеток крови (нейтропения при Х-АГГ, тромбоцитопения при СВО, дефиците АДА) • Гемофагоцитарные синдромы

Селективный дефицит ИТП, АГА Ig. A, ОВИН Гем. васкулит Гипер-Ig. M синдром ИТП АЛПС ИТП, АГА Нейтропения Синдром Вискотта- АГА Олдрича ИТП Дефекты ИЛ 12ИФНg Гем. васкулит
. 2004; 83(4): 254 -63 21 больной")
ОВИН Michel M et al, Medicine (Baltimore). 2004; 83(4): 254 -63 21 больной с ОВИН и ИТП Диагноз ИТП - 23 г. Диагноз ОВИН – 27 л.

Цитопении при ПИДC • Часто резистенты к терапии первой линии • Часто рецидивируют • Возможно урежаются при регуляно проводимой заместительной терапии ВВИГ • ГКС, иммунодепрессанты и спленэктомия усиливают инфекционные проявления • Использование средств «резерва»

ГЕМАТОЛОГИЧЕСКИЕ МАСКИ • 30 % больных с синдромом Фишера- Эванса – соматические мутации Fas. L • Дефект CD 27 и Coronin – 60% лимфомы без других признаков (X)LP
 • В основе заболевания - врожденные дефекты апоптоза •")
АУТОИММУННЫЙ ЛИМФОПРОЛИФЕРАТИВНЫЙ СИНДРОМ (АЛПС) • В основе заболевания - врожденные дефекты апоптоза • Хроническая незлокачественнная неинфекционная лимфопролиферация • Гипергаммаглобулинемия • Аутоиммунные нарушения • ↑ CD 3+ TCRα/β+CD 4 -CD 8 -

АЛПС ALPS-FASLG - <5% ALPS-FAS – 65 -70% ALPS-s. FAS – 15 -20% ALPS-CASP 10 - <5% ALPS-? – 10%

АЛПС • Лимфопролиферация 100% • Увеличение лимфоузлов 87% • Спленомегалия 90% • Гепатомегалия 45% • АГА 53% • Тромбоцитопения 44% • Нейтропения 31% • Лимфома и др 15%

Семейный анамнез Мутация гена TNFRSF 6 АГА Т-клеточная лимфома? Лимфогранулематоз

КОНТРОЛЬ ПАЦИЕНТ 1 ПАЦИЕНТ 2
 • Эверолимус? ? ? • Цитостатическая терапия")
АЛПС - ТЕРАПИЯ • Сиролимус (рапамун) • Эверолимус? ? ? • Цитостатическая терапия посиндромно • Улучшение с возрастом
 • Цитопении вследствие нарушения структуры,")
ОНКОГЕМАТОЛОГИЧЕСКИЕ МАСКИ • Аутоиммунные цитопении (анемия, тромбоцитопения, нейтропения) • Цитопении вследствие нарушения структуры, дифференцировки/пролиферации, либо метаболизма клеток крови (нейтропения при HIGM, тромбоцитопения при СВО, анемия при дефиците АДА)
 ¨Частота 1 : 250, 000 рожденных мальчиков. ¨СВО возникает вследствие мутаций")
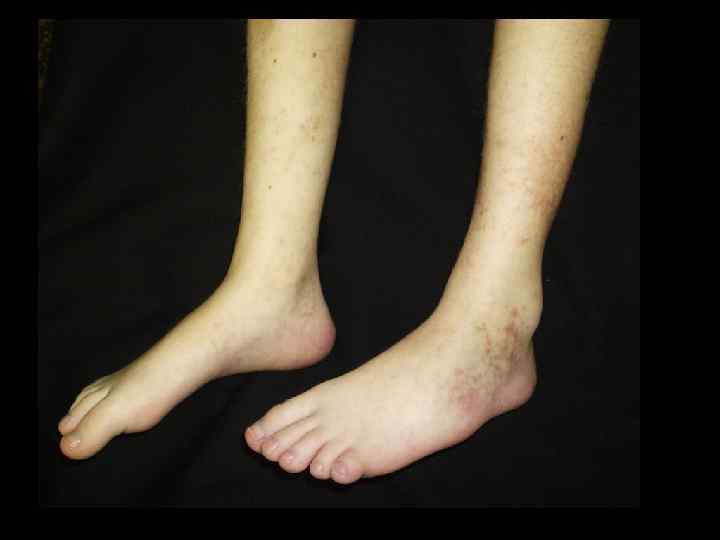
Синдром Вискотта-Олдрича (СВО) ¨Частота 1 : 250, 000 рожденных мальчиков. ¨СВО возникает вследствие мутаций гена WASP, локализованного на Хр11. 22.

Частота основных проявлений СВО Тромбоцитопения 100% Уменьшение диаметра тромбоцитов 100% Геморрагический синдром 95% Инфекции 63% Дерматит 78% Аутоиммунная патология 38% Злокачественные заболевания 8%

ДИАГНОСТИКА СВО • Экспрессия белка WASP • Генетика

Число лимфоцитов Концентрация белка WASP
 • Вариабельность тяжести • Необходимость ТГСК")
Синдром Вискотта-Олдрича (СВО) • Вариабельность тяжести • Необходимость ТГСК

ЛЕЧЕНИЕ СВО • ТГСК • Заместительная терапия • Посиндромная терапия • Агонисты тромбопоэтиного рецептора • Спленэктомия?
 • Цитопении вследствие нарушения структуры,")
ОНКОГЕМАТОЛОГИЧЕСКИЕ МАСКИ • Аутоиммунные цитопении (анемия, тромбоцитопения, нейтропения) • Цитопении вследствие нарушения структуры, дифференцировки/пролиферации, либо метаболизма клеток крови (нейтропения при HIGM, тромбоцитопения при СВО, анемия при дефиците АДА) • Гемафагоцитарные синдромы
 • Цитопении вследствие нарушения структуры,")
ОНКОГЕМАТОЛОГИЧЕСКИЕ МАСКИ • Аутоиммунные цитопении (анемия, тромбоцитопения, нейтропения) • Цитопении вследствие нарушения структуры, дифференцировки/пролиферации, либо метаболизма клеток крови (нейтропения при HIGM, тромбоцитопения при СВО, анемия при дефиците АДА) • Гемафагоцитарные синдромы • Повышенная склонность к опухолям
")
X-сцепленный лимфопролиферативный синдром (ХLP)
, BIRC, Сoronin,")
Патогенез XLP • Мутации генов SAP (SH 2 D 1 A), BIRC, Сoronin, локализующихся на Х-хромосоме • Другие (CD 27, MAGT) • Нарушение функции Т лимфоцитов (CD 8)

Клиника XLP До инфекции EBV – практически норма. Редко: колит, нейсерийный менингит, коревая пневмония, В лимфома

Клиника XLP: EBV инфекция • Фульминантный ИМ 58% • ИДС 31% • Лимфопролиф. состояние 30% • Апластическая анемия 3% • Васкулит, лимфоматоидный грануломатоз 3%

Фульминантный ИМ=HLH Дебют в ср. 5 лет Поликлональная Т активация • Гепатит, печеночная недостаточность • Тромбоцитопения • Коагулопатия • Энцефалопатия • Аплазия КМ • Миокарит • Нефрит

ИДС • Некроз лимфоретикулярной системы • К. п. гипогамаглобулинемия(иногда повышение Ig. A, Ig. M) • Иногда поражение Т и NK клеток, клиника КИН

МРТ: Крупный очаг в печени, который биопсировали, а

МРТ: Поражение головки бедренной

Терапия XLP Ритукcимаб, другие? Заместительная терапия ВВИГ Необходимость ранней ТГСК
 • Цитопении вследствие нарушения структуры,")
ОНКОГЕМАТОЛОГИЧЕСКИЕ МАСКИ • Аутоиммунные цитопении (анемия, тромбоцитопения, нейтропения) • Цитопении вследствие нарушения структуры, дифференцировки/пролиферации, либо метаболизма клеток крови (нейтропения при HIGM, тромбоцитопения при СВО, анемия при дефиците АДА) • Гемафагоцитарные синдромы • Повышенная склонность к опухолям • Атипичный ответ на терапию, вторичные опухоли
 АЛПС С-м Ниймеген СВО АТ")
Встречаемость лимфом при различных формах иммунодефицитов (регистр ФНКЦ ДГОИ) АЛПС С-м Ниймеген СВО АТ ОВИН

ИДС с нарушением репарации ДНК • Атаксия-тельангиоэктазия • Синдром Ниймеген • Другие

События при повреждении ДНК 1. В ответ на повреждение ДНК происходит аутофосфорилирование неактивного мультимера АТМ и он распадается на активные фосфорилированные мономеры. 2. Одновременно гистоновый белок, находящийся внутри хроматина , становится страховочной платформой для повреждённой молекулы ДНК и к месту повреждения ДНК прикрепляется белковый комплекс MRE 11 -RAD 50 -NBS 1, а также p 53(супрессор опухоли, предотвращает переход клетки от. G 1 до фазы S и вызывает апоптоз) и BRAC-1 3. Все компоненты этого мультбелкового комплекса фосфорилируются с помощью активных ATM-мономеров

Нарушение контроля репарации DSB Норма: АТМ р53/р21 Сd 2/cyclin блок G 1/S NBS p 95/RAD 50/MRE 11 репарация ДНК А-Т: АТМ р53/р21 Сd 2/cyclin блок G 1/S NBS p 95/RAD 50/MRE 11 репарация ДНК NBS: АТМ р53/р21 Сd 2/cyclin блок G 1/S NBS p 95/RAD 50/MRE 11 репарация ДНК

ИДС с нарушением репарации ДНК • Не всегда инфекции • Высокая частота опухолей • Особенности терапии

Особенности химиотерапии: с осторожностью агенты, вызывающих повреждения ДНК

Синдром Луи-Барр Ген ATM на 11 q 23 Аутосомно-рецессивный тип наследования Популяционная частота заболевания - 1 на 1 000000

Синдром Луи-Барр • Атаксия, прогрессирующая нейродегенерация • Тельангиэктазии • Радиочувствительность • Иммунодефицит разл. выраженности • Предрасположенность к опухолям • Стерильность • Повышение альфа-фетопротеина
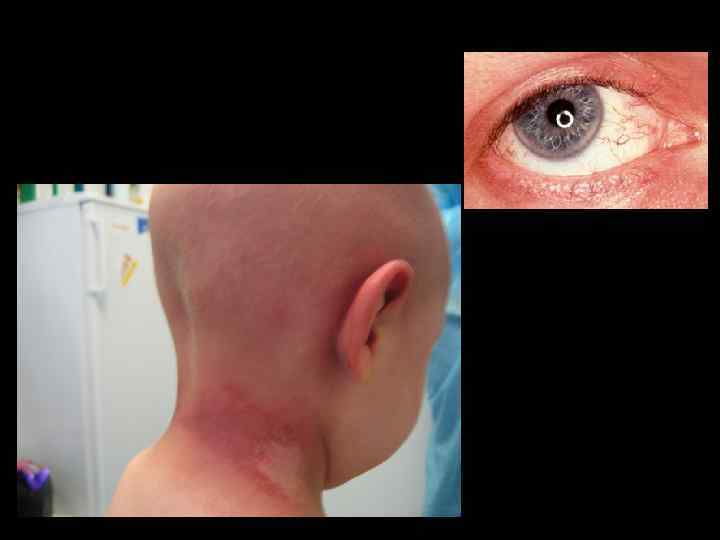

Синдром Луи-Барр • Совокупность клинических признаков • Ig. A дефицит, м. б. и Ig. Е • Ig. М м. б. повышен, дефект субклассов Ig. G • М. б. снижение количества Т-лимфоцитов, инверсия соотношения СD 4+/CD 8+ • Повышение альфа-фетопротеина • Спонтанные хромосомные аберрации

Синдром Ниймеген • Аутосомно-рецессивное заболевание • Ген NBS на 8 q 21

Клинические проявления • Микроцефалия • «Птичье» лицо • Поли-синдактилия • Атрезии ЖКТ • Гидронефроз • Пигментные пятна • Редко - задержка умств. развития

Клинические проявления • Инфекции легких, бронхоэктазы • Аутоимм. цитопении • Гранулематозное поражение кожи • ИЛБЛ
 • Снижение одного из классов/субклассов • Лимфопения •")
Лабораторные показатели • Агаммаглобулинемия (30%) • Снижение одного из классов/субклассов • Лимфопения • Снижение митогенного ответа • Хромосомная нестабильность

СИНДРОМ НИЙМЕГЕН

ИДС с нарушением репарации ДНК • Минимизировать облучение • Меньшие дозы алкилирующих агентов • ТГСК?

Наиболее тяжелые ПИДС

ТКИН Пораженные Заболевание Частота клетки Т В NK Ретикулярная < 1% дизгенезия Дефицит АДА, ПНФ 10% T- B- NK- Дефицит гама-цепи 50% T- B+ Дефицит JAK 3 5 -10% RAG 1, RAG 2, 20% T- B- NK+ Artemis Дефицит ИЛ-7 Р 5 -10% T- B+ NK+ Дефицит CD 3 d ? Дефицит CD 45 <1%

Основные клинические проявления • Персистирующая диарея, с-м мальабсорбции • Грибковые инфекции кожи и слизистых • Прогрессирующее поражение респираторного тракта • Отставание в физическом развитии • Гипоплазия лимфоидной ткани • Кожные сыпи, поражение печени вследствие РТПХ • Посттрансфузионная РТПХ • Регионарный или генерализованный БЦЖ-ит
 • Pneumocystis carinea • Вирусы: РС, парагрипп, EBV,")
Инфекционные агенты • Бактерии (любые) • Pneumocystis carinea • Вирусы: РС, парагрипп, EBV, CMV (интерстициальная пневмония, гепатит), энтеровирусы, аденовирусы (менингоэнцефалит) • Грибы (кандидоз кожи, слизитых, ногтей, другие локализации реже) • Микобактерии (БЦЖ-ит)
 • Отсутствие специфических АТ (вакцинальные,")
Лабораторные данные • Лимфопения • Агаммаглобулинемия (не всегда) • Отсутствие специфических АТ (вакцинальные, инфекционные) • Снижение пролиферативной активности лимфоцитов • М. б. гематологические нарушения (тромбоцитопения, анемия, нейтропения)
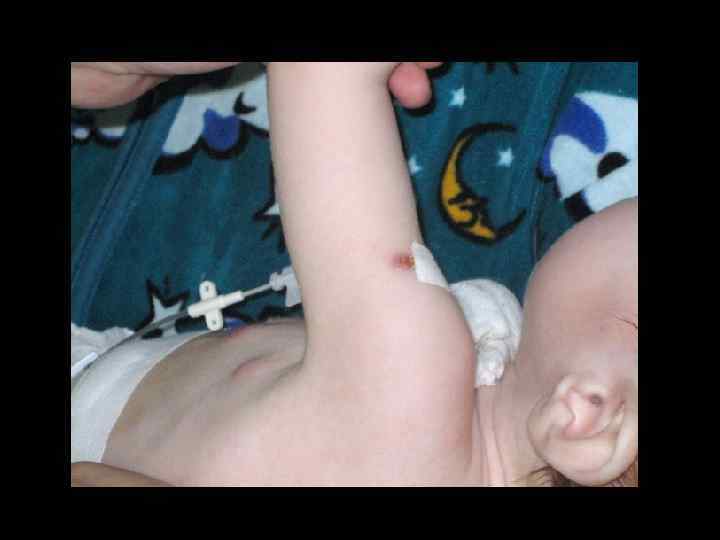
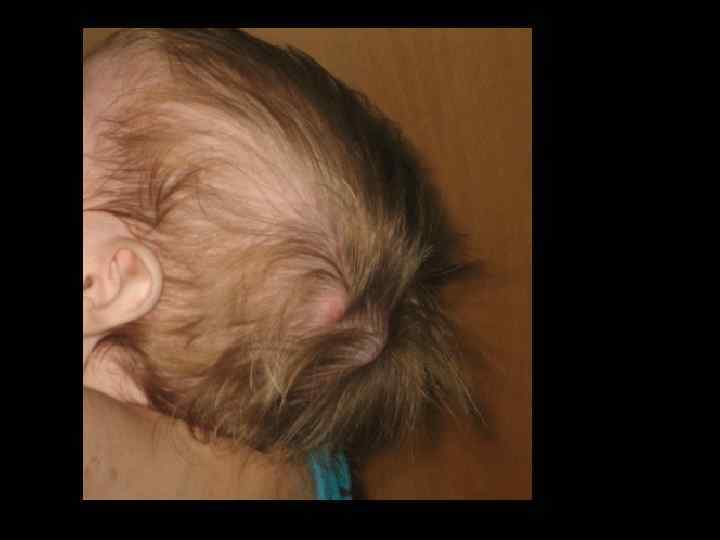
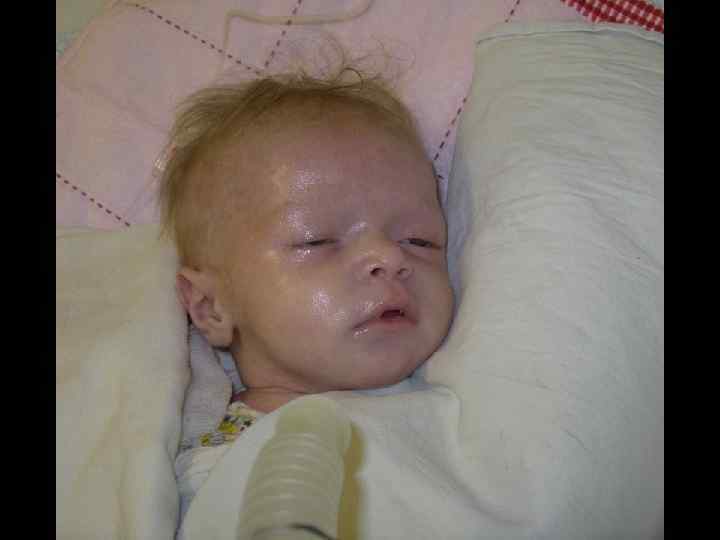
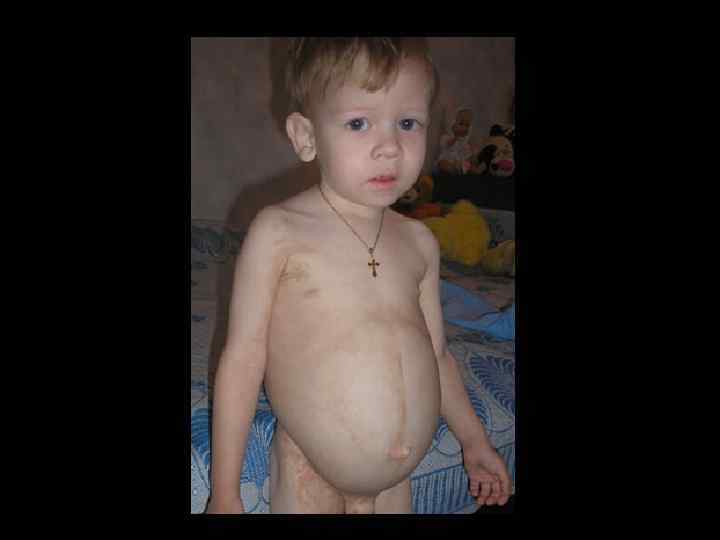

ЛЕЧЕНИЕ ТКИН • ТГСК экстренно! • Заместительная терапия ВВИГ – еженедельно • Профилактикатерапия ПЦП • Профилактикатерапия ЦМВ • Профилактическая противогрибковая терапия

Частые ПИДС

Синдром Ди. Джорджи • Самая частая хромосомная аберрация 1: 4000 • Характерная делеция 22 q 11. 2
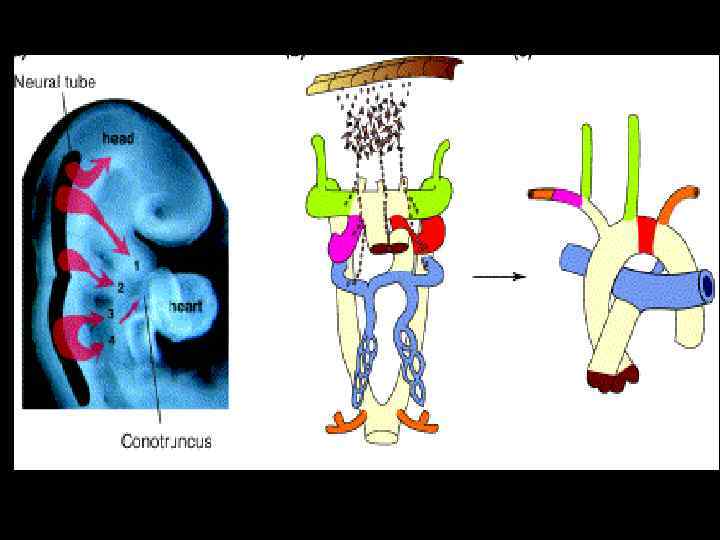

Синдром Ди. Джорджи • Выраженная гипоплазия тимуса Т-клеточный ИДС • Порок сердца • Патология паращит. желез Гипокальцемия • Патология лицевого скелета

Синдром Ди. Джорджи с делецией 22 q 11. 2 • Синуситы, отиты • Легочные инфекции • Аутоиммунные проявления (цитопении, ревматоидный артрит, тиреоидит) • Оухоли (лимфомы)

СИНДРОМ ДИДЖОРДЖИ

СИНДРОМ ДИДЖОРДЖИ
ПИДС диагностика и терапия.ppt