

СН и сосудистый тонус.pptx
- Количество слайдов: 91
 ПАТОФИЗИОЛОГИЯ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ
ПАТОФИЗИОЛОГИЯ СЕРДЕЧНО-СОСУДИСТОЙ СИСТЕМЫ
 Определение понятия СН. • В самом общем виде сердечная недостаточность – состояние, при котором сердце не обеспечивает адекватного кровоснабжения органов и тканей (адекватного метаболическим потребностям). Следствием неадекватного кровоснабжения органов и тканей является циркуляторная гипоксия.
Определение понятия СН. • В самом общем виде сердечная недостаточность – состояние, при котором сердце не обеспечивает адекватного кровоснабжения органов и тканей (адекватного метаболическим потребностям). Следствием неадекватного кровоснабжения органов и тканей является циркуляторная гипоксия.
 функции сердца. Производительная функция сердца") Этиология СН. В основе СН лежит снижение производительной (насосной) функции сердца. Производительная функция сердца обеспечивается 4 составляющими: 1. сократимость, 2. работа клапанного аппарата, 3. сердечный ритм, 4. наполнение полостей сердца.
Этиология СН. В основе СН лежит снижение производительной (насосной) функции сердца. Производительная функция сердца обеспечивается 4 составляющими: 1. сократимость, 2. работа клапанного аппарата, 3. сердечный ритм, 4. наполнение полостей сердца.
 4 группы причин СН • • 1 группа - непосредственное поражение миокарда, приводящее к снижению сократимости: некроз участка миокарда (инфаркт миокарда). постинфарктный кардиосклероз, атеросклеротический кардиосклероз, миокардит любой этиологии, миокардиодистрофия; дилатационная кардиомиопатия, • 2 группа – пороки сердца (врожденные и приобретенные), при которых возникает гемодинамическая перегрузка сердца при интактном миокарде; • 3 группа – гемодинамически значимые нарушения ритма (при интактном миокарде); • • • 4 группа – уменьшение наполнения желудочков: гипертрофическая кардиомиопатия, гипертоническая болезнь, перикардит, ТЭЛА, напряженный пневмоторакс.
4 группы причин СН • • 1 группа - непосредственное поражение миокарда, приводящее к снижению сократимости: некроз участка миокарда (инфаркт миокарда). постинфарктный кардиосклероз, атеросклеротический кардиосклероз, миокардит любой этиологии, миокардиодистрофия; дилатационная кардиомиопатия, • 2 группа – пороки сердца (врожденные и приобретенные), при которых возникает гемодинамическая перегрузка сердца при интактном миокарде; • 3 группа – гемодинамически значимые нарушения ритма (при интактном миокарде); • • • 4 группа – уменьшение наполнения желудочков: гипертрофическая кардиомиопатия, гипертоническая болезнь, перикардит, ТЭЛА, напряженный пневмоторакс.
; 2. хроническая") Классификации СН. По скорости развития и течению: 1. острая (мин, часы, дни); 2. хроническая (недели, месяцы, годы). По преимущественному поражению того или иного отдела сердца: 1. преимущественно правожелудочковая (недостаточность по б. к. к. ); 2. преимущественно левожелудочковая (недостаточность по м. к. к. ); 3. тотальная.
Классификации СН. По скорости развития и течению: 1. острая (мин, часы, дни); 2. хроническая (недели, месяцы, годы). По преимущественному поражению того или иного отдела сердца: 1. преимущественно правожелудочковая (недостаточность по б. к. к. ); 2. преимущественно левожелудочковая (недостаточность по м. к. к. ); 3. тотальная.
 миокардиальная (от повреждения). В основе – первичное повреждение миокарда:") Классификации СН По происхождению: а) миокардиальная (от повреждения). В основе – первичное повреждение миокарда: некроз, склероз, воспаление, кардиомиопатия; б) перегрузочная (от перегрузки). Миокард интактен. Однако сердце вынуждено работать в условиях повышенной нагрузки. Чрезмерная нагрузка может предъявляться сердцу избыточным объемом и избыточным сопротивлением (давлением). В соответствии с этим различают 2 варианта перегрузочной СН: • от перегрузки объемом (клапанные пороки по типу недостаточности, внутрисердечные шунты), • от перегрузки сопротивлением (клапанные пороки по типу стеноза, гипертензия большого или малого круга кровообращения); в) смешанная. Характеризуется сочетанием разных видов СН. Например, ревматическое поражение сердца (панкардит): миокардит + клапанный порок, миокардиальная + перегрузочная СН. Или ОИМ: некроз сердечной мышцы + относительная недостаточность митрального клапана из-за зоны акинезии, миокардиальная + перегрузочная СН.
Классификации СН По происхождению: а) миокардиальная (от повреждения). В основе – первичное повреждение миокарда: некроз, склероз, воспаление, кардиомиопатия; б) перегрузочная (от перегрузки). Миокард интактен. Однако сердце вынуждено работать в условиях повышенной нагрузки. Чрезмерная нагрузка может предъявляться сердцу избыточным объемом и избыточным сопротивлением (давлением). В соответствии с этим различают 2 варианта перегрузочной СН: • от перегрузки объемом (клапанные пороки по типу недостаточности, внутрисердечные шунты), • от перегрузки сопротивлением (клапанные пороки по типу стеноза, гипертензия большого или малого круга кровообращения); в) смешанная. Характеризуется сочетанием разных видов СН. Например, ревматическое поражение сердца (панкардит): миокардит + клапанный порок, миокардиальная + перегрузочная СН. Или ОИМ: некроз сердечной мышцы + относительная недостаточность митрального клапана из-за зоны акинезии, миокардиальная + перегрузочная СН.
 систолическая, • б) диастолическая,") Классификации СН В зависимости от фазы сердечного цикла: • а) систолическая, • б) диастолическая, • в) смешанная (например, при ГБ вначале развивается диастолическая СН, а затем к ней присоединяется систолическая). Систолическая СН. Страдает систола. Сердце не может обеспечить нормальный сердечный выброс. Причины – снижение сократимости миокарда (инфаркт миокарда, миокардит, дилатационная кардиомиопатия, некоторые пороки сердца), отражением чего является уменьшение фракции выброса ФВ = УО: КДО (в норме 55 -70%). Характеризуется клиническими признаками застоя на фоне сниженной сократительной функции (ФВ меньше нормы). Встречается в 70 -80% всех случаев СН.
Классификации СН В зависимости от фазы сердечного цикла: • а) систолическая, • б) диастолическая, • в) смешанная (например, при ГБ вначале развивается диастолическая СН, а затем к ней присоединяется систолическая). Систолическая СН. Страдает систола. Сердце не может обеспечить нормальный сердечный выброс. Причины – снижение сократимости миокарда (инфаркт миокарда, миокардит, дилатационная кардиомиопатия, некоторые пороки сердца), отражением чего является уменьшение фракции выброса ФВ = УО: КДО (в норме 55 -70%). Характеризуется клиническими признаками застоя на фоне сниженной сократительной функции (ФВ меньше нормы). Встречается в 70 -80% всех случаев СН.
 Диастолическая СН. Страдает диастола. Сердце не может полноценно расслабиться, а значит, нарушается наполнение желудочков. Это возникает из-за ухудшения их растяжимости или внешнего сдавления. Причины: 1. гипертрофия миокарда (артериальная гипертензия, гипертрофическая кардиомиопатия); 2. увеличение массы миокарда и утолщение стенки желудочков (амилоидоз, саркоидоз, гемохроматоз, эндомиокардиальный фиброз); 3. внешнее ограничение увеличения объема желудочков (выпот в перикард, констриктивный перикардит, опухоль перикарда) и др. Характеризуется наличием признаков застоя при нормальной сократительной функции (ФВ существенно не меняется). Встречается в 20 -30% всех случаев СН.
Диастолическая СН. Страдает диастола. Сердце не может полноценно расслабиться, а значит, нарушается наполнение желудочков. Это возникает из-за ухудшения их растяжимости или внешнего сдавления. Причины: 1. гипертрофия миокарда (артериальная гипертензия, гипертрофическая кардиомиопатия); 2. увеличение массы миокарда и утолщение стенки желудочков (амилоидоз, саркоидоз, гемохроматоз, эндомиокардиальный фиброз); 3. внешнее ограничение увеличения объема желудочков (выпот в перикард, констриктивный перикардит, опухоль перикарда) и др. Характеризуется наличием признаков застоя при нормальной сократительной функции (ФВ существенно не меняется). Встречается в 20 -30% всех случаев СН.
 с низким МОС (подавляющее число случаев СН),") Классификации СН По величине МОС: • а) с низким МОС (подавляющее число случаев СН), • б) с высоким МОС (как исключение в ситуациях, когда СН развивается на фоне тяжелого тиреотоксикоза, выраженной анемии, феохромоцитомы и др. ). По степени тяжести выделяют I, II (А и Б) и III стадии ХСН (см. пропедевтику). В зависимости от выраженности симптомов декомпенсации, их стойкости, связи с физической нагрузкой, а также функционального состояния органов и дистрофических изменений в них.
Классификации СН По величине МОС: • а) с низким МОС (подавляющее число случаев СН), • б) с высоким МОС (как исключение в ситуациях, когда СН развивается на фоне тяжелого тиреотоксикоза, выраженной анемии, феохромоцитомы и др. ). По степени тяжести выделяют I, II (А и Б) и III стадии ХСН (см. пропедевтику). В зависимости от выраженности симптомов декомпенсации, их стойкости, связи с физической нагрузкой, а также функционального состояния органов и дистрофических изменений в них.
 Патогенез ХСН В основе СН могут лежать различные этиологические факторы. Одни вызывают непосредственное повреждение миокарда, другие – увеличение нагрузки на сердце. В обоих случаях развивается дисфункция миокарда (снижение производительной, или насосной, функции сердца), вначале бессимптомная. Для предотвращения ее прогрессирования с развертыванием полной клинической картины СН запускаются различные компенсаторные механизмы. Однако со временем мощность их оказывается недостаточной, и, более того, при длительном течении процесса они начинают приобретать отрицательное значение, приводя к прогрессированию СН. Включение компенсаторных механизмов и постепенная трансформация их роли из положительной в отрицательную – и есть основа патогенеза ХСН.
Патогенез ХСН В основе СН могут лежать различные этиологические факторы. Одни вызывают непосредственное повреждение миокарда, другие – увеличение нагрузки на сердце. В обоих случаях развивается дисфункция миокарда (снижение производительной, или насосной, функции сердца), вначале бессимптомная. Для предотвращения ее прогрессирования с развертыванием полной клинической картины СН запускаются различные компенсаторные механизмы. Однако со временем мощность их оказывается недостаточной, и, более того, при длительном течении процесса они начинают приобретать отрицательное значение, приводя к прогрессированию СН. Включение компенсаторных механизмов и постепенная трансформация их роли из положительной в отрицательную – и есть основа патогенеза ХСН.
 Компенсаторные механизмы при ХСН являются по сути своей гемодинамическими, т. е. направлены на поддержание адекватной гемодинамики 1. Механизм Франка-Старлинга. Принцип работы «длина-сила» (чем больше мышечное волокно растягивается в диастолу, тем сильнее оно сокращается в систолу). Механизм включается при значительном наполнении полости ЛЖ в диастолу, т. е. при увеличении преднагрузки и встречается при перегрузке объемом. Дилатация соответствующей полости выражена. Само сокращение волокна характеризуется уменьшением его длины без изменения напряжения, что получило название изотонической гиперфункции. Сопровождается незначительным увеличением коронарного кровотока и потребления кислорода. Амплитуда сокращений миокарда значительно увеличена. Работа сердца возрастает за счет увеличения МОС без существенного увеличения систолического давления.
Компенсаторные механизмы при ХСН являются по сути своей гемодинамическими, т. е. направлены на поддержание адекватной гемодинамики 1. Механизм Франка-Старлинга. Принцип работы «длина-сила» (чем больше мышечное волокно растягивается в диастолу, тем сильнее оно сокращается в систолу). Механизм включается при значительном наполнении полости ЛЖ в диастолу, т. е. при увеличении преднагрузки и встречается при перегрузке объемом. Дилатация соответствующей полости выражена. Само сокращение волокна характеризуется уменьшением его длины без изменения напряжения, что получило название изотонической гиперфункции. Сопровождается незначительным увеличением коронарного кровотока и потребления кислорода. Амплитуда сокращений миокарда значительно увеличена. Работа сердца возрастает за счет увеличения МОС без существенного увеличения систолического давления.
 2. Механизм Анрепа. Суть: увеличение силы сокращения на фоне возрастания сопротивления этому сокращению. Включается при значительном возрастании ОПСС, т. е. при перегрузке сопротивлением, а значит - при увеличении постнагрузки. Выражена гипертрофия стенок соответствующей полости сердца. Само сокращение характеризуется увеличением напряжения без изменения длины мышечного волокна, что получило название изометрической гиперфункции. Сопровождается значительным усилением коронарного кровотока и потребления кислорода; амплитуда сердечных сокращений и МОС существенно не меняются. Работа сердца возрастает за счет увеличения систолического давления;
2. Механизм Анрепа. Суть: увеличение силы сокращения на фоне возрастания сопротивления этому сокращению. Включается при значительном возрастании ОПСС, т. е. при перегрузке сопротивлением, а значит - при увеличении постнагрузки. Выражена гипертрофия стенок соответствующей полости сердца. Само сокращение характеризуется увеличением напряжения без изменения длины мышечного волокна, что получило название изометрической гиперфункции. Сопровождается значительным усилением коронарного кровотока и потребления кислорода; амплитуда сердечных сокращений и МОС существенно не меняются. Работа сердца возрастает за счет увеличения систолического давления;
. Суть: увеличение силы сокращения на фоне возрастания ЧСС") 3. Феномен Боудича (феномен ритмо-инотропной зависимости). Суть: увеличение силы сокращения на фоне возрастания ЧСС вследствие энергозависимого процесса изменения концентрации Са 2+ в цитозоле. • Любая гиперфункция приводит к увеличению интенсивности функционирования структур (ИФС): увеличивается потребление кислорода на единицу массы миокарда, возрастает окислительное фосфорилирование. Больше ресинтезируется АТФ. Все это требует развития компенсаторной гипертрофии.
3. Феномен Боудича (феномен ритмо-инотропной зависимости). Суть: увеличение силы сокращения на фоне возрастания ЧСС вследствие энергозависимого процесса изменения концентрации Са 2+ в цитозоле. • Любая гиперфункция приводит к увеличению интенсивности функционирования структур (ИФС): увеличивается потребление кислорода на единицу массы миокарда, возрастает окислительное фосфорилирование. Больше ресинтезируется АТФ. Все это требует развития компенсаторной гипертрофии.
 • Гипертрофия миокарда – увеличение массы миокардиальных клеток без увеличения их числа. • Направлена на приведение структуры в соответствие возросшей функции. • Если между структурой и функцией имеется соответствие, ИФС=1. Компенсаторная гипертрофия миокарда протекает в 3 стадии: • 1) аварийная. Активация генетического аппарата КМЦ—усиление синтеза структурных и сократительных белков—увеличение массы миокарда. Но ИФС еще не достиг нормы (больше 1), так как массы недостаточно; работа/масса =1, 5/1 • 2) Стадия завершившейся гипертрофии и относительно устойчивой гиперфункции. Полное соответствие массы миокарда и возросшей функции. ИФС=1; работа/масса =1, 5/1, 5 • 3) стадия прогрессирующего кардиосклероза. Характеризуется замещением сократительных кардиомиоцитов соединительной тканью. ИФС<1 ; работа/масса =0, 5/1, 5 • Причина – нарушение энергетического обмена в КМЦ→активация гликолиза→накопление недоокисленных продуктов цикла Кребса→ацидоз→развитие соединительной ткани, которая не может выполнять сократительную функцию.
• Гипертрофия миокарда – увеличение массы миокардиальных клеток без увеличения их числа. • Направлена на приведение структуры в соответствие возросшей функции. • Если между структурой и функцией имеется соответствие, ИФС=1. Компенсаторная гипертрофия миокарда протекает в 3 стадии: • 1) аварийная. Активация генетического аппарата КМЦ—усиление синтеза структурных и сократительных белков—увеличение массы миокарда. Но ИФС еще не достиг нормы (больше 1), так как массы недостаточно; работа/масса =1, 5/1 • 2) Стадия завершившейся гипертрофии и относительно устойчивой гиперфункции. Полное соответствие массы миокарда и возросшей функции. ИФС=1; работа/масса =1, 5/1, 5 • 3) стадия прогрессирующего кардиосклероза. Характеризуется замещением сократительных кардиомиоцитов соединительной тканью. ИФС<1 ; работа/масса =0, 5/1, 5 • Причина – нарушение энергетического обмена в КМЦ→активация гликолиза→накопление недоокисленных продуктов цикла Кребса→ацидоз→развитие соединительной ткани, которая не может выполнять сократительную функцию.
 Нейрогуморальные механизмы: 1. Активация САС. Механизмы активации САС при СН до конца не ясны. Вероятно, связаны с гипоперфузией тканей и повышением венозного давления (патологические барорецепторные рефлексы). Активация САС приводит к реализации сосудосуживающих эффектов катехоламинов (норадреналина и адреналина). Цель – поддержание АД и перераспределение кровотока в пользу так называемых жизненно важных органов (сердце, мозг), то есть централизация кровообращения. Это возможно потому, что в названных органах не происходит сужения сосудов под действием КХА. Кроме того, под действием КХА происходит усиление и учащение сердечной деятельности, но эти компенсаторные эффекты относят к сердечным, и о них уже говорилось.
Нейрогуморальные механизмы: 1. Активация САС. Механизмы активации САС при СН до конца не ясны. Вероятно, связаны с гипоперфузией тканей и повышением венозного давления (патологические барорецепторные рефлексы). Активация САС приводит к реализации сосудосуживающих эффектов катехоламинов (норадреналина и адреналина). Цель – поддержание АД и перераспределение кровотока в пользу так называемых жизненно важных органов (сердце, мозг), то есть централизация кровообращения. Это возможно потому, что в названных органах не происходит сужения сосудов под действием КХА. Кроме того, под действием КХА происходит усиление и учащение сердечной деятельности, но эти компенсаторные эффекты относят к сердечным, и о них уже говорилось.
 2. Активация РААС. Осуществляется 2 путями. Во-первых, снижение АД, снижение МОС и рефлекторное сужение почечных артериол приводят к гипоперфузии почек → происходит возбуждение волюморецепторов клеток ЮГА и → усиление выработки ренина. Во-вторых, высокие концентрации КХА в крови вызывают возбуждение -адренорецепторов клеток ЮГА → усиление выработки ренина. Далее запускается вся РААС: ренин → ангиотензин 1 → ангиотензин 2 → альдостерон → усиление реабсорбции натрия → гипернатриемия → повышение Росм. крови → возбуждение осморецепторов гипоталамуса → усиление выработки и освобождения через гипофиз АДГ → усиление реабсорбции воды. Цель активации РААС: поддержание МОС за счет увеличения ОЦК и вазоконстрикторные эффекты ангиотензина 2.
2. Активация РААС. Осуществляется 2 путями. Во-первых, снижение АД, снижение МОС и рефлекторное сужение почечных артериол приводят к гипоперфузии почек → происходит возбуждение волюморецепторов клеток ЮГА и → усиление выработки ренина. Во-вторых, высокие концентрации КХА в крови вызывают возбуждение -адренорецепторов клеток ЮГА → усиление выработки ренина. Далее запускается вся РААС: ренин → ангиотензин 1 → ангиотензин 2 → альдостерон → усиление реабсорбции натрия → гипернатриемия → повышение Росм. крови → возбуждение осморецепторов гипоталамуса → усиление выработки и освобождения через гипофиз АДГ → усиление реабсорбции воды. Цель активации РААС: поддержание МОС за счет увеличения ОЦК и вазоконстрикторные эффекты ангиотензина 2.
. 4. Увеличение выработки предсердного и мозгового натрийуретического") 3. Увеличение продукции эндотелина (суживает периферические сосуды). 4. Увеличение выработки предсердного и мозгового натрийуретического пептида (НУП). Он является эндогенным диуретиком и позволяет удерживать адекватную преднагрузку. 5. Увеличение выработки оксида азота эндотелиальными клетками резистивных сосудов. Он эти сосуды расширяет, обеспечивая тем самым адекватную постнагрузку.
3. Увеличение продукции эндотелина (суживает периферические сосуды). 4. Увеличение выработки предсердного и мозгового натрийуретического пептида (НУП). Он является эндогенным диуретиком и позволяет удерживать адекватную преднагрузку. 5. Увеличение выработки оксида азота эндотелиальными клетками резистивных сосудов. Он эти сосуды расширяет, обеспечивая тем самым адекватную постнагрузку.
 Если степень снижения насосной функции сердца продолжает нарастать, развивается феномен нейрогуморальной гиперкомпенсации. В первую очередь, он касается РААС и САС. Суть феномена сводится к тому, что на 1 план начинают выступать неблагоприятные эффекты компенсаторных механизмов, т. е. они становятся неадекватными. Фактически это ведет к декомпенсации сердечной деятельности.
Если степень снижения насосной функции сердца продолжает нарастать, развивается феномен нейрогуморальной гиперкомпенсации. В первую очередь, он касается РААС и САС. Суть феномена сводится к тому, что на 1 план начинают выступать неблагоприятные эффекты компенсаторных механизмов, т. е. они становятся неадекватными. Фактически это ведет к декомпенсации сердечной деятельности.
 заключается в укорочении") Механизмы декомпенсации 1. Неадекватность тахикардии (которая исходно служит для увеличения МОС) заключается в укорочении диастолы и, как следствие, ухудшении коронарной перфузии и увеличению потребности миокарда в кислороде. 2. Неадекватность задержки натрия и жидкости (исходно направленной на поддержание МОС за счет ОЦК) приводит к развитию отеков и увеличению нагрузки на сердце (ОЦК). 3. Неадекватность вазоконстрикции (исходно направленной на поддержание АД и сохранение перфузии жизненно важных органов) приводит к увеличению постнагрузки на сердце (ОПСС) и ухудшению кровоснабжения органов и тканей, в частности почек.
Механизмы декомпенсации 1. Неадекватность тахикардии (которая исходно служит для увеличения МОС) заключается в укорочении диастолы и, как следствие, ухудшении коронарной перфузии и увеличению потребности миокарда в кислороде. 2. Неадекватность задержки натрия и жидкости (исходно направленной на поддержание МОС за счет ОЦК) приводит к развитию отеков и увеличению нагрузки на сердце (ОЦК). 3. Неадекватность вазоконстрикции (исходно направленной на поддержание АД и сохранение перфузии жизненно важных органов) приводит к увеличению постнагрузки на сердце (ОПСС) и ухудшению кровоснабжения органов и тканей, в частности почек.
, как ни парадоксально,") 4. Неадекватность гипертрофии миокарда (изначально направленной на материальное обеспечение возросшей функции), как ни парадоксально, неизбежно ведет к декомпенсации сердечной деятельности. Причина в том, что гипертрофия миокарда при ХСН с самого начала ведет себя как несбалансированная форма роста. Несбалансированность прослеживается на всех уровнях организации (органном, тканевом, клеточном, субклеточном, молекулярном). • • • На уровне органа. Масса сердца увеличивается быстрее, чем образовываются новые нервные окончания. В результате снижается плотность симпатической иннервации. На уровне ткани. Происходит отставание роста массы капилляров от массы кардиомиоцитов. В результате страдает питание сердечной мышцы. На уровне клетки. Площадь сарколеммы увеличивается пропорционально квадрату, а объем клетки – пропорционально кубу, то есть площадь мембраны относительно уменьшается. В результате снижается мощность мембранных ионообменных насосов, мембранных ферментных систем, страдают другие функции мембран. На уровне внутриклеточных органелл. Темпы увеличения массы митохондрий отстают от темпов увеличения массы миофибрилл, что закономерно ухудшает энергообеспечение кардиомиоцитов. На уровне молекулярных структур. Снижается способность миозиновых волокон использовать энергию АТФ, так как нарушается структура миозина.
4. Неадекватность гипертрофии миокарда (изначально направленной на материальное обеспечение возросшей функции), как ни парадоксально, неизбежно ведет к декомпенсации сердечной деятельности. Причина в том, что гипертрофия миокарда при ХСН с самого начала ведет себя как несбалансированная форма роста. Несбалансированность прослеживается на всех уровнях организации (органном, тканевом, клеточном, субклеточном, молекулярном). • • • На уровне органа. Масса сердца увеличивается быстрее, чем образовываются новые нервные окончания. В результате снижается плотность симпатической иннервации. На уровне ткани. Происходит отставание роста массы капилляров от массы кардиомиоцитов. В результате страдает питание сердечной мышцы. На уровне клетки. Площадь сарколеммы увеличивается пропорционально квадрату, а объем клетки – пропорционально кубу, то есть площадь мембраны относительно уменьшается. В результате снижается мощность мембранных ионообменных насосов, мембранных ферментных систем, страдают другие функции мембран. На уровне внутриклеточных органелл. Темпы увеличения массы митохондрий отстают от темпов увеличения массы миофибрилл, что закономерно ухудшает энергообеспечение кардиомиоцитов. На уровне молекулярных структур. Снижается способность миозиновых волокон использовать энергию АТФ, так как нарушается структура миозина.
 5. Неадекватная нейрогормональная регуляция (первоначально направлена на увеличение силы и частоты сердечных сокращений, а значит МОС, и поддержание АД). Однако со временем она приводит к следующим неблагоприятным последствиям: • а) увеличение потребления кислорода сердцем из-за тахикардии ( «виноваты» катехоламины); • б) увеличение нагрузки на сердце из-за вазоконстрикции и задержки жидкости ( «виноваты» катехоламины, ангиотензин 2, альдостерон, вазопрессин); • в) развитие отеков из-за активации РААС и задержки воды; • г) непосредственное неблагоприятное воздействие нейрогормонов на миокард: риск развития аритмий, некрозов миокарда, фиброзирование, стимуляция ремоделирования сердца.
5. Неадекватная нейрогормональная регуляция (первоначально направлена на увеличение силы и частоты сердечных сокращений, а значит МОС, и поддержание АД). Однако со временем она приводит к следующим неблагоприятным последствиям: • а) увеличение потребления кислорода сердцем из-за тахикардии ( «виноваты» катехоламины); • б) увеличение нагрузки на сердце из-за вазоконстрикции и задержки жидкости ( «виноваты» катехоламины, ангиотензин 2, альдостерон, вазопрессин); • в) развитие отеков из-за активации РААС и задержки воды; • г) непосредственное неблагоприятное воздействие нейрогормонов на миокард: риск развития аритмий, некрозов миокарда, фиброзирование, стимуляция ремоделирования сердца.
 • Ремоделирование – неотъемлемая часть патогенеза ХСН. Под ремоделированием понимают процесс комплексного нарушения структуры и функции сердца в ответ на перегрузку или утрату части жизнеспособного миокарда. • Процесс ремоделирования при ХСН включает в себя прогрессирующее увеличение массы миокарда, дилатацию полостей и изменение геометрических характеристик желудочков.
• Ремоделирование – неотъемлемая часть патогенеза ХСН. Под ремоделированием понимают процесс комплексного нарушения структуры и функции сердца в ответ на перегрузку или утрату части жизнеспособного миокарда. • Процесс ремоделирования при ХСН включает в себя прогрессирующее увеличение массы миокарда, дилатацию полостей и изменение геометрических характеристик желудочков.
 В норме в систолу сердце принимает более элипсоидную форму, а в диастолу – более сферическую. Подобное изменение геометрии во время сердечного цикла обеспечивает нормальную систолическую и диастолическую функции. Нарушение этих геометрических характеристик при ремоделировании приводит к снижению ФВ, нарушениям системной гемодинамики и появлению клинических проявлений СН (предшествует им и сопровождает их). • Вначале ремоделирование при СН носит компенсаторный характер, однако ведет к ряду негативных последствий и, в конце концов, к срыву компенсации. Сердце вступает в фазу «прогрессирующего кардиосклероза и изнашивания структур» (Ф. З. Меерсон), которая характеризуется: 1. гибелью клеток, 2. нарушением обновления структур, 3. развитием склероза органа, 4. увеличением риска развития аритмий, 5. прогрессирующим нарушением биоэнергетики, 6. систолической и диастолической дисфункцией
В норме в систолу сердце принимает более элипсоидную форму, а в диастолу – более сферическую. Подобное изменение геометрии во время сердечного цикла обеспечивает нормальную систолическую и диастолическую функции. Нарушение этих геометрических характеристик при ремоделировании приводит к снижению ФВ, нарушениям системной гемодинамики и появлению клинических проявлений СН (предшествует им и сопровождает их). • Вначале ремоделирование при СН носит компенсаторный характер, однако ведет к ряду негативных последствий и, в конце концов, к срыву компенсации. Сердце вступает в фазу «прогрессирующего кардиосклероза и изнашивания структур» (Ф. З. Меерсон), которая характеризуется: 1. гибелью клеток, 2. нарушением обновления структур, 3. развитием склероза органа, 4. увеличением риска развития аритмий, 5. прогрессирующим нарушением биоэнергетики, 6. систолической и диастолической дисфункцией
") Нарушения энергетического обмена при ХСН • Энергетический обмен принципиально складывается из процессов синтеза (ресинтеза) АТФ, транспорта его энергии к эффекторным структурам кардиомиоцитов и утилизации ими энергии макроэргических фосфатов. • Нарушение биоэнергетики может происходить на любом из этапов.
Нарушения энергетического обмена при ХСН • Энергетический обмен принципиально складывается из процессов синтеза (ресинтеза) АТФ, транспорта его энергии к эффекторным структурам кардиомиоцитов и утилизации ими энергии макроэргических фосфатов. • Нарушение биоэнергетики может происходить на любом из этапов.
 • I этап – синтез Е и кумуляция ее в фосфатных связях. В норме образование химической энергии (окисление) происходит на 95% за счет аэробного окисления (обеспечивает сократимость) и на 5% за счет гликолиза (обеспечивает пластические процессы). Субстратами окисления на 80% являются жирные кислоты, на 20% - глюкоза. Необходим О 2 и нормальная структура и функция митохондрий. Возможные нарушения энергетического обмена на I этапе: 1. недостаток О 2 или субстратов, повреждение митохондрий ведет к снижению интенсивности аэробного окисления и усилению гликолиза, который, как известно, в 18 раз менее эффективен, чем митохондриальное окисление, нарушение доставки О 2, 2. процесс кумуляции (фосфорилирования) может страдать из-за разобщения ОФ. Разобщители: избыток ионов Са 2+ , Н+, НЭЖК, производные фенола, некоторые лекарственные препараты (грамицидин, антикоагулянты), микробные токсины, гормоны щитовидной железы, алкоголь.
• I этап – синтез Е и кумуляция ее в фосфатных связях. В норме образование химической энергии (окисление) происходит на 95% за счет аэробного окисления (обеспечивает сократимость) и на 5% за счет гликолиза (обеспечивает пластические процессы). Субстратами окисления на 80% являются жирные кислоты, на 20% - глюкоза. Необходим О 2 и нормальная структура и функция митохондрий. Возможные нарушения энергетического обмена на I этапе: 1. недостаток О 2 или субстратов, повреждение митохондрий ведет к снижению интенсивности аэробного окисления и усилению гликолиза, который, как известно, в 18 раз менее эффективен, чем митохондриальное окисление, нарушение доставки О 2, 2. процесс кумуляции (фосфорилирования) может страдать из-за разобщения ОФ. Разобщители: избыток ионов Са 2+ , Н+, НЭЖК, производные фенола, некоторые лекарственные препараты (грамицидин, антикоагулянты), микробные токсины, гормоны щитовидной железы, алкоголь.
 к местам") • II этап - транспорт Е АТФ (Е концевых фосфатных связей) к местам утилизации осуществляется благодаря работе так называемого «челночного» механизма (Розенштраух, Сакс, Чазов; 1977).
• II этап - транспорт Е АТФ (Е концевых фосфатных связей) к местам утилизации осуществляется благодаря работе так называемого «челночного» механизма (Розенштраух, Сакс, Чазов; 1977).
 механизм транспорта энергии Участники «челночного» механизма: • а) АДФ/АТФ-транслоказа. Локализация") • «Челночный» (креатинфосфокиназный) механизм транспорта энергии Участники «челночного» механизма: • а) АДФ/АТФ-транслоказа. Локализация – подмембранное пространство митохондрий. Функция – перенос макроэргов из внутреннего пространства в межмембранное и обратно; • б) Креатинфосфокиназа (КФК, митохондриальная фракция). Локализация – наружная поверхность внутренней мембраны митохондрий. Контролирует реакцию: АТФ + К КФ + АДФ, после чего АДФ отправляется обратно в митохондрии (с помощью фермента АДФ/АТФ-транслоказы) на реутилизацию (рефосфорилирование), а КФ (макроэрг, более мобильный, чем АТФ) – через внешнюю мембрану к местам утилизации энергии, например, миофибриллам. • В миофибриллах есть своя фракция КФК (миофибриллярная), которая контролирует обратную реакцию: АДФ + КФ АТФ + К, то есть КФ отдает энергию концевой фосфатной связи на рефосфорилирование АДФ (от прежнего сокращения), после чего креатин отправляется в митохондрии за новой порцией энергии (отсюда «челнок» ). Возможные нарушения в работе «челночного» механизма: 1. количественные или качественные нарушения ферментов (транслоказы, КФК); 2. дефицит креатина.
• «Челночный» (креатинфосфокиназный) механизм транспорта энергии Участники «челночного» механизма: • а) АДФ/АТФ-транслоказа. Локализация – подмембранное пространство митохондрий. Функция – перенос макроэргов из внутреннего пространства в межмембранное и обратно; • б) Креатинфосфокиназа (КФК, митохондриальная фракция). Локализация – наружная поверхность внутренней мембраны митохондрий. Контролирует реакцию: АТФ + К КФ + АДФ, после чего АДФ отправляется обратно в митохондрии (с помощью фермента АДФ/АТФ-транслоказы) на реутилизацию (рефосфорилирование), а КФ (макроэрг, более мобильный, чем АТФ) – через внешнюю мембрану к местам утилизации энергии, например, миофибриллам. • В миофибриллах есть своя фракция КФК (миофибриллярная), которая контролирует обратную реакцию: АДФ + КФ АТФ + К, то есть КФ отдает энергию концевой фосфатной связи на рефосфорилирование АДФ (от прежнего сокращения), после чего креатин отправляется в митохондрии за новой порцией энергии (отсюда «челнок» ). Возможные нарушения в работе «челночного» механизма: 1. количественные или качественные нарушения ферментов (транслоказы, КФК); 2. дефицит креатина.
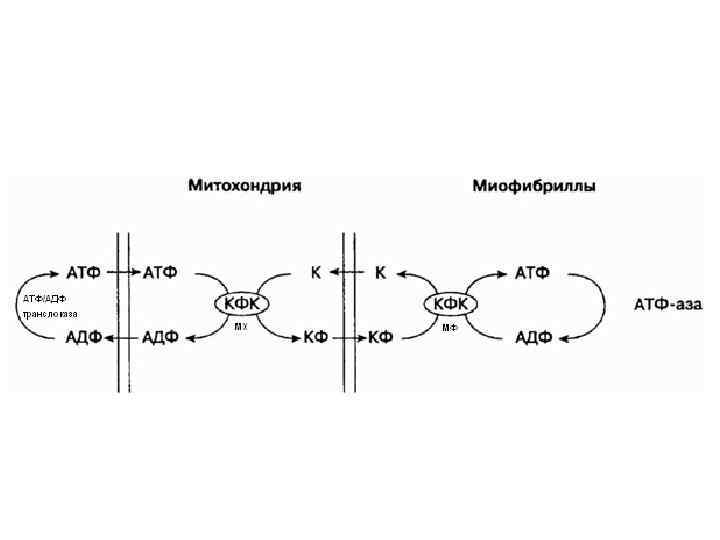
 III этап – этап утилизации Е. Энергия идет на сокращение за счет образования актин/миозиновых мостиков. Возможные нарушения энергетического обмена на III этапе: 1. недостаток сократительных белков (актин, миозин); 2. снижение АТФазной активности миозина.
III этап – этап утилизации Е. Энергия идет на сокращение за счет образования актин/миозиновых мостиков. Возможные нарушения энергетического обмена на III этапе: 1. недостаток сократительных белков (актин, миозин); 2. снижение АТФазной активности миозина.
 Нарушения гемодинамики и микроциркуляции при ХСН Снижение насосной функции сердца при ХСН закономерно ведет к определенным нарушениям кровообращения (гемодинамическим сдвигам). 1. во-первых, снижается УО, а значит и МОС, и АД (несмотря на рефлекторное увеличение ЧСС; 2. вследствие рефлекторного сужения периферических артериол возрастает ОПСС; 3. несостоятельность сердца как насоса ведет к скоплению избыточных объемов крови в венозном отделе сосудистого русла, то есть к формированию венозного застоя (вот почему ХСН часто называют ХЗСН). Венозное давление повышается (более 160 мм вод. ст. ). 4. Застойные явления в м. к. к. приводят к развитию одышки, отека легких, цианоза. Застойные явления в б. к. к. проявляются тканевыми и полостными отеками, набуханием шейных вен, увеличением печени, цианозом, нарушением функций почек, ж. к. т. и др. 5. нарушения гемодинамики (снижение МОС, АД, сужение периферических артериол, венозный застой) неизбежно ведут к снижению перфузии органов и тканей, нарушению микроциркуляции и развитию гипоксии.
Нарушения гемодинамики и микроциркуляции при ХСН Снижение насосной функции сердца при ХСН закономерно ведет к определенным нарушениям кровообращения (гемодинамическим сдвигам). 1. во-первых, снижается УО, а значит и МОС, и АД (несмотря на рефлекторное увеличение ЧСС; 2. вследствие рефлекторного сужения периферических артериол возрастает ОПСС; 3. несостоятельность сердца как насоса ведет к скоплению избыточных объемов крови в венозном отделе сосудистого русла, то есть к формированию венозного застоя (вот почему ХСН часто называют ХЗСН). Венозное давление повышается (более 160 мм вод. ст. ). 4. Застойные явления в м. к. к. приводят к развитию одышки, отека легких, цианоза. Застойные явления в б. к. к. проявляются тканевыми и полостными отеками, набуханием шейных вен, увеличением печени, цианозом, нарушением функций почек, ж. к. т. и др. 5. нарушения гемодинамики (снижение МОС, АД, сужение периферических артериол, венозный застой) неизбежно ведут к снижению перфузии органов и тканей, нарушению микроциркуляции и развитию гипоксии.
 Патогенез основных клинических проявлений СН • Одышка – наиболее частое и раннее проявление СН, в 1 -ую очередь, левожелудочковой. Это компенсаторный механизм, направленный на увеличение оксигенации крови в легких за счет увеличения объема вентиляции и, следовательно, на борьбу с гипоксией. • Механизм: застой крови в сосудах м. к. к. нарушает функцию внешнего дыхания → снижается РО 2, увеличивается РСО 2, накапливается молочная кислота. Гипоксемия, гиперкапния и ацидоз → возбуждаются хеморецепторы → рефлекторное возбуждение ДЦ → увеличение частоты и глубины дыхании, т. е. одышка.
Патогенез основных клинических проявлений СН • Одышка – наиболее частое и раннее проявление СН, в 1 -ую очередь, левожелудочковой. Это компенсаторный механизм, направленный на увеличение оксигенации крови в легких за счет увеличения объема вентиляции и, следовательно, на борьбу с гипоксией. • Механизм: застой крови в сосудах м. к. к. нарушает функцию внешнего дыхания → снижается РО 2, увеличивается РСО 2, накапливается молочная кислота. Гипоксемия, гиперкапния и ацидоз → возбуждаются хеморецепторы → рефлекторное возбуждение ДЦ → увеличение частоты и глубины дыхании, т. е. одышка.
. Механизмы: •") • Тахикардия (вначале только при физической нагрузке, затем и в покое). Механизмы: • а) рефлекс Бейнбриджа с растягивающихся вследствие венозного застоя устьев полых вен; • б) активация САС, (+) хронотропное действие КХА. Цель тахикардии – поддержание МОС за счет ЧСС.
• Тахикардия (вначале только при физической нагрузке, затем и в покое). Механизмы: • а) рефлекс Бейнбриджа с растягивающихся вследствие венозного застоя устьев полых вен; • б) активация САС, (+) хронотропное действие КХА. Цель тахикардии – поддержание МОС за счет ЧСС.
. Отеки могут быть тканевые (стопы,") Отеки (вначале «скрытые» – до 5 литров, потом явные). Отеки могут быть тканевые (стопы, голени, половые органы, передняя брюшная стенка, поясница) и полостные (асцит, гидроперикард, плевральный выпот) в зависимости от причины и тяжести ХСН. Механизмы: • 1) гидростатический: снижение насосной функции сердца → венозный застой → увеличение гидростатического давления в капиллярах (более 20 -25 мм); • 2) онкотический: снижение онкотического давления плазмы из-за алиментарного дефицита белка, снижения белоксинтезирующей функции печени, альбуминурии; • 3) мембраногенный: увеличение проницаемости сосудистой стенки из-за гипоксемии, ацидоза, снижения скорости кровотока; • 4) осмотический: активная задержка натрия и воды вследствие альдостеронизма. Повышение уровня альдостерона происходит 2 путями: 1) активация РААС из-за ухудшения центральной, а следовательно, внутрипочечной гемодинамики; 2) нарушение инактивации альдостерона в печени из-за застоя. • 5) лимфатический (абсолютная и относительная лимфатическая недостаточность). Абсолютная лимфатическая недостаточность из-за рефлекторного спазма грудного лимфатического протока вследствие повышения давления в полых венах и сдавления мелких лимфатических сосудов отечным интерстицием. Относительная лимфатическая недостаточность развивается при быстром накоплении значительных объемов воды в тканях.
Отеки (вначале «скрытые» – до 5 литров, потом явные). Отеки могут быть тканевые (стопы, голени, половые органы, передняя брюшная стенка, поясница) и полостные (асцит, гидроперикард, плевральный выпот) в зависимости от причины и тяжести ХСН. Механизмы: • 1) гидростатический: снижение насосной функции сердца → венозный застой → увеличение гидростатического давления в капиллярах (более 20 -25 мм); • 2) онкотический: снижение онкотического давления плазмы из-за алиментарного дефицита белка, снижения белоксинтезирующей функции печени, альбуминурии; • 3) мембраногенный: увеличение проницаемости сосудистой стенки из-за гипоксемии, ацидоза, снижения скорости кровотока; • 4) осмотический: активная задержка натрия и воды вследствие альдостеронизма. Повышение уровня альдостерона происходит 2 путями: 1) активация РААС из-за ухудшения центральной, а следовательно, внутрипочечной гемодинамики; 2) нарушение инактивации альдостерона в печени из-за застоя. • 5) лимфатический (абсолютная и относительная лимфатическая недостаточность). Абсолютная лимфатическая недостаточность из-за рефлекторного спазма грудного лимфатического протока вследствие повышения давления в полых венах и сдавления мелких лимфатических сосудов отечным интерстицием. Относительная лимфатическая недостаточность развивается при быстром накоплении значительных объемов воды в тканях.
, затем диффузный.") Цианоз кожи и слизистых. Холодный. Вначале акроцианоз (руки, ноги, мочки ушей, губы), затем диффузный. Это виды цианоза. У каждого вида свой механизм. Механизмы: • а) периферический: замедление скорости кровотока в расширенных капиллярах и венулах (застой) → усиление утилизации О 2 и повышение содержания в крови восстановленного Hb. Вид цианоза – акроцианоз. • б) центральный: нарушение оксигенации крови в легких из-за застоя. Вид цианоза – диффузный.
Цианоз кожи и слизистых. Холодный. Вначале акроцианоз (руки, ноги, мочки ушей, губы), затем диффузный. Это виды цианоза. У каждого вида свой механизм. Механизмы: • а) периферический: замедление скорости кровотока в расширенных капиллярах и венулах (застой) → усиление утилизации О 2 и повышение содержания в крови восстановленного Hb. Вид цианоза – акроцианоз. • б) центральный: нарушение оксигенации крови в легких из-за застоя. Вид цианоза – диффузный.
. Возникает из-за переполнения кровью печеночных вен и капилляров.") • Увеличение печени (при ПЖСН). Возникает из-за переполнения кровью печеночных вен и капилляров. При длительном застое нарушается функция печени, затем развиваются морфологические изменения – цирроз печени и, как следствие, синдром портальной гипертензии (гепатоспленомегалия, асцит, симптом “головы медузы”). • Набухание шейных вен (в обе фазы дыхания) из-за венозного застоя. • Нарушение функции ж. к. т. также связано с застойными явлениями и, как следствие, нарушением трофики органов пищеварения. Выражается снижением тонуса и перистальтики желудка и кишечника, атрофией пищеварительных желез, нарушением всасывания. Клинически проявляется диспепсией и ведет к развитию сердечной кахексии.
• Увеличение печени (при ПЖСН). Возникает из-за переполнения кровью печеночных вен и капилляров. При длительном застое нарушается функция печени, затем развиваются морфологические изменения – цирроз печени и, как следствие, синдром портальной гипертензии (гепатоспленомегалия, асцит, симптом “головы медузы”). • Набухание шейных вен (в обе фазы дыхания) из-за венозного застоя. • Нарушение функции ж. к. т. также связано с застойными явлениями и, как следствие, нарушением трофики органов пищеварения. Выражается снижением тонуса и перистальтики желудка и кишечника, атрофией пищеварительных желез, нарушением всасывания. Клинически проявляется диспепсией и ведет к развитию сердечной кахексии.
 • Нарушение функции почек. Гемодинамические сдвиги при ХСН, а также увеличение секреции альдостерона и АДГ ведет к снижению фильтрации и усилению реабсорбции, то есть к развитию почечной недостаточности. • Хрипы в легких – следствие и признак застоя в м. к. к. (при ЛЖСН). • Снижение толерантности к физическим нагрузкам. Происходит из-за а) развития дистрофических процессов в скелетных мышцах; б) изменения соотношения разных типов мышечных волокон. У больных ХСН преобладают «быстрые» (анаэробные, гликолитические) быстро утомляемые МВ над «медленными» (аэробные, окислительные) устойчивыми к утомлению.
• Нарушение функции почек. Гемодинамические сдвиги при ХСН, а также увеличение секреции альдостерона и АДГ ведет к снижению фильтрации и усилению реабсорбции, то есть к развитию почечной недостаточности. • Хрипы в легких – следствие и признак застоя в м. к. к. (при ЛЖСН). • Снижение толерантности к физическим нагрузкам. Происходит из-за а) развития дистрофических процессов в скелетных мышцах; б) изменения соотношения разных типов мышечных волокон. У больных ХСН преобладают «быстрые» (анаэробные, гликолитические) быстро утомляемые МВ над «медленными» (аэробные, окислительные) устойчивыми к утомлению.
 –") ОСТРАЯ СЕРДЕЧНАЯ НЕДОСТАТОЧНОСТЬ • В зависимости от причины может быть: 1. левожелудочковая (ОЛЖН) – встречается значительно чаще ОПЖН; 2. правожелудочковая (ОПЖН); 3. тотальная (неспецифический миокардит=Абрамова-Фидлера).
ОСТРАЯ СЕРДЕЧНАЯ НЕДОСТАТОЧНОСТЬ • В зависимости от причины может быть: 1. левожелудочковая (ОЛЖН) – встречается значительно чаще ОПЖН; 2. правожелудочковая (ОПЖН); 3. тотальная (неспецифический миокардит=Абрамова-Фидлера).
 • Причины ОПЖН: 1. ТЭЛА, 2. астматический статус, 3. пневмоторакс, 4. тяжелая пневмония, 5. инфаркт миокарда правого желудочка (редко) и др. ситуации, при которых остро развивается гипертензия малого круга кровообращения. • Патогенез ОПЖН: непосредственное поражение миокарда правого желудочка (ИМ) или значительная перегрузка ПЖ сопротивлением (в случае повышения давления в м. к. к. ) ведут к несостоятельности ПЖ как насоса. Вследствие этого остро развиваются застойные явления в большом круге кровообращения.
• Причины ОПЖН: 1. ТЭЛА, 2. астматический статус, 3. пневмоторакс, 4. тяжелая пневмония, 5. инфаркт миокарда правого желудочка (редко) и др. ситуации, при которых остро развивается гипертензия малого круга кровообращения. • Патогенез ОПЖН: непосредственное поражение миокарда правого желудочка (ИМ) или значительная перегрузка ПЖ сопротивлением (в случае повышения давления в м. к. к. ) ведут к несостоятельности ПЖ как насоса. Вследствие этого остро развиваются застойные явления в большом круге кровообращения.
 • Клинические проявления ОПЖН – и есть признаки застоя в б. к. к. : • тахикардия, • одышка, • отеки, • увеличение печени, • набухание шейных вен, • эпигастральная пульсация, • цианоз, • нарушение функции почек, ц. н. с. и других систем органов
• Клинические проявления ОПЖН – и есть признаки застоя в б. к. к. : • тахикардия, • одышка, • отеки, • увеличение печени, • набухание шейных вен, • эпигастральная пульсация, • цианоз, • нарушение функции почек, ц. н. с. и других систем органов
 • Причины ОЛЖН: 1. инфаркт миокарда ЛЖ, 2. обратимая ишемия миокарда ЛЖ, 3. гипертонический криз, 4. остро развившиеся гемодинамически значимые нарушения ритма, 5. остро развившаяся декомпенсация митрального стеноза, 6. тяжелый миокардит. • Патогенез ОЛЖН в самом общем виде связан с резким снижением насосной функции ЛЖ вследствие либо непосредственного поражения миокарда (ИМ), либо вследствие его гемодинамической перегрузки (аритмии, гипертонический криз, декомпенсация порока). • Клиника ОЛЖН варьирует, в связи с чем выделяют клинические варианты ОЛЖН: 1. сердечная астма и отек легких; 2. кардиогенный шок.
• Причины ОЛЖН: 1. инфаркт миокарда ЛЖ, 2. обратимая ишемия миокарда ЛЖ, 3. гипертонический криз, 4. остро развившиеся гемодинамически значимые нарушения ритма, 5. остро развившаяся декомпенсация митрального стеноза, 6. тяжелый миокардит. • Патогенез ОЛЖН в самом общем виде связан с резким снижением насосной функции ЛЖ вследствие либо непосредственного поражения миокарда (ИМ), либо вследствие его гемодинамической перегрузки (аритмии, гипертонический криз, декомпенсация порока). • Клиника ОЛЖН варьирует, в связи с чем выделяют клинические варианты ОЛЖН: 1. сердечная астма и отек легких; 2. кардиогенный шок.
 • Сердечная астма и отек легких. • Непосредственное повреждение миокарда или остро возникшая гемодинамическая перегрузка приводят к несостоятельности ЛЖ как насоса растет КДО и КДД в ЛЖ, а потом и в ЛП. Это, в свою очередь, приводит к увеличению Р гидр. в легочных капиллярах. Увеличение Р гидр. в легочных капиллярах – главный патогенетический фактор в развитии этого клинического варианта ОЛЖН (сердечной астмы и отека легких). Он обусловливает выход жидкой части крови из сосудов легких в интерстиций легких и альвеолы. Дополнительное значение имеет увеличение проницаемости стенок легочных капилляров из-за снижения скорости кровотока, гипоксемии и ацидоза.
• Сердечная астма и отек легких. • Непосредственное повреждение миокарда или остро возникшая гемодинамическая перегрузка приводят к несостоятельности ЛЖ как насоса растет КДО и КДД в ЛЖ, а потом и в ЛП. Это, в свою очередь, приводит к увеличению Р гидр. в легочных капиллярах. Увеличение Р гидр. в легочных капиллярах – главный патогенетический фактор в развитии этого клинического варианта ОЛЖН (сердечной астмы и отека легких). Он обусловливает выход жидкой части крови из сосудов легких в интерстиций легких и альвеолы. Дополнительное значение имеет увеличение проницаемости стенок легочных капилляров из-за снижения скорости кровотока, гипоксемии и ацидоза.
 • Жидкая часть крови вначале наводняет легочный интерстиций формируется интерстициальный отек легких (клинически – сердечная астма). По мере прогрессирования патологического процесса отечная жидкость (транссудат) начинает «затоплять» альвеолы. Формируется альвеолярный отек легких. Его еще называют истинным. Важно понимать, что такое разделение весьма условно. • Сердечная астма и отек легких – стадии одного процесса, имеют общий патогенез.
• Жидкая часть крови вначале наводняет легочный интерстиций формируется интерстициальный отек легких (клинически – сердечная астма). По мере прогрессирования патологического процесса отечная жидкость (транссудат) начинает «затоплять» альвеолы. Формируется альвеолярный отек легких. Его еще называют истинным. Важно понимать, что такое разделение весьма условно. • Сердечная астма и отек легких – стадии одного процесса, имеют общий патогенез.
 • Клиника сердечной астмы и отека легких: одышка до степени удушья, вынужденное положение сидя с опущенными ногами, беспокойство, страх, кашель, вначале сухой, затем с мокротой, которая на этапе альвеолярного отека становится жидкой, пенистой, может быть окрашена в розовый цвет из-за примеси эритроцитов. При нарастании отека – клокочущее дыхание.
• Клиника сердечной астмы и отека легких: одышка до степени удушья, вынужденное положение сидя с опущенными ногами, беспокойство, страх, кашель, вначале сухой, затем с мокротой, которая на этапе альвеолярного отека становится жидкой, пенистой, может быть окрашена в розовый цвет из-за примеси эритроцитов. При нарастании отека – клокочущее дыхание.
 • Принципы патогенетической терапии сердечной астмы и отека легких: 1. Разгрузка малого круга кровообращения (жгуты на конечности, диуретики, периферические вазодилататоры, наркотические анальгетики); 2. Пеногашение (спирт, антифомсилан); 3. Оксигенотерапия.
• Принципы патогенетической терапии сердечной астмы и отека легких: 1. Разгрузка малого круга кровообращения (жгуты на конечности, диуретики, периферические вазодилататоры, наркотические анальгетики); 2. Пеногашение (спирт, антифомсилан); 3. Оксигенотерапия.
 • Кардиогенный шок – другой клинический вариант ОЛЖН. Наиболее частой причиной его развития является ИМ, реже – разрыв МЖП, тампонада сердца, гемодинамически значимые нарушения ритма, острая митральная регургитация. • Некроз значительного по площади участка миокарда ЛЖ (как правило, более 40%) ведет к формированию зоны акинезии или гипокинезии, то есть часть миокарда не участвует в сокращении. • Вследствие этого сократимость ЛЖ резко падает, и неизбежно развиваются нарушения центральной гемодинамики, в рамках которых можно выделить систолический и диастолический компоненты. Систолический компонент: падение УО, МОС, АД и, как следствие, признаки ухудшения тканевой перфузии: нарушения сознания, холодные конечности, снижение пульсации периферических артерий, олигоанурия. Диастолический компонент: увеличение КДД - отек легких – гипоксемия – усугубление гипоксии всех органов и тканей, в том числе миокарда.
• Кардиогенный шок – другой клинический вариант ОЛЖН. Наиболее частой причиной его развития является ИМ, реже – разрыв МЖП, тампонада сердца, гемодинамически значимые нарушения ритма, острая митральная регургитация. • Некроз значительного по площади участка миокарда ЛЖ (как правило, более 40%) ведет к формированию зоны акинезии или гипокинезии, то есть часть миокарда не участвует в сокращении. • Вследствие этого сократимость ЛЖ резко падает, и неизбежно развиваются нарушения центральной гемодинамики, в рамках которых можно выделить систолический и диастолический компоненты. Систолический компонент: падение УО, МОС, АД и, как следствие, признаки ухудшения тканевой перфузии: нарушения сознания, холодные конечности, снижение пульсации периферических артерий, олигоанурия. Диастолический компонент: увеличение КДД - отек легких – гипоксемия – усугубление гипоксии всех органов и тканей, в том числе миокарда.
 • Сразу же включаются рефлекторные компенсаторные механизмы, направленные на поддержание гемодинамики и кровотока прежде всего в жизненно важных органах: активируется САС и выбрасываются катехоламины. Эффектами КХА являются: 1. увеличение сократимости, а значит УО и 2. увеличение ЧСС, направленные на поддержание МОС, 3. а также сужение периферических артериол и венул. Артериолы кожи, мышц, органов брюшной полости и др. суживаются, а в мозге и сердце – нет. Тем самым достигается временное перераспределение кровотока в пользу жизненно важных органов (феномен централизации кровообращения). Цель сужения венул – оптимизация венозного возврата, ликвидация венозного застоя.
• Сразу же включаются рефлекторные компенсаторные механизмы, направленные на поддержание гемодинамики и кровотока прежде всего в жизненно важных органах: активируется САС и выбрасываются катехоламины. Эффектами КХА являются: 1. увеличение сократимости, а значит УО и 2. увеличение ЧСС, направленные на поддержание МОС, 3. а также сужение периферических артериол и венул. Артериолы кожи, мышц, органов брюшной полости и др. суживаются, а в мозге и сердце – нет. Тем самым достигается временное перераспределение кровотока в пользу жизненно важных органов (феномен централизации кровообращения). Цель сужения венул – оптимизация венозного возврата, ликвидация венозного застоя.
 • Однако компенсаторные механизмы быстро истощаются, и, несмотря на тахикардию, МОС и АД прогрессивно падают, что в сочетании с сужением артериол ведет к ухудшению перфузии всех органов и тканей. • Доставка О 2 и субстратов ухудшается. В условиях гипоксии активируется гликолиз, как следствие, формируется энергодефицит и метаболический ацидоз. • Метаболический ацидоз – ключевой биохимический феномен любого шока, поскольку играет решающую роль в развитии микроциркуляторных нарушений (непосредственной причины смерти при шоке).
• Однако компенсаторные механизмы быстро истощаются, и, несмотря на тахикардию, МОС и АД прогрессивно падают, что в сочетании с сужением артериол ведет к ухудшению перфузии всех органов и тканей. • Доставка О 2 и субстратов ухудшается. В условиях гипоксии активируется гликолиз, как следствие, формируется энергодефицит и метаболический ацидоз. • Метаболический ацидоз – ключевой биохимический феномен любого шока, поскольку играет решающую роль в развитии микроциркуляторных нарушений (непосредственной причины смерти при шоке).
 • Прогрессирующий метаболический ацидоз расслабляет прекапиллярные сфинктеры, следовательно, вызывает расширение прекапилляров, по времени опережающее расширение посткапилляров (для расслабления посткапиллярных сфинктеров нужны более низкие значения р. Н). • Вазодилатация при КШ на почве ИМ усугубляется избытком оксида азота (из-за патологической активации NO-синтазы в рамках системного воспалительного ответа). Следствием этого является переполнение кровью сосудов микроциркуляторного русла и нарастание Р гидр. .
• Прогрессирующий метаболический ацидоз расслабляет прекапиллярные сфинктеры, следовательно, вызывает расширение прекапилляров, по времени опережающее расширение посткапилляров (для расслабления посткапиллярных сфинктеров нужны более низкие значения р. Н). • Вазодилатация при КШ на почве ИМ усугубляется избытком оксида азота (из-за патологической активации NO-синтазы в рамках системного воспалительного ответа). Следствием этого является переполнение кровью сосудов микроциркуляторного русла и нарастание Р гидр. .
 • Под действием того же ацидоза, гипоксемии и некоторых БАВ увеличивается проницаемость сосудистой стенки, что в сочетании с увеличением Р гидр. приводит к выходу жидкой части крови в интерстиций. Экстравазация жидкости обусловливает гиповолемию (снижение ОЦК), а значит, усугубляет уменьшение МОС, а также нарушает реологические свойства крови (сгущение). В сочетании с увеличением активности свертывающей системы крови это создает предпосылки для микротромбообразования, то есть для развития ДВС-синдрома.
• Под действием того же ацидоза, гипоксемии и некоторых БАВ увеличивается проницаемость сосудистой стенки, что в сочетании с увеличением Р гидр. приводит к выходу жидкой части крови в интерстиций. Экстравазация жидкости обусловливает гиповолемию (снижение ОЦК), а значит, усугубляет уменьшение МОС, а также нарушает реологические свойства крови (сгущение). В сочетании с увеличением активности свертывающей системы крови это создает предпосылки для микротромбообразования, то есть для развития ДВС-синдрома.
 КОРОНАРНАЯ НЕДОСТАТОЧНОСТЬ • Коронарная недостаточность – типовая форма патологии сердца, характеризующаяся несоответствием потребности миокарда в кислороде и субстратах и реальным притоком крови по коронарным сосудам.
КОРОНАРНАЯ НЕДОСТАТОЧНОСТЬ • Коронарная недостаточность – типовая форма патологии сердца, характеризующаяся несоответствием потребности миокарда в кислороде и субстратах и реальным притоком крови по коронарным сосудам.
 • Подобная ситуация несоответствия может возникнуть в 2 случаях: 1. существенное повышение запроса при нормальном или даже повышенном притоке крови по коронарам; 2. абсолютное снижение притока крови по коронарам. • Соответственно различают виды КН: относительная и абсолютная.
• Подобная ситуация несоответствия может возникнуть в 2 случаях: 1. существенное повышение запроса при нормальном или даже повышенном притоке крови по коронарам; 2. абсолютное снижение притока крови по коронарам. • Соответственно различают виды КН: относительная и абсолютная.
 повышение в крови и миокарде уровня") • • • Причины относительной КН: 1) повышение в крови и миокарде уровня КХА, которые: усиливают функции сердца, а значит потребность в О 2, а также делают расход кислорода и субстратов непроизводительным; 2) значительное возрастание работы сердца, что бывает при физической нагрузке, длительной тахикардии, остром повышении АД, выраженной гиперволемии. • • • Причины абсолютной КН: 1) атеросклеротическое поражение коронаров, 2) тромбоз коронаров, 3) спазм коронаров, 4) значительное снижение перфузионного давления в коронарах вследствие системных гемодинамических нарушений (острая гиповолемия, острая гипотензия).
• • • Причины относительной КН: 1) повышение в крови и миокарде уровня КХА, которые: усиливают функции сердца, а значит потребность в О 2, а также делают расход кислорода и субстратов непроизводительным; 2) значительное возрастание работы сердца, что бывает при физической нагрузке, длительной тахикардии, остром повышении АД, выраженной гиперволемии. • • • Причины абсолютной КН: 1) атеросклеротическое поражение коронаров, 2) тромбоз коронаров, 3) спазм коронаров, 4) значительное снижение перфузионного давления в коронарах вследствие системных гемодинамических нарушений (острая гиповолемия, острая гипотензия).
 • Абсолютная КН ведет к развитию ишемии миокарда в виде 1 из 5 вариантов: 1. Стенокардия - болевая форма ишемии. Приступ загрудинных болей с характерной иррадиацией. Восстановление коронарного кровотока приводит к купированию приступа. Некроз миокарда не развивается (обратимая форма). 2. Безболевая форма ишемии миокарда. Прогностически неблагоприятна, поскольку может привести к развитию ИМ или внезапной смерти. 3. Оглушенный миокард – сохраняющееся в течение нескольких часов локальное снижение сократимости миокарда, несмотря на восстановление кровотока после короткого эпизода ишемии. Существенные черты: - нарушение сократимости носит локальный, временный, полностью обратимый характер; - несоответствие кровотока и функции (кровоток N или почти, а функции снижена); - макро- и микроскопически – N.
• Абсолютная КН ведет к развитию ишемии миокарда в виде 1 из 5 вариантов: 1. Стенокардия - болевая форма ишемии. Приступ загрудинных болей с характерной иррадиацией. Восстановление коронарного кровотока приводит к купированию приступа. Некроз миокарда не развивается (обратимая форма). 2. Безболевая форма ишемии миокарда. Прогностически неблагоприятна, поскольку может привести к развитию ИМ или внезапной смерти. 3. Оглушенный миокард – сохраняющееся в течение нескольких часов локальное снижение сократимости миокарда, несмотря на восстановление кровотока после короткого эпизода ишемии. Существенные черты: - нарушение сократимости носит локальный, временный, полностью обратимый характер; - несоответствие кровотока и функции (кровоток N или почти, а функции снижена); - макро- и микроскопически – N.
. Длительное (хроническое), обратимое (если будет восстановлен коронарный кровоток) снижение сократимости") 4. «Уснувший» миокард (гибернирующий). Длительное (хроническое), обратимое (если будет восстановлен коронарный кровоток) снижение сократимости миокарда в условиях выраженного и продолжительного снижения коронарного кровотока (т. е. может проснуться). Существенные черты: • - соответствие кровотока и функции (оба снижены); • - длительное состояние (до года и более); • - потенциально обратимо (если восстановится кровоток и когда он восстановится); • - морфологические изменения есть (характерные для эмбриональной ткани: деградация миофибрилл, накопление гликогена); • - рассматривается как адаптивный механизм (сердце приспосабливается к новым условиям, т. е. к снижению кровотока). 5. Инфаркт миокарда – необратимая ишемия миокарда с развитием некроза участка сердечной мышцы. Самостоятельная нозологическая единица (вариант ИБС).
4. «Уснувший» миокард (гибернирующий). Длительное (хроническое), обратимое (если будет восстановлен коронарный кровоток) снижение сократимости миокарда в условиях выраженного и продолжительного снижения коронарного кровотока (т. е. может проснуться). Существенные черты: • - соответствие кровотока и функции (оба снижены); • - длительное состояние (до года и более); • - потенциально обратимо (если восстановится кровоток и когда он восстановится); • - морфологические изменения есть (характерные для эмбриональной ткани: деградация миофибрилл, накопление гликогена); • - рассматривается как адаптивный механизм (сердце приспосабливается к новым условиям, т. е. к снижению кровотока). 5. Инфаркт миокарда – необратимая ишемия миокарда с развитием некроза участка сердечной мышцы. Самостоятельная нозологическая единица (вариант ИБС).
 Этиология ИМ: атеросклероз, тромбоз, спазм коронаров. Патогенез ИМ: Схематично можно выделить несколько блоков: • 1 блок. Повреждение миокарда. а) Редукция коронарного кровотока ведет к ишемии и гипоксии участка миокарда, следствием чего является некроз и апоптоз. б) В зоне ишемии и некроза из-за недостатка О 2 активируется гликолиз, вследствие чего накапливаются продукты гликолиза (лактат, кетоновые тела), а также ионы Са 2+, К+, простагландины, кинины. Все эти субстанции раздражают нервные окончания и возникает боль. В рамках стрессовой реакции выбрасываются КХА, ГК. в) КХА, с одной стороны, обеспечивают компенсаторные реакции, направленные на поддержание МОС (инотропные резервы, хронотропные резервы, сужение периферических сосудов). С другой – оказывают кардиотоксическое действие на миокард
Этиология ИМ: атеросклероз, тромбоз, спазм коронаров. Патогенез ИМ: Схематично можно выделить несколько блоков: • 1 блок. Повреждение миокарда. а) Редукция коронарного кровотока ведет к ишемии и гипоксии участка миокарда, следствием чего является некроз и апоптоз. б) В зоне ишемии и некроза из-за недостатка О 2 активируется гликолиз, вследствие чего накапливаются продукты гликолиза (лактат, кетоновые тела), а также ионы Са 2+, К+, простагландины, кинины. Все эти субстанции раздражают нервные окончания и возникает боль. В рамках стрессовой реакции выбрасываются КХА, ГК. в) КХА, с одной стороны, обеспечивают компенсаторные реакции, направленные на поддержание МОС (инотропные резервы, хронотропные резервы, сужение периферических сосудов). С другой – оказывают кардиотоксическое действие на миокард
 Кардиотоксические эффекты КХА разобщают ОФ, увеличивают потребность в кислороде, активируют ПОЛ, активируют липазы, фосфолипазы мембран, гидролазы лизосом, что ведет к повреждению биомембран и, следовательно, увеличению зоны некроза (замыкается порочный круг), • инактивируют ферментные системы (тканевого дыхания, гликолиза, пентозофосфатного шунта). • укорачивают диастолу, следовательно, ухудшают кровоснабжение миокарда, • усиливают агрегацию форменных элементов крови. • •
Кардиотоксические эффекты КХА разобщают ОФ, увеличивают потребность в кислороде, активируют ПОЛ, активируют липазы, фосфолипазы мембран, гидролазы лизосом, что ведет к повреждению биомембран и, следовательно, увеличению зоны некроза (замыкается порочный круг), • инактивируют ферментные системы (тканевого дыхания, гликолиза, пентозофосфатного шунта). • укорачивают диастолу, следовательно, ухудшают кровоснабжение миокарда, • усиливают агрегацию форменных элементов крови. • •
: супероксидный анион О") дополнительное повреждение миокарда в зоне ишемии вызывают активные формы кислорода (АФК): супероксидный анион О 2 -; Н 2 О 2; гидроксильный радикал ОН ; синглетный кислород (кислород с 1 возбужденным электроном 1 О 2), основным источником которых являются лейкоциты, мигрирующие в зону ишемии и некроза для фагоцитоза детрита. Механизмы повреждающего действия АФК: • инактивация ферментов, гормонов, рецепторов и др. белков, • изменение их структуры, • деградация гиалуроновой кислоты, коллагена, • изменение структуры ДНК, • усиление ПОЛ
дополнительное повреждение миокарда в зоне ишемии вызывают активные формы кислорода (АФК): супероксидный анион О 2 -; Н 2 О 2; гидроксильный радикал ОН ; синглетный кислород (кислород с 1 возбужденным электроном 1 О 2), основным источником которых являются лейкоциты, мигрирующие в зону ишемии и некроза для фагоцитоза детрита. Механизмы повреждающего действия АФК: • инактивация ферментов, гормонов, рецепторов и др. белков, • изменение их структуры, • деградация гиалуроновой кислоты, коллагена, • изменение структуры ДНК, • усиление ПОЛ
 • 2 блок. Гемодинамические нарушения. Зона ишемии и некроза теряет способность сокращаться, вследствие чего ухудшается насосная функция сердца. Величины УО, МОС и АД снижаются, рефлекторно суживаются периферические сосуды, следовательно, возрастает ОПСС. Подобные гемодинамические нарушения укладываются в синдром острой сердечной недостаточности вплоть до развития кардиогенного шока. • 3 блок. Нарушения ритма и проводимости. Аритмии вносят существенный вклад в патогенез ИМ. Они возникают из-за изменения электрических свойств кардиомиоцитов и клеток проводящей системы (подробнее механизмы – на соответствующей лекции). Негативная роль аритмий определяется прежде всего нарушениями гемодинамики, а именно – снижением МОС. Брадиаритмии уменьшают МОС за счет снижения ЧСС, а тахиаритмии – за счет укорочения диастолы и, следовательно, снижения УО.
• 2 блок. Гемодинамические нарушения. Зона ишемии и некроза теряет способность сокращаться, вследствие чего ухудшается насосная функция сердца. Величины УО, МОС и АД снижаются, рефлекторно суживаются периферические сосуды, следовательно, возрастает ОПСС. Подобные гемодинамические нарушения укладываются в синдром острой сердечной недостаточности вплоть до развития кардиогенного шока. • 3 блок. Нарушения ритма и проводимости. Аритмии вносят существенный вклад в патогенез ИМ. Они возникают из-за изменения электрических свойств кардиомиоцитов и клеток проводящей системы (подробнее механизмы – на соответствующей лекции). Негативная роль аритмий определяется прежде всего нарушениями гемодинамики, а именно – снижением МОС. Брадиаритмии уменьшают МОС за счет снижения ЧСС, а тахиаритмии – за счет укорочения диастолы и, следовательно, снижения УО.
 • Основные клинические проявления ИМ: боль, аритмии, признаки СН. Их патогенез разобрали. Кроме того, нейтрофильный лейкоцитоз, повышение температуры тела, ускорение СОЭ, связанные с резорбцией некротических масс, и гиперферментемия (повышение активности КФК, Ал. АТ, Ас. АТ, ЛДГ в крови вследствие выхода этих ферментов из разрушенных кардиомиоцитов). • Смерть может наступить в разные сроки от таких осложнений, как кардиогенный шок, разрыв сердца, ТЭЛА, тяжелые нарушения ритма (фибрилляция желудочков). • Если больной не погибает, исходом ИМ является формирование рубца, постинфарктного кардиосклероза и ХСН.
• Основные клинические проявления ИМ: боль, аритмии, признаки СН. Их патогенез разобрали. Кроме того, нейтрофильный лейкоцитоз, повышение температуры тела, ускорение СОЭ, связанные с резорбцией некротических масс, и гиперферментемия (повышение активности КФК, Ал. АТ, Ас. АТ, ЛДГ в крови вследствие выхода этих ферментов из разрушенных кардиомиоцитов). • Смерть может наступить в разные сроки от таких осложнений, как кардиогенный шок, разрыв сердца, ТЭЛА, тяжелые нарушения ритма (фибрилляция желудочков). • Если больной не погибает, исходом ИМ является формирование рубца, постинфарктного кардиосклероза и ХСН.
 РЕПЕРФУЗИОННОЕ ПОВРЕЖДЕНИЕ МИОКАРДА В ряде случаев реперфузия сама вызывает дополнительные повреждения. Основные механизмы реперфузионного повреждения сердца являются так называемый кислородный парадокс и кальциевый парадокс. • Кислородный парадокс – токсическое действие кислорода в период реперфузии (реоксигенации) после ишемии. В период ишемии дефицит кислорода ведет к восстановлению переносчиков электронов (цитохромы, НАДН-дегидрогеназа, убихинон) в дыхательной цепи митохондрий. На этапе реоксигенации эти переносчики становятся донорами электронов для молекул кислорода, вследствие чего молекулы кислорода превращаются в АФК (свободные радикалы). Механизмы повреждающего действия АФК рассмотрены выше.
РЕПЕРФУЗИОННОЕ ПОВРЕЖДЕНИЕ МИОКАРДА В ряде случаев реперфузия сама вызывает дополнительные повреждения. Основные механизмы реперфузионного повреждения сердца являются так называемый кислородный парадокс и кальциевый парадокс. • Кислородный парадокс – токсическое действие кислорода в период реперфузии (реоксигенации) после ишемии. В период ишемии дефицит кислорода ведет к восстановлению переносчиков электронов (цитохромы, НАДН-дегидрогеназа, убихинон) в дыхательной цепи митохондрий. На этапе реоксигенации эти переносчики становятся донорами электронов для молекул кислорода, вследствие чего молекулы кислорода превращаются в АФК (свободные радикалы). Механизмы повреждающего действия АФК рассмотрены выше.
 • Кальциевый парадокс – перегрузка кардиомиоцитов ионами Са 2+ на этапе реперфузии. Дефицит кислорода на этапе ишемии ведет к энергодефициту, а значит, к нарушению работы энергозависимых ионных насосов, обеспечивающих нормальное трансмембранное распределение ионов. В частности, снижается активность К+/Na+- АТФазы, в результате кардиомиоциты перегружаются натрием. Резко активируется Na+/Ca 2+ обменник с целью удаления избытка натрия. Однако удаление натрия происходит в обмен на Са 2+ (у них общий белок-переносчик), вследствие чего перегрузка натрием сменяется перегрузкой кальцием. Избыток кальция разобщает ОФ (развивается энергодефицит), приводит к нарушению диастолической функции и развитию контрактур, активирует Са 2+-зависимые протеазы и липазы, что ведет к повреждению мембран кардиомиоцитов.
• Кальциевый парадокс – перегрузка кардиомиоцитов ионами Са 2+ на этапе реперфузии. Дефицит кислорода на этапе ишемии ведет к энергодефициту, а значит, к нарушению работы энергозависимых ионных насосов, обеспечивающих нормальное трансмембранное распределение ионов. В частности, снижается активность К+/Na+- АТФазы, в результате кардиомиоциты перегружаются натрием. Резко активируется Na+/Ca 2+ обменник с целью удаления избытка натрия. Однако удаление натрия происходит в обмен на Са 2+ (у них общий белок-переносчик), вследствие чего перегрузка натрием сменяется перегрузкой кальцием. Избыток кальция разобщает ОФ (развивается энергодефицит), приводит к нарушению диастолической функции и развитию контрактур, активирует Са 2+-зависимые протеазы и липазы, что ведет к повреждению мембран кардиомиоцитов.
 Проявления реперфузионного повреждения сердца 1. 2. 3. 4. реперфузионную сократительную дисфункцию – нарушение как систолической, так и в большей степени диастолической функции сердца ( «оглушенный» миокард). реперфузионные нарушения ритма. феномен невосстановленного кровотока. (no reflow) – сохраняющийся дефицит микроциркуляторного кровотока, несмотря на восстановление перфузии по коронарным артериям. Патогенетические факторы: повреждение эндотелиоцитов, агрегация форменных элементов крови, микротромбообразование. «Виновниками» этих нарушений являются, прежде всего, мигрирующие в зону повреждения лейкоциты, генерирующие АФК, тромбоксаны и лейкотриены. еще один реперфузионный феномен - прекондиционирование (или метаболическая адаптация) – феномен адаптации к ишемии. Суть заключается в том, что повторные эпизоды ишемии переживаются лучше. Механизм: накапливающиеся во время ишемии БАВ (аденозин, ангиотензин 2, брадикинин, опиоидные пептиды) активируют работу АТФ-зависимых К+каналов (через протеинкиназу С). Дефицит АТФ при ишемии также активирует эти каналы. В результате в кардиомиоцитах нормализуется содержание К+ и баланс ионов вообще.
Проявления реперфузионного повреждения сердца 1. 2. 3. 4. реперфузионную сократительную дисфункцию – нарушение как систолической, так и в большей степени диастолической функции сердца ( «оглушенный» миокард). реперфузионные нарушения ритма. феномен невосстановленного кровотока. (no reflow) – сохраняющийся дефицит микроциркуляторного кровотока, несмотря на восстановление перфузии по коронарным артериям. Патогенетические факторы: повреждение эндотелиоцитов, агрегация форменных элементов крови, микротромбообразование. «Виновниками» этих нарушений являются, прежде всего, мигрирующие в зону повреждения лейкоциты, генерирующие АФК, тромбоксаны и лейкотриены. еще один реперфузионный феномен - прекондиционирование (или метаболическая адаптация) – феномен адаптации к ишемии. Суть заключается в том, что повторные эпизоды ишемии переживаются лучше. Механизм: накапливающиеся во время ишемии БАВ (аденозин, ангиотензин 2, брадикинин, опиоидные пептиды) активируют работу АТФ-зависимых К+каналов (через протеинкиназу С). Дефицит АТФ при ишемии также активирует эти каналы. В результате в кардиомиоцитах нормализуется содержание К+ и баланс ионов вообще.
 НЕКОРОНАРОГЕННЫЕ НЕКРОЗЫ МИОКАРДА • 1. 2. 3. 4. Некроз участка миокарда в клинике чаще всего вызывается редукцией коронарного кровотока и называется инфарктом миокарда. Наряду с этим некроз миокарда может формироваться при неповрежденных коронарах. Такие некрозы называются некоронарогенными. Речь идет в первую очередь об экспериментальных моделях. Однако и в клинике имеют место подобные механизмы развития некрозов. Катехоламиновая модель. Высокие концентрации норадреналина и адреналина вызывают резкое возрастание функций сердца. Возникает несоответствие между возросшей метаболической потребностью и неизмененной реальной доставкой крови по коронарам (относительная КН). Дополнительное значение имеет прямое повреждающее действие токсических концентраций катехоламинов на миокард (см. выше). Гипоксическая модель. Гипоксия различного генеза (экзогенная, дыхательная, гемическая) может привести к развитию некроза из-за недостатка кислорода. Электролитно-стероидная модель. При введении крысам значительного количества солей натрия и стероидных гормонов развиваются дегенеративно-некротическе очаги в миокарде. Иммунные повреждения сердца. Развиваются при введении в организм экспериментального животного гетерогенной сыворотки, содержащей антитела к белкам сердца животного данного вида. В клинике подобная ситуация может возникнуть при выработке аутоантител к кардиомиоцитам.
НЕКОРОНАРОГЕННЫЕ НЕКРОЗЫ МИОКАРДА • 1. 2. 3. 4. Некроз участка миокарда в клинике чаще всего вызывается редукцией коронарного кровотока и называется инфарктом миокарда. Наряду с этим некроз миокарда может формироваться при неповрежденных коронарах. Такие некрозы называются некоронарогенными. Речь идет в первую очередь об экспериментальных моделях. Однако и в клинике имеют место подобные механизмы развития некрозов. Катехоламиновая модель. Высокие концентрации норадреналина и адреналина вызывают резкое возрастание функций сердца. Возникает несоответствие между возросшей метаболической потребностью и неизмененной реальной доставкой крови по коронарам (относительная КН). Дополнительное значение имеет прямое повреждающее действие токсических концентраций катехоламинов на миокард (см. выше). Гипоксическая модель. Гипоксия различного генеза (экзогенная, дыхательная, гемическая) может привести к развитию некроза из-за недостатка кислорода. Электролитно-стероидная модель. При введении крысам значительного количества солей натрия и стероидных гормонов развиваются дегенеративно-некротическе очаги в миокарде. Иммунные повреждения сердца. Развиваются при введении в организм экспериментального животного гетерогенной сыворотки, содержащей антитела к белкам сердца животного данного вида. В клинике подобная ситуация может возникнуть при выработке аутоантител к кардиомиоцитам.
 Причиной развития АМД является этанол и продукт его метаболизма") АЛКОГОЛЬНОЕ ПОРАЖЕНИЕ СЕРДЦА (алкогольная миокардиодистрофия) Причиной развития АМД является этанол и продукт его метаболизма ацетальдегид. Способствующие факторы: 1. стрессы, 2. недостаточность питания, 3. наследственная предрасположенность, 4. вирусные инфекции, 5. изменение исходного состояния миокарда. Патогенез АМД. Можно выделить 3 основных направления: 1. нарушения биоэнергетики, 2. воздействие избытка симпатических нейрогормонов, 3. расстройства микроциркуляци.
АЛКОГОЛЬНОЕ ПОРАЖЕНИЕ СЕРДЦА (алкогольная миокардиодистрофия) Причиной развития АМД является этанол и продукт его метаболизма ацетальдегид. Способствующие факторы: 1. стрессы, 2. недостаточность питания, 3. наследственная предрасположенность, 4. вирусные инфекции, 5. изменение исходного состояния миокарда. Патогенез АМД. Можно выделить 3 основных направления: 1. нарушения биоэнергетики, 2. воздействие избытка симпатических нейрогормонов, 3. расстройства микроциркуляци.
 этанол и ацетальдегид нарушают процесс синтеза") Нарушения биоэнергетики происходят по разным механизмам: • а) этанол и ацетальдегид нарушают процесс синтеза энергии путем угнетения НАД-зависимых дегидрогеназ, нарушают активность ферментов дыхательной цепи и активируют гликолиз. • б) кроме того, этанол ингибирует основные пути утилизации энергии в кардиомиоцитах. Этанол взаимодействует с гидрофобными участками белковых молекул, изменяя их свойства (транспортных белков, белков-ферментов, сократительных белков). • в) ацетальдегид конкурирует с жирными кислотами за включение в ЦТК, вследствие чего ослабляется окисление ЖК (основной энергетический субстрат в сердце). • г) этанол растворяет липиды биомембран. В результате: нарушается работа ионных насосов (К+/Na+-АТФаза и Сa 2+/Mg 2+-АТФаза) и, как следствие, нарушается электролитный баланс (снижается концентрация K+ и увеличивается концентрация Na+ в КМЦ) и функции сердца (возбудимость, электромеханическое сопряжение).
Нарушения биоэнергетики происходят по разным механизмам: • а) этанол и ацетальдегид нарушают процесс синтеза энергии путем угнетения НАД-зависимых дегидрогеназ, нарушают активность ферментов дыхательной цепи и активируют гликолиз. • б) кроме того, этанол ингибирует основные пути утилизации энергии в кардиомиоцитах. Этанол взаимодействует с гидрофобными участками белковых молекул, изменяя их свойства (транспортных белков, белков-ферментов, сократительных белков). • в) ацетальдегид конкурирует с жирными кислотами за включение в ЦТК, вследствие чего ослабляется окисление ЖК (основной энергетический субстрат в сердце). • г) этанол растворяет липиды биомембран. В результате: нарушается работа ионных насосов (К+/Na+-АТФаза и Сa 2+/Mg 2+-АТФаза) и, как следствие, нарушается электролитный баланс (снижается концентрация K+ и увеличивается концентрация Na+ в КМЦ) и функции сердца (возбудимость, электромеханическое сопряжение).
 Воздействие избытка катехоламинов. Этанол усиливает биосинтез и высвобождение адреналина в надпочечниках и высвобождение норадреналина в симпатических окончаниях. Патогенное действие избытка катехоламинов - см. патогенез ИМ. Расстройства микроциркуляции включают в себя: 1. агрегацию тромбоцитов, сладжирование форменных элементов, стаз, 2. повышение проницаемости стенок микрососудов, отек эндотелия, 3. тромбоз сосудов, 4. аневризмы и разрывы сосудов.
Воздействие избытка катехоламинов. Этанол усиливает биосинтез и высвобождение адреналина в надпочечниках и высвобождение норадреналина в симпатических окончаниях. Патогенное действие избытка катехоламинов - см. патогенез ИМ. Расстройства микроциркуляции включают в себя: 1. агрегацию тромбоцитов, сладжирование форменных элементов, стаз, 2. повышение проницаемости стенок микрососудов, отек эндотелия, 3. тромбоз сосудов, 4. аневризмы и разрывы сосудов.
 Морфологические изменения при АМД: 1. жировая инфильтрация миокарда, 2. склероз сосудов, 3. гипертрофия и дилатация полостей, 4. рубцы в миокарде. Клинические проявления АМД включают в себя: 1. вегетативные расстройства, 2. нарушения ритма и проводимости (нередко после однократного употребления большой дозы алкоголя – синдром «праздничного сердца» ), 3. симптомы сердечной недостаточности, 4. нередко тромбоэмболические осложнения.
Морфологические изменения при АМД: 1. жировая инфильтрация миокарда, 2. склероз сосудов, 3. гипертрофия и дилатация полостей, 4. рубцы в миокарде. Клинические проявления АМД включают в себя: 1. вегетативные расстройства, 2. нарушения ритма и проводимости (нередко после однократного употребления большой дозы алкоголя – синдром «праздничного сердца» ), 3. симптомы сердечной недостаточности, 4. нередко тромбоэмболические осложнения.
 Нарушения сосудистого тонуса • Механизмы регуляции АД Уровень АД зависит от 2 групп факторов • I группа – гемодинамические факторы (непосредственно формирующие гидродинамическое давление, т. е. АД): • 1) УО (его величина определяется насосной функцией ЛЖ); • 2) ОЦК; • 3) ОПСС (сопротивление резистивных сосудов (артериол и терминальных артерий с прекапиллярными сфинктерами); • 4) эластическое сопротивление стенок аорты и ее крупных ветвей; • 5) вязкость крови.
Нарушения сосудистого тонуса • Механизмы регуляции АД Уровень АД зависит от 2 групп факторов • I группа – гемодинамические факторы (непосредственно формирующие гидродинамическое давление, т. е. АД): • 1) УО (его величина определяется насосной функцией ЛЖ); • 2) ОЦК; • 3) ОПСС (сопротивление резистивных сосудов (артериол и терминальных артерий с прекапиллярными сфинктерами); • 4) эластическое сопротивление стенок аорты и ее крупных ветвей; • 5) вязкость крови.
, меняющие уровень") • II группа факторов – собственно регуляторные механизмы (нервные и гуморальные), меняющие уровень АД в зависимости от обстоятельств. 1. барорецепторный рефлекс. 2. САС. 3. тканевые системы РААС (в сосудах, сердце, мозге); 4. почечная система РААС; 5. вазоконстрикторные вещества эндотелия (эндотелины 1, 2, 3) и др. 6. депрессорная система стенок сосудов (оксид азота, натрийуретический пептид С, адреномедуллин и др. ); 7. депрессорная система мозгового слоя почек: ПГ E 2, I 2; калликреин-кининовая система почек; 8. натрийуретические пептиды (2 предсердных и 3 мозговых субтипа); 9. депрессорные механизмы ЦНС (упомянутые мозговые НУП, а также допаминергические, серотониновые, имидазолиновые и α-адренорецепторы).
• II группа факторов – собственно регуляторные механизмы (нервные и гуморальные), меняющие уровень АД в зависимости от обстоятельств. 1. барорецепторный рефлекс. 2. САС. 3. тканевые системы РААС (в сосудах, сердце, мозге); 4. почечная система РААС; 5. вазоконстрикторные вещества эндотелия (эндотелины 1, 2, 3) и др. 6. депрессорная система стенок сосудов (оксид азота, натрийуретический пептид С, адреномедуллин и др. ); 7. депрессорная система мозгового слоя почек: ПГ E 2, I 2; калликреин-кининовая система почек; 8. натрийуретические пептиды (2 предсердных и 3 мозговых субтипа); 9. депрессорные механизмы ЦНС (упомянутые мозговые НУП, а также допаминергические, серотониновые, имидазолиновые и α-адренорецепторы).
 • Нарушения системного уровня АД делятся на 2 вида: 1. артериальные гипертензии; 2. артериальные гипотензии.
• Нарушения системного уровня АД делятся на 2 вида: 1. артериальные гипертензии; 2. артериальные гипотензии.
 – стойкое повышение АД выше нормы. АГ") • Артериальные гипертензии Артериальная гипертензия (АГ) – стойкое повышение АД выше нормы. АГ по происхождению: 1. - первичная (эссенциальная) гипертензия, или ГБ; 2. - вторичные (симптоматические) АГ.
• Артериальные гипертензии Артериальная гипертензия (АГ) – стойкое повышение АД выше нормы. АГ по происхождению: 1. - первичная (эссенциальная) гипертензия, или ГБ; 2. - вторичные (симптоматические) АГ.
 • Общая характеристика: – самостоятельное заболевание; – важное значение имеют") Гипертоническая болезнь (эссенциальная гипертензия) • Общая характеристика: – самостоятельное заболевание; – важное значение имеют наследственные факторы (генетические дефекты клеточных мембран и механизмов регуляции АД); – нередко семейный характер; – этиология и патогенез изучены недостаточно. Факторы риска развития ГБ (вместо этиологии) делятся на основные и дополнительные. Основные: • - возраст (мужчины старше 55, женщины старше 65); • - курение; • - холестерин выше 6, 5 ммоль/л; • - семейный анамнез ранних сердечно-сосудистых заболеваний) • - сахарный диабет. Дополнительные: • - снижение Хс ЛПВП и повышение Хс ЛПНП; • - микроальбуминурия при СД; • - увеличение содержания фибриногена; • - нарушение толерантности к глюкозе; • - ожирение; • - гиподинамия; • - социально-экономическая группа риска (длительное психоэмоциональное перенапряжение, стрессы) •
Гипертоническая болезнь (эссенциальная гипертензия) • Общая характеристика: – самостоятельное заболевание; – важное значение имеют наследственные факторы (генетические дефекты клеточных мембран и механизмов регуляции АД); – нередко семейный характер; – этиология и патогенез изучены недостаточно. Факторы риска развития ГБ (вместо этиологии) делятся на основные и дополнительные. Основные: • - возраст (мужчины старше 55, женщины старше 65); • - курение; • - холестерин выше 6, 5 ммоль/л; • - семейный анамнез ранних сердечно-сосудистых заболеваний) • - сахарный диабет. Дополнительные: • - снижение Хс ЛПВП и повышение Хс ЛПНП; • - микроальбуминурия при СД; • - увеличение содержания фибриногена; • - нарушение толерантности к глюкозе; • - ожирение; • - гиподинамия; • - социально-экономическая группа риска (длительное психоэмоциональное перенапряжение, стрессы) •
 1. Основной плацдарм повышения АД при АГ– резистивные сосуды. Здесь развиваются 3 группы структурно-функциональных изменений: Ремоделирование сосудов, т. е. изменение их структуры и геометрии из-за пролиферации ГМК и наработки коллагена. Пусковые факторы ремоделирования сосудов при АГ: гемодинамические (повышение внутрисосудистого давления) и нейрогуморальные (РААС, САС, дисбаланс эндотелиальных факторов эндотелина и оксида азота). Ремоделирование по сути – адаптивный процесс. • 2. Повышение реактивности ремоделированных сосудов (в основе – эндотелиальная дисфункция), т. е. нарушение регуляции сосудистого тонуса с преобладанием прессорных влияний. Такие сосуды еще больше повышают ОПСС, т. к. не могут адекватно отвечать на сосудосуживающие импульсы. Это приводит к тому, что адаптивный по сути процесс ремоделирования приобретает патологические черты. • 3. Рарефикация (разрежение), т. е. уменьшение плотности МЦР (уменьшение числа «рабочих» микрососудов). Эти микроциркуляторные нарушения играют важную роль в развитии и прогрессировании поражения органов-мишеней при АГ: - поражение коронаров ведет к развитию различных вариантов ишемии миокарда, вплоть до ИМ; - поражение капилляров почечных клубочков - к развитию ХПН; - поражение церебральных сосудов – к различным вариантам нарушений мозгового кровообращения, в том числе ишемического и геморрагического инсультов, а также к дисциркуляторной энцефалопатии с исходом в деменцию. - поражение сосудов сетчатки – к гипертонической ретинопатии.
1. Основной плацдарм повышения АД при АГ– резистивные сосуды. Здесь развиваются 3 группы структурно-функциональных изменений: Ремоделирование сосудов, т. е. изменение их структуры и геометрии из-за пролиферации ГМК и наработки коллагена. Пусковые факторы ремоделирования сосудов при АГ: гемодинамические (повышение внутрисосудистого давления) и нейрогуморальные (РААС, САС, дисбаланс эндотелиальных факторов эндотелина и оксида азота). Ремоделирование по сути – адаптивный процесс. • 2. Повышение реактивности ремоделированных сосудов (в основе – эндотелиальная дисфункция), т. е. нарушение регуляции сосудистого тонуса с преобладанием прессорных влияний. Такие сосуды еще больше повышают ОПСС, т. к. не могут адекватно отвечать на сосудосуживающие импульсы. Это приводит к тому, что адаптивный по сути процесс ремоделирования приобретает патологические черты. • 3. Рарефикация (разрежение), т. е. уменьшение плотности МЦР (уменьшение числа «рабочих» микрососудов). Эти микроциркуляторные нарушения играют важную роль в развитии и прогрессировании поражения органов-мишеней при АГ: - поражение коронаров ведет к развитию различных вариантов ишемии миокарда, вплоть до ИМ; - поражение капилляров почечных клубочков - к развитию ХПН; - поражение церебральных сосудов – к различным вариантам нарушений мозгового кровообращения, в том числе ишемического и геморрагического инсультов, а также к дисциркуляторной энцефалопатии с исходом в деменцию. - поражение сосудов сетчатки – к гипертонической ретинопатии.
 АГ Общая характеристика. Являются следствием первичного поражения каких-либо органов или систем, участвующих") Вторичные (симптоматические) АГ Общая характеристика. Являются следствием первичного поражения каких-либо органов или систем, участвующих в поддержании системного уровня АД. Иными словами, это АГ, этиология которых может быть установлена. Нередко характеризуются тяжелым и даже злокачественным течением, рефрактерностью к терапии. Классификация вторичных форм АГ: 1. почечные АГ (вазоренальные и ренопривные) 2. эндокринные АГ 3. центрогенные.
Вторичные (симптоматические) АГ Общая характеристика. Являются следствием первичного поражения каких-либо органов или систем, участвующих в поддержании системного уровня АД. Иными словами, это АГ, этиология которых может быть установлена. Нередко характеризуются тяжелым и даже злокачественным течением, рефрактерностью к терапии. Классификация вторичных форм АГ: 1. почечные АГ (вазоренальные и ренопривные) 2. эндокринные АГ 3. центрогенные.
 В основе ВРАГ – снижение перфузионного давления крови в сосудах почек") Вазоренальные АГ (ВРАГ) В основе ВРАГ – снижение перфузионного давления крови в сосудах почек различного генеза. Причины снижения перфузионного давления: 1. - сдавление магистральных почечных артерий извне (опухоль, рубец); 2. - уменьшение или полное закрытие просвета сосуда изнутри (тромб, эмбол, атеросклеротическая бляшка, прорастание опухоли); 3. - гиповолемия (после кровотечения, при ожоговой болезни); 4. - сдавление ветвей почечной артерии при воспалении паренхимы почки (диффузный гломерулонефрит). Механизм развития ВРАГ. Снижение перфузионного давления и объема протекающей крови ведет к возбуждению волюморецепторов клеток ЮГА и запуску всей РААС, отдельные компоненты которой обладают прессорными эффектами.
Вазоренальные АГ (ВРАГ) В основе ВРАГ – снижение перфузионного давления крови в сосудах почек различного генеза. Причины снижения перфузионного давления: 1. - сдавление магистральных почечных артерий извне (опухоль, рубец); 2. - уменьшение или полное закрытие просвета сосуда изнутри (тромб, эмбол, атеросклеротическая бляшка, прорастание опухоли); 3. - гиповолемия (после кровотечения, при ожоговой болезни); 4. - сдавление ветвей почечной артерии при воспалении паренхимы почки (диффузный гломерулонефрит). Механизм развития ВРАГ. Снижение перфузионного давления и объема протекающей крови ведет к возбуждению волюморецепторов клеток ЮГА и запуску всей РААС, отдельные компоненты которой обладают прессорными эффектами.
 Ангиотензин II: • - вызывает сокращение ГМК артериол, • - активирует освобождение КХА из аксонов симпатических нервов, • - повышает чувствительностьстенки к КХА и др. вазоконстрикторам, • - продукт его метаболизма АТ III обладает положительным хронотропным действием, что ведет к увеличению ЧСС, СВ и АД; • - стимулирует синтез и освобождение в кровь (из клубочковой зоны надпочечников) альдостерона; Альдостерон: • - усиливает реабсорбцию Na в почках, что ведет к повышаению Росм. крови, возбуждению осморецепторов сосудистого русла и нейросекреции АДГ, АДГ (вазопрессин): • - усиливает реабсорбцию воды и, следовательно, увеличение ОЦК в уже суженном сосудистом русле; • - стимулирует транспорт ионов Na в клетки, в т. ч. в сосудистую стенку (экстраренальный эффект), в связи с чем стенки артериол набухают, их тонус повышается, повышается чувствительность к вазоконстрикторам (КХА, АТ II, вазопрессин, ПГ).
Ангиотензин II: • - вызывает сокращение ГМК артериол, • - активирует освобождение КХА из аксонов симпатических нервов, • - повышает чувствительностьстенки к КХА и др. вазоконстрикторам, • - продукт его метаболизма АТ III обладает положительным хронотропным действием, что ведет к увеличению ЧСС, СВ и АД; • - стимулирует синтез и освобождение в кровь (из клубочковой зоны надпочечников) альдостерона; Альдостерон: • - усиливает реабсорбцию Na в почках, что ведет к повышаению Росм. крови, возбуждению осморецепторов сосудистого русла и нейросекреции АДГ, АДГ (вазопрессин): • - усиливает реабсорбцию воды и, следовательно, увеличение ОЦК в уже суженном сосудистом русле; • - стимулирует транспорт ионов Na в клетки, в т. ч. в сосудистую стенку (экстраренальный эффект), в связи с чем стенки артериол набухают, их тонус повышается, повышается чувствительность к вазоконстрикторам (КХА, АТ II, вазопрессин, ПГ).
 Причина РАГ –") • Ренопривная АГ (ren – почка; privo – лишать, уменьшать) Причина РАГ – уменьшение массы почечной паренхимы вследствие: • - удаления части почки, 1 или обеих почек; • - некроза паренхимы; • - гидронефроза, • - поликистоза почек и др. ; Механизм: уменьшение массы паренхимы ведет к снижению синтеза депрессивных веществ. • 1) ПГ • 2) кининов. Их дефицит ведет к снижению кровотока в почках, а значит к активации РААС. Кроме того, дефицит их ведет к реабсорбции натрия, к гипернатриемии, значит – гиперволемии.
• Ренопривная АГ (ren – почка; privo – лишать, уменьшать) Причина РАГ – уменьшение массы почечной паренхимы вследствие: • - удаления части почки, 1 или обеих почек; • - некроза паренхимы; • - гидронефроза, • - поликистоза почек и др. ; Механизм: уменьшение массы паренхимы ведет к снижению синтеза депрессивных веществ. • 1) ПГ • 2) кининов. Их дефицит ведет к снижению кровотока в почках, а значит к активации РААС. Кроме того, дефицит их ведет к реабсорбции натрия, к гипернатриемии, значит – гиперволемии.
 Эндокринные АГ • Минералокортикоидные АГ. Обусловлены гиперпродукцией альдостерона. Причина последней – опухоль клубочковой зоны коры надпочечников. Такой альдостеронизм называют первичным, или синдромом Конна. Механизмы повышения АД при первичном альдостеронизме: А) почечный механизм: гиперпродукция альдостерона - усиление реабсорбции Na – гиперосмия плазмы – активация осмо. RR сосудов – нейросекреция АДГ – реабсорбция воды – увеличение ОЦК – увеличение СВ – повышение АД. Б) внепочечный механизм: гиперпродукция альдостерона – усиление транспорта ионов Na через мембраны клеток, в т. ч. ГМК и КМЦ – набухание клеток – уменьшение просвета сосудов, повышение их тонуса, увеличение чувствительности к вазоконстрикторам (КХА, вазопрессин, АТ II, ПГ) – повышение АД.
Эндокринные АГ • Минералокортикоидные АГ. Обусловлены гиперпродукцией альдостерона. Причина последней – опухоль клубочковой зоны коры надпочечников. Такой альдостеронизм называют первичным, или синдромом Конна. Механизмы повышения АД при первичном альдостеронизме: А) почечный механизм: гиперпродукция альдостерона - усиление реабсорбции Na – гиперосмия плазмы – активация осмо. RR сосудов – нейросекреция АДГ – реабсорбция воды – увеличение ОЦК – увеличение СВ – повышение АД. Б) внепочечный механизм: гиперпродукция альдостерона – усиление транспорта ионов Na через мембраны клеток, в т. ч. ГМК и КМЦ – набухание клеток – уменьшение просвета сосудов, повышение их тонуса, увеличение чувствительности к вазоконстрикторам (КХА, вазопрессин, АТ II, ПГ) – повышение АД.
. Причины гиперпродукции – аденома гипофиза (избыток") Глюкокортикоидные АГ. Обусловлены гиперпродукцией ГК (гиперкортизолизм, синдром Иценко-Кушинга). Причины гиперпродукции – аденома гипофиза (избыток АКТГ и избыток кортизола в коре надпочечников - болезнь Иценко-Кушинга), гормоноактивная опухоль коры надпочечников (кортикостерома), длительный прием ГК. Механизмы повышения АД при гиперкортизолизме: А) ГК пермиссируют действие КХА (повышают чувствительность стенок сосудов к КХА); Б) ГК стимулируют глюконеогенез (из АК). При этом образуется аммиак, который повышает тонус ГМК сосудов (Селье: ГК – дача нашатыря изнутри) В) ГК стимулируют продукцию ангиотензиногена в печени; Г) ГК стимулируют синтез прессорного амина серотонина; Д) ГК обладают минералокортикоидным действием (типа альдостерона).
Глюкокортикоидные АГ. Обусловлены гиперпродукцией ГК (гиперкортизолизм, синдром Иценко-Кушинга). Причины гиперпродукции – аденома гипофиза (избыток АКТГ и избыток кортизола в коре надпочечников - болезнь Иценко-Кушинга), гормоноактивная опухоль коры надпочечников (кортикостерома), длительный прием ГК. Механизмы повышения АД при гиперкортизолизме: А) ГК пермиссируют действие КХА (повышают чувствительность стенок сосудов к КХА); Б) ГК стимулируют глюконеогенез (из АК). При этом образуется аммиак, который повышает тонус ГМК сосудов (Селье: ГК – дача нашатыря изнутри) В) ГК стимулируют продукцию ангиотензиногена в печени; Г) ГК стимулируют синтез прессорного амина серотонина; Д) ГК обладают минералокортикоидным действием (типа альдостерона).
 Катехоламиновые АГ. Обусловлены увеличением уровня КХА. Причина гиперкатехоламинемии – гормоноактивная опухоль мозгового вещества надпочечников – феохромоцитома. Механизмы повышения АД при феохромоцитоме: А) увеличение тонуса сосудов и работы сердца, следовательно, повышение ОПСС и СВ; Б) активация РААС вследствие сужения приносящих почечных артериол, а значит, снижения перфузионного давления в почках. Кроме того, КХА прямо возбуждают -адренорецепторы в сосудах почек, следствием чего является увеличение синтеза ренина и запуск всей РААС.
Катехоламиновые АГ. Обусловлены увеличением уровня КХА. Причина гиперкатехоламинемии – гормоноактивная опухоль мозгового вещества надпочечников – феохромоцитома. Механизмы повышения АД при феохромоцитоме: А) увеличение тонуса сосудов и работы сердца, следовательно, повышение ОПСС и СВ; Б) активация РААС вследствие сужения приносящих почечных артериол, а значит, снижения перфузионного давления в почках. Кроме того, КХА прямо возбуждают -адренорецепторы в сосудах почек, следствием чего является увеличение синтеза ренина и запуск всей РААС.
 и/или") Гипертиреоидные АГ. Обусловлены длительным увеличением содержания в крови гормонов щитовидной железы (тироксина, трийодтиронина) и/или повышением чувствительности тканей к ним. Причины повышения уровня гормонов - гиперплазия или опухоль щитовидной железы. Механизм повышения САД связан с кардиотоническим действием гормонов, следовательно, увеличением СВ. ДАД при этом, как правило, в норме или понижено вследствие компенсаторного (в ответ на увеличение СВ) расширения резистивных сосудов, а также вследствие прямого повреждающего действия гормонов на стенки сосудов и снижения их тонуса по этой причине.
Гипертиреоидные АГ. Обусловлены длительным увеличением содержания в крови гормонов щитовидной железы (тироксина, трийодтиронина) и/или повышением чувствительности тканей к ним. Причины повышения уровня гормонов - гиперплазия или опухоль щитовидной железы. Механизм повышения САД связан с кардиотоническим действием гормонов, следовательно, увеличением СВ. ДАД при этом, как правило, в норме или понижено вследствие компенсаторного (в ответ на увеличение СВ) расширения резистивных сосудов, а также вследствие прямого повреждающего действия гормонов на стенки сосудов и снижения их тонуса по этой причине.
 Нейрогенные АГ условно делят на центрогенные и рефлекторные. Центрогенные АГ. Причины: функциональные нарушения ВНД или органические поражения структур, регулирующих системную гемодинамику (в 1 -ую очередь, сосудодвигательного центра). Механизмы. Повышение АД связано с возбуждением: 1. - симпатических ядер заднего гипоталамуса, 2. адренергических структур ретикулярной формации, 3. сосудодвигательного центра, 4. центров, регулирующих активность системы «гипоталамусгипофиз-надпочечники» 5. и, как следствие, усиление симпатических влияний на органы и ткани, т. е. : 6. повышение тонуса артериальных сосудов и работы сердца, а значит, повышение АД.
Нейрогенные АГ условно делят на центрогенные и рефлекторные. Центрогенные АГ. Причины: функциональные нарушения ВНД или органические поражения структур, регулирующих системную гемодинамику (в 1 -ую очередь, сосудодвигательного центра). Механизмы. Повышение АД связано с возбуждением: 1. - симпатических ядер заднего гипоталамуса, 2. адренергических структур ретикулярной формации, 3. сосудодвигательного центра, 4. центров, регулирующих активность системы «гипоталамусгипофиз-надпочечники» 5. и, как следствие, усиление симпатических влияний на органы и ткани, т. е. : 6. повышение тонуса артериальных сосудов и работы сердца, а значит, повышение АД.
 условнорефлекторные; б) безусловнорефлекторные. Условнорефлекторные АГ развиваются при") Рефлекторные АГ делятся на 2 группы: а) условнорефлекторные; б) безусловнорефлекторные. Условнорефлекторные АГ развиваются при неоднократном сочетании действия индифферентных ( «условных» ) факторов с факторами, повышающими АД. В дальнейшем повышение АД возникает при действии только индифферентного раздражителя. Например, прием кофеина, алкоголя, психостимуляторов и др. перед публичным выступлением, соревнованием, лекцией. В дальнейшем только мысль о лекции вызывает повышение АД. Безусловнорефлекторные АГ могут развиваться по 2 механизмам: • а) в результате хронического раздражения интеро- и экстеро. RR или нервных стволов (при каузалгии, патологии тройничного, лицевого, седалищного нервов, т. е. при длительном болевом синдроме); • б) вследствие прекращения афферентной импульсации, оказывающей в норме тормозящее (депрессорное) действие на сосудодвигательный центр.
Рефлекторные АГ делятся на 2 группы: а) условнорефлекторные; б) безусловнорефлекторные. Условнорефлекторные АГ развиваются при неоднократном сочетании действия индифферентных ( «условных» ) факторов с факторами, повышающими АД. В дальнейшем повышение АД возникает при действии только индифферентного раздражителя. Например, прием кофеина, алкоголя, психостимуляторов и др. перед публичным выступлением, соревнованием, лекцией. В дальнейшем только мысль о лекции вызывает повышение АД. Безусловнорефлекторные АГ могут развиваться по 2 механизмам: • а) в результате хронического раздражения интеро- и экстеро. RR или нервных стволов (при каузалгии, патологии тройничного, лицевого, седалищного нервов, т. е. при длительном болевом синдроме); • б) вследствие прекращения афферентной импульсации, оказывающей в норме тормозящее (депрессорное) действие на сосудодвигательный центр.
, выделяют еще ряд категорий (встречаются") Кроме 3 основных групп симптоматических АГ (почечные, эндокринные, нейрогенные), выделяют еще ряд категорий (встречаются реже), в частности: • Гемические АГ вследствие увеличения массы циркулирующей крови и/или ее вязкости, например, при полицитемиях или гиперпротеинемиях. • Алкогольная АГ (5 -25% всех АГ). Вследствие стимуляция этанолом и ацетальдегидом синтеза и секреции КХА и ГК. • Лекарственные АГ, в частности: q адреномиметики суживают сосуды и увеличивают работу сердца; q пероральные контрацептивы за счет содержания эстрогенов активируют РААС и задерживают жидкость, следовательно, увеличивают ОЦК; q НПВС снижают синтез ПГ, обладающих сосудорасширяющим действием; q ГК повышают реактивность сосудов по отношению к АТ II и НА, кроме того, задерживают жидкость, следовательно, увеличивают ОЦК;
Кроме 3 основных групп симптоматических АГ (почечные, эндокринные, нейрогенные), выделяют еще ряд категорий (встречаются реже), в частности: • Гемические АГ вследствие увеличения массы циркулирующей крови и/или ее вязкости, например, при полицитемиях или гиперпротеинемиях. • Алкогольная АГ (5 -25% всех АГ). Вследствие стимуляция этанолом и ацетальдегидом синтеза и секреции КХА и ГК. • Лекарственные АГ, в частности: q адреномиметики суживают сосуды и увеличивают работу сердца; q пероральные контрацептивы за счет содержания эстрогенов активируют РААС и задерживают жидкость, следовательно, увеличивают ОЦК; q НПВС снижают синтез ПГ, обладающих сосудорасширяющим действием; q ГК повышают реактивность сосудов по отношению к АТ II и НА, кроме того, задерживают жидкость, следовательно, увеличивают ОЦК;
 АРТЕРИАЛЬНЫЕ ГИПОТЕНЗИИ Артериальная гипотензия – снижение АД ниже нормы. Различают 2 вида артериальной гипотензии: физиологическая и патологическая. Физиологическая артериальная гипотензия, в свою очередь, делится на 3 категории: 1. Индивидуальный вариант нормы (так называемое нормальное низкое АД); 2. Спортивная артериальная гипотензия (артериальная гипотензия высокой тренированности); 3. Адаптивная артериальная гипотензия (компенсаторная) – у жителей высокогорья, тропиков, Заполярья.
АРТЕРИАЛЬНЫЕ ГИПОТЕНЗИИ Артериальная гипотензия – снижение АД ниже нормы. Различают 2 вида артериальной гипотензии: физиологическая и патологическая. Физиологическая артериальная гипотензия, в свою очередь, делится на 3 категории: 1. Индивидуальный вариант нормы (так называемое нормальное низкое АД); 2. Спортивная артериальная гипотензия (артериальная гипотензия высокой тренированности); 3. Адаптивная артериальная гипотензия (компенсаторная) – у жителей высокогорья, тропиков, Заполярья.
 и хроническую.") • Патологическая артериальная гипотензия делится на: острую (при шоке и коллапсе) и хроническую. • Хроническая делится на • первичную (гипотоническая болезнь) и • вторичные (симптоматические) артериальные гипотензии. Среди вторичных артериальных гипотензий выделяют: нейрогенные, эндокринные, метаболические.
• Патологическая артериальная гипотензия делится на: острую (при шоке и коллапсе) и хроническую. • Хроническая делится на • первичную (гипотоническая болезнь) и • вторичные (симптоматические) артериальные гипотензии. Среди вторичных артериальных гипотензий выделяют: нейрогенные, эндокринные, метаболические.
 Коллапс – острая сосудистая недостаточность Причин резкого снижения сосудистого тонуса и ОЦК довольно много. В соответствии с причиной различают следующие виды коллапса: 1. Токсико-инфекционный. Развивается при кишечных инфекциях, возбудители которых выделяют эндотоксин, поражающий нервно-мышечный аппарат стенок сосудов. 2. Гипоксический (аноксический). Развивается при быстром снижении парциального давления кислорода во вдыхаемом воздухе и, как следствие, снижения тонуса сосудистой стенки. 3. Постгеморрагический. Развивается при острой массивной кровопотере по гипоксическому механизму (см выше), а также вследствие резкого уменьшения ОЦК. 4. Панкреатический. Развивается при панкреонекрозе. Трипсин непосредственно снижает тонус ГМК сосудов. 5. Ортостатический. Развивается при резком переходе из горизонтального в вертикальное положение после операций на органах брюшной полости, при опухолях и некоторых заболеваниях головного мозга (нейросифилисе, паркинсонизме, сирингомиелии), при диабетической нейропатии, синдроме каротидного синуса и др. В этих случаях не успевает сработать барорецепторный рефлекс. 6. Коллапс при неправильном назначении лекарств, понижающих АД (нейролептиков, периферических вазодилататоров, адреноблокаторов, ганглиоблокаторов и др. ).
Коллапс – острая сосудистая недостаточность Причин резкого снижения сосудистого тонуса и ОЦК довольно много. В соответствии с причиной различают следующие виды коллапса: 1. Токсико-инфекционный. Развивается при кишечных инфекциях, возбудители которых выделяют эндотоксин, поражающий нервно-мышечный аппарат стенок сосудов. 2. Гипоксический (аноксический). Развивается при быстром снижении парциального давления кислорода во вдыхаемом воздухе и, как следствие, снижения тонуса сосудистой стенки. 3. Постгеморрагический. Развивается при острой массивной кровопотере по гипоксическому механизму (см выше), а также вследствие резкого уменьшения ОЦК. 4. Панкреатический. Развивается при панкреонекрозе. Трипсин непосредственно снижает тонус ГМК сосудов. 5. Ортостатический. Развивается при резком переходе из горизонтального в вертикальное положение после операций на органах брюшной полости, при опухолях и некоторых заболеваниях головного мозга (нейросифилисе, паркинсонизме, сирингомиелии), при диабетической нейропатии, синдроме каротидного синуса и др. В этих случаях не успевает сработать барорецепторный рефлекс. 6. Коллапс при неправильном назначении лекарств, понижающих АД (нейролептиков, периферических вазодилататоров, адреноблокаторов, ганглиоблокаторов и др. ).
 • Гипотоническая болезнь - самостоятельное заболевание. Причины и механизмы ее развития изучены недостаточно. Предположительно повторный стресс ведет к развитию невроза с формированием корково-подкорковой доминанты возбуждения, распространяющегося на передние ядра гипоталамуса и другие структуры парасимпатической нервной системы, что ведет к снижению тонуса резистивных сосудов, снижению сократимости миокарда и, следовательно, МОС. Расширение сосудов и снижение МОС являются непосредственными причинами снижения АД. • Клинически гипотоническая болезнь проявляется стойким снижением АД, астеническим синдромом, нарушениями сна, эмоциональной и сексуальной сферы, диспепсическим синдромом, нередко кардиалгией, миалгией, артралгией, и комплексом вегетативных нарушений (гипергидроз, зябкость, парестезии, лабильность температуры тела, ЧСС и АД, судороги и др. ). Фактически это клиника вегето-сосудистой дистонии.
• Гипотоническая болезнь - самостоятельное заболевание. Причины и механизмы ее развития изучены недостаточно. Предположительно повторный стресс ведет к развитию невроза с формированием корково-подкорковой доминанты возбуждения, распространяющегося на передние ядра гипоталамуса и другие структуры парасимпатической нервной системы, что ведет к снижению тонуса резистивных сосудов, снижению сократимости миокарда и, следовательно, МОС. Расширение сосудов и снижение МОС являются непосредственными причинами снижения АД. • Клинически гипотоническая болезнь проявляется стойким снижением АД, астеническим синдромом, нарушениями сна, эмоциональной и сексуальной сферы, диспепсическим синдромом, нередко кардиалгией, миалгией, артралгией, и комплексом вегетативных нарушений (гипергидроз, зябкость, парестезии, лабильность температуры тела, ЧСС и АД, судороги и др. ). Фактически это клиника вегето-сосудистой дистонии.
 • Нейрогенные артериальные гипотензии делятся на центрогенные и рефлекторные. • Центрогенные артериальные гипотензии развиваются при органическом поражении головного мозга (черепно-мозговая травма, нарушения мозгового кровообращения, дегенеративные заболевания нервной системы и др. ) с вовлечением структур, участвующих в регуляции АД. Механизм снижения АД при этом связан с абсолютным или относительным преобладанием эффектов парасимпатической нервной системы на сердце и сосуды, следовательно, снижением МОС и тонуса сосудов. • Рефлекторные артериальные гипотензии связаны с нарушением проведения эфферентных сосудосуживающих импульсов от сосудодвигательного центра к стенкам сосудов и кардиотонических импульсов к сердцу (бывает при нейросифилисе, сирингомиелии, диабетической полинейропатии и др. ), что ведет к снижению ОПСС и МОС, а значит, к снижению АД.
• Нейрогенные артериальные гипотензии делятся на центрогенные и рефлекторные. • Центрогенные артериальные гипотензии развиваются при органическом поражении головного мозга (черепно-мозговая травма, нарушения мозгового кровообращения, дегенеративные заболевания нервной системы и др. ) с вовлечением структур, участвующих в регуляции АД. Механизм снижения АД при этом связан с абсолютным или относительным преобладанием эффектов парасимпатической нервной системы на сердце и сосуды, следовательно, снижением МОС и тонуса сосудов. • Рефлекторные артериальные гипотензии связаны с нарушением проведения эфферентных сосудосуживающих импульсов от сосудодвигательного центра к стенкам сосудов и кардиотонических импульсов к сердцу (бывает при нейросифилисе, сирингомиелии, диабетической полинейропатии и др. ), что ведет к снижению ОПСС и МОС, а значит, к снижению АД.
 • Эндокринные артериальные гипотензии • В основе эндокринных артериальных гипотензий лежит дефицит гормонов, поддерживающих в норме уровень АД. • Это может наблюдаться при патологии надпочечников, гипофиза, щитовидной железы. Гипоплазия (недоразвитие) или разрушение паренхимы надпочечников (при опухоли, туберкулезе надпочечников, кровоизлиянии в ткань железы и др. ). ведет к недостаточной выработке КХА, минерало- и глюкокортикоидов, следовательно, к снижению тонуса сосудов, ОЦК и МОС, а значит – к снижению АД. Механизмы понижения АД при гипофункции гипофиза связаны с дефицитом АДГ, АКТГ, ТТГ, СТГ. Снижение АД при гипотиреозе связано с дефицитом Т 3, Т 4, а значит недостатком эффектов этих гормонов на ино- и хронотропную функции сердца и, как следствие, снижением МОС. Дополнительное значение имеет снижение тонуса сосудов из-за развития дистрофических процессов в них.
• Эндокринные артериальные гипотензии • В основе эндокринных артериальных гипотензий лежит дефицит гормонов, поддерживающих в норме уровень АД. • Это может наблюдаться при патологии надпочечников, гипофиза, щитовидной железы. Гипоплазия (недоразвитие) или разрушение паренхимы надпочечников (при опухоли, туберкулезе надпочечников, кровоизлиянии в ткань железы и др. ). ведет к недостаточной выработке КХА, минерало- и глюкокортикоидов, следовательно, к снижению тонуса сосудов, ОЦК и МОС, а значит – к снижению АД. Механизмы понижения АД при гипофункции гипофиза связаны с дефицитом АДГ, АКТГ, ТТГ, СТГ. Снижение АД при гипотиреозе связано с дефицитом Т 3, Т 4, а значит недостатком эффектов этих гормонов на ино- и хронотропную функции сердца и, как следствие, снижением МОС. Дополнительное значение имеет снижение тонуса сосудов из-за развития дистрофических процессов в них.
 • Метаболические артериальные гипотензии • Встречаются редко, связаны с нарушением метаболизма, точнее – со снижением синтеза веществ, обладающих гипертензивным действием: ангиотензина II, эндотелина, тромбоксана А 2. Причинами снижения синтеза прессорных веществ могут быть гипогидратация (обезвоживание), дистрофические изменения в тканях при хронических интоксикациях, инфекциях, голодании. Механизмы снижения АД при этом связаны со снижением тонуса сосудов, и следовательно, ОПСС; снижением сократимости миокарда, и следовательно, МОС; снижением ОЦК.
• Метаболические артериальные гипотензии • Встречаются редко, связаны с нарушением метаболизма, точнее – со снижением синтеза веществ, обладающих гипертензивным действием: ангиотензина II, эндотелина, тромбоксана А 2. Причинами снижения синтеза прессорных веществ могут быть гипогидратация (обезвоживание), дистрофические изменения в тканях при хронических интоксикациях, инфекциях, голодании. Механизмы снижения АД при этом связаны со снижением тонуса сосудов, и следовательно, ОПСС; снижением сократимости миокарда, и следовательно, МОС; снижением ОЦК.