

Острые лейкозы 2013.ppt
- Количество слайдов: 89
 Острые лейкозы Лисуков Игорь Андреевич д. м. н. профессор кафедра госпитальной терапии СПГМУ им. академика И. П. Павлова
Острые лейкозы Лисуков Игорь Андреевич д. м. н. профессор кафедра госпитальной терапии СПГМУ им. академика И. П. Павлова
 Определение лейкоза • Лейкозы – группа неопластических клональных заболеваний, при которых мутантный опухолевый клон происходит из клеток гемопоэтического ряда и первично возникает в костном мозге; • Острые лейкозы: опухолевые клетки теряют способность к дифференцировке; • Хронические лейкозы: способность опухолевых клеток к дифференцировке сохраняется;
Определение лейкоза • Лейкозы – группа неопластических клональных заболеваний, при которых мутантный опухолевый клон происходит из клеток гемопоэтического ряда и первично возникает в костном мозге; • Острые лейкозы: опухолевые клетки теряют способность к дифференцировке; • Хронические лейкозы: способность опухолевых клеток к дифференцировке сохраняется;
 Острые лейкозы • Злокачественные клональные опухоли кроветворной ткани, субстратом которых являются клетки-предшественницы гемопоэза
Острые лейкозы • Злокачественные клональные опухоли кроветворной ткани, субстратом которых являются клетки-предшественницы гемопоэза
 патолог больницы Шарите в Берлине.") Рудольф Вирхов (1821 -1902) патолог больницы Шарите в Берлине.
Рудольф Вирхов (1821 -1902) патолог больницы Шарите в Берлине.
 • 1847 г. R. Virchow , термин «лейкемия» , описание морфологических вариантов • 1891 г. P. Erlich, дифференцированная окраска бластных клеток, цитохимические исследования • 1902 г. А. А. Максимов, клональность лейкозов, лейкемические клоногенные предшественники
• 1847 г. R. Virchow , термин «лейкемия» , описание морфологических вариантов • 1891 г. P. Erlich, дифференцированная окраска бластных клеток, цитохимические исследования • 1902 г. А. А. Максимов, клональность лейкозов, лейкемические клоногенные предшественники
 • 1921 г. – Эллерман предложил более точный термин «лейкоз» , который означает системную пролиферацию кроветворной ткани, вне зависимости от показателей периферической крови;
• 1921 г. – Эллерман предложил более точный термин «лейкоз» , который означает системную пролиферацию кроветворной ткани, вне зависимости от показателей периферической крови;
 Острые лейкозы ОЛ - гетерогенная группа опухолевых заболеваний системы крови. Характеризуются первичным поражением костного мозга морфологически незрелыми (бластными) клетками, вытеснением нормального гемопоэза и инфильтрацией различных органов и тканей. ОЛ составляют 3% злокачественных опухолей человека, 5 случаев на 100 000 населения в год. 75% заболевших – взрослые пациенты.
Острые лейкозы ОЛ - гетерогенная группа опухолевых заболеваний системы крови. Характеризуются первичным поражением костного мозга морфологически незрелыми (бластными) клетками, вытеснением нормального гемопоэза и инфильтрацией различных органов и тканей. ОЛ составляют 3% злокачественных опухолей человека, 5 случаев на 100 000 населения в год. 75% заболевших – взрослые пациенты.
 Острые лейкозы ОЛ - клональные заболевания крови. Лейкозные клетки часто экспрессируют на своей поверхности маркеры, характеризующие определенные этапы дифференцировки нормальных гемопоэтических клеток. Часто отмечается аберрантная экспрессия дифференцировочных антигенов. Встречаются ОЛ, клетки которых несут маркеры разных линий гемопоэза или различных уровней дифференцировки.
Острые лейкозы ОЛ - клональные заболевания крови. Лейкозные клетки часто экспрессируют на своей поверхности маркеры, характеризующие определенные этапы дифференцировки нормальных гемопоэтических клеток. Часто отмечается аберрантная экспрессия дифференцировочных антигенов. Встречаются ОЛ, клетки которых несут маркеры разных линий гемопоэза или различных уровней дифференцировки.
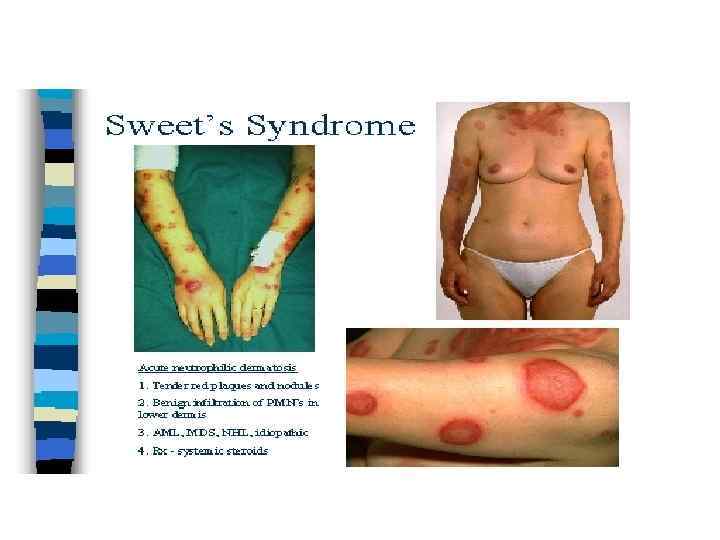
 Клинические проявления острых лейкозов 1. 2. 3. 4. 5. 6. 7. 8. Цитопенические синдромы (анемия, тромбоцитопения, нейтропения) Инфильтрация органов и тканей лейкозными клетками (лимфоузы, печень, селезенка, лейкемиды на коже, мозговые оболочки, ЦНС, яички при ОЛЛ) Синдром Свита (острый фебрильный дерматоз) Инфекционные осложнения (бактериальные: кожные покровы, легкие, глотка; микозы; вирусные инфекции) Геморрагический синдром (тяжелое течение при ОМЛ М 3) Боли в костях, артралгии Нейролейкоз (головная боль, ригидность затылочных мышц, парезы ЧМН, парезы нижних конечностей) Гипертрофия десен ( ОМЛ М 4, 5)
Клинические проявления острых лейкозов 1. 2. 3. 4. 5. 6. 7. 8. Цитопенические синдромы (анемия, тромбоцитопения, нейтропения) Инфильтрация органов и тканей лейкозными клетками (лимфоузы, печень, селезенка, лейкемиды на коже, мозговые оболочки, ЦНС, яички при ОЛЛ) Синдром Свита (острый фебрильный дерматоз) Инфекционные осложнения (бактериальные: кожные покровы, легкие, глотка; микозы; вирусные инфекции) Геморрагический синдром (тяжелое течение при ОМЛ М 3) Боли в костях, артралгии Нейролейкоз (головная боль, ригидность затылочных мышц, парезы ЧМН, парезы нижних конечностей) Гипертрофия десен ( ОМЛ М 4, 5)
 Нормальное количество") Диагностика ОЛ. Клинический анализ крови. • • • Лейкоцитоз (55 -65% больных) Нормальное количество лейкоцитов (15%) Нейтропения (30 -40%) Анемия (85 -90%) Тромбоцитопения (80 -90%) Бластемия (85 -95%)
Диагностика ОЛ. Клинический анализ крови. • • • Лейкоцитоз (55 -65% больных) Нормальное количество лейкоцитов (15%) Нейтропения (30 -40%) Анемия (85 -90%) Тромбоцитопения (80 -90%) Бластемия (85 -95%)
 Дифференциальный диагноз • • Апластическая анемия ЛПЗ, МПЗ МДС Гиперспленизм Иммунные цитопении (коллагенозы, АГА, ИТП) Инфекции Мегалобластные анемии ОЛБ
Дифференциальный диагноз • • Апластическая анемия ЛПЗ, МПЗ МДС Гиперспленизм Иммунные цитопении (коллагенозы, АГА, ИТП) Инфекции Мегалобластные анемии ОЛБ
 Взаимосвязь синдромов недостаточности костного мозга БГЛ ПНГ МДС/ПНГ АФ ВД АА/ПНГ АА АА/МДС ОМЛ АДБ Конституциональные Иммуноопосредованные Неопластические
Взаимосвязь синдромов недостаточности костного мозга БГЛ ПНГ МДС/ПНГ АФ ВД АА/ПНГ АА АА/МДС ОМЛ АДБ Конституциональные Иммуноопосредованные Неопластические
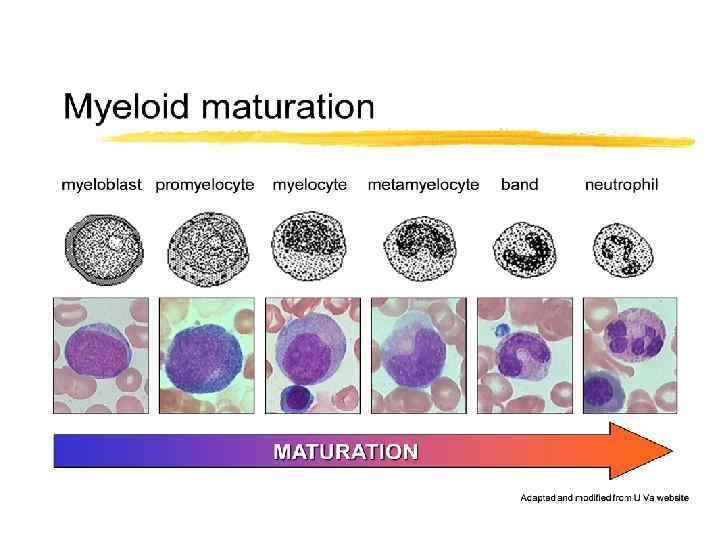
 Принципы диагностики острых лейкозов • 1. Морфологическое исследование периферической крови и костного мозга – бластоз. • 2. Цитохимические характеристики бластных клеток. • 3. Иммунофенотипирование бластных клеток. • 4. Цитогенетические исследования. • 5. Молекулярно-биологические исследования.
Принципы диагностики острых лейкозов • 1. Морфологическое исследование периферической крови и костного мозга – бластоз. • 2. Цитохимические характеристики бластных клеток. • 3. Иммунофенотипирование бластных клеток. • 4. Цитогенетические исследования. • 5. Молекулярно-биологические исследования.
 Критерии диагностики ОЛ • Количество бластов в КМ: FAB классификация > 30% ВОЗ > 20% Клональные цитогенетические нарушения могут верифицироваться как ОЛ независимо от количества бластных клеток в костном мозге и периферической крови
Критерии диагностики ОЛ • Количество бластов в КМ: FAB классификация > 30% ВОЗ > 20% Клональные цитогенетические нарушения могут верифицироваться как ОЛ независимо от количества бластных клеток в костном мозге и периферической крови
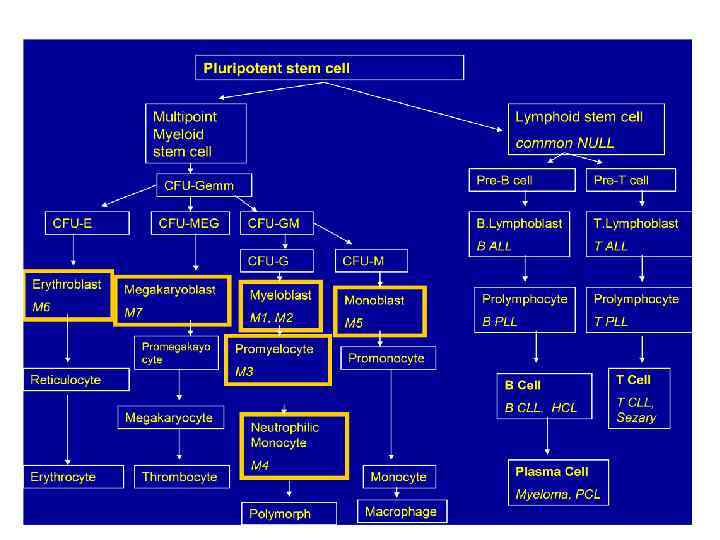
 Острые лейкозы миелоидные варианты : лимфоидные варианты 6: 1 Медиана возраста: ОМЛ 60 -65 лет ОЛЛ 10 лет
Острые лейкозы миелоидные варианты : лимфоидные варианты 6: 1 Медиана возраста: ОМЛ 60 -65 лет ОЛЛ 10 лет
 Острые миелоидные лейкозы • Термин ОМЛ объединяет группу ОЛ, возникших из клетки-предшественницы миелопоэза и различающихся между собой определенными морфологическими, цитохимическими, иммунофенотипическими и цитогенетическими характеристиками.
Острые миелоидные лейкозы • Термин ОМЛ объединяет группу ОЛ, возникших из клетки-предшественницы миелопоэза и различающихся между собой определенными морфологическими, цитохимическими, иммунофенотипическими и цитогенетическими характеристиками.
 • M 0 - острый недифференцированный лейкоз inv(3 q 26), t(3;") Классификация ОМЛ (FAB) • M 0 - острый недифференцированный лейкоз inv(3 q 26), t(3; 3) • М 1 - острый миелобластный лейкоз без признаков созревания • М 2 -острый миелобластный лейкоз с признаками созревания t(8; 21), t(6; 9) • М 3 - острый промиелоцитарный лейкоз t(15, 17) 98% t(11; 17) 1% t(5; 17)1% • М 4 - острый миеломонобластный лейкоз • М 5 - острый монобластный лейкоз • М 6 - острый эритробластный лейкоз • М 7 - острый мегакариобластный лейкоз
Классификация ОМЛ (FAB) • M 0 - острый недифференцированный лейкоз inv(3 q 26), t(3; 3) • М 1 - острый миелобластный лейкоз без признаков созревания • М 2 -острый миелобластный лейкоз с признаками созревания t(8; 21), t(6; 9) • М 3 - острый промиелоцитарный лейкоз t(15, 17) 98% t(11; 17) 1% t(5; 17)1% • М 4 - острый миеломонобластный лейкоз • М 5 - острый монобластный лейкоз • М 6 - острый эритробластный лейкоз • М 7 - острый мегакариобластный лейкоз
 (1) • 1. ОМЛ с цитогенетическими транслокациями ОМЛ с t") Классификация ОМЛ ВОЗ (2006) (1) • 1. ОМЛ с цитогенетическими транслокациями ОМЛ с t (8; 21)(q 22; q 22), (AML 1/ETO) ОМЛ с нарушениями эозинофилопоэза inv(16) или t(16; 16) Острый промиелоцитарный лейкоз с t(15; 17) • 2. ОМЛ с миелоидной дисплазией • 3. ОМЛ, связанный с предшествующей терапией Алкилирующими препаратами Ингибиторы топоизомеразы II типа Другие
Классификация ОМЛ ВОЗ (2006) (1) • 1. ОМЛ с цитогенетическими транслокациями ОМЛ с t (8; 21)(q 22; q 22), (AML 1/ETO) ОМЛ с нарушениями эозинофилопоэза inv(16) или t(16; 16) Острый промиелоцитарный лейкоз с t(15; 17) • 2. ОМЛ с миелоидной дисплазией • 3. ОМЛ, связанный с предшествующей терапией Алкилирующими препаратами Ингибиторы топоизомеразы II типа Другие
 (2) • 4. ОМЛ, не попадающий под вышеперечисленные категории ОМЛ") Классификация ОМЛ ВОЗ (2006) (2) • 4. ОМЛ, не попадающий под вышеперечисленные категории ОМЛ с минимальной дифференцировкой ОМЛ без созревания ОМЛ с созреванием О. миеломоноцитарный лейкоз О. эритроцитарный лейкоз О. мегакариоцитарный лейкоз О. базофильный лейкоз О. панмиелоз с миелофиброзом О. бифенонотипический лейкоз
Классификация ОМЛ ВОЗ (2006) (2) • 4. ОМЛ, не попадающий под вышеперечисленные категории ОМЛ с минимальной дифференцировкой ОМЛ без созревания ОМЛ с созреванием О. миеломоноцитарный лейкоз О. эритроцитарный лейкоз О. мегакариоцитарный лейкоз О. базофильный лейкоз О. панмиелоз с миелофиброзом О. бифенонотипический лейкоз
 Цитохимические реакции в диагностике ОЛ Цитохимия M 1 -M 3 M 4 -M 5 M 6 -M 7 ОЛЛ МПА +/++ + - - Судан черный +/++ + - - Неспецифич. эстераза - ++ +(очаговая) - ШИК-реакция - + +(мелкие +(глыбки) - + (диффузная) +(очаговая) Кислая фосфатаза гранулы) -
Цитохимические реакции в диагностике ОЛ Цитохимия M 1 -M 3 M 4 -M 5 M 6 -M 7 ОЛЛ МПА +/++ + - - Судан черный +/++ + - - Неспецифич. эстераза - ++ +(очаговая) - ШИК-реакция - + +(мелкие +(глыбки) - + (диффузная) +(очаговая) Кислая фосфатаза гранулы) -
 11 13 14 15 M") Дифференцировочные антигены при ОМЛ Вариант Антигены кластеров дифференцировки (CD) 11 13 14 15 M 0 - + M 1 - + M 2 + + +/- M 3 + + - M 4 + + M 5 +/- M 6 - - M 7 - - 33 34 41 + + + + - +/_ + + + - - - - + - - - + + + 42 b - - - + HLA-DR - - - + - +
Дифференцировочные антигены при ОМЛ Вариант Антигены кластеров дифференцировки (CD) 11 13 14 15 M 0 - + M 1 - + M 2 + + +/- M 3 + + - M 4 + + M 5 +/- M 6 - - M 7 - - 33 34 41 + + + + - +/_ + + + - - - - + - - - + + + 42 b - - - + HLA-DR - - - + - +
 Бласты характеризуются") Иммунофенотипирование костного мозга методом проточной цитометрии Острый нелимфобластный лейкоз (М 3 вариант) Бласты характеризуются снижением экспрессии панлейкоцитарного антигена CD 45, гранулярностью, экспрессией миелоидных антигенов CD 13, CD 33, CD 15, МПО, CD 117, CD 64. Особенностью острого промиелоцитарного лейкоза является отсутствие экспрессии HLA -DR, CD 11 a, CD 11 b, CD 11 c, характерна гетерогенность экспрессии CD 13, коэкспрессия лимфоидных маркеров CD 2, CD 9
Иммунофенотипирование костного мозга методом проточной цитометрии Острый нелимфобластный лейкоз (М 3 вариант) Бласты характеризуются снижением экспрессии панлейкоцитарного антигена CD 45, гранулярностью, экспрессией миелоидных антигенов CD 13, CD 33, CD 15, МПО, CD 117, CD 64. Особенностью острого промиелоцитарного лейкоза является отсутствие экспрессии HLA -DR, CD 11 a, CD 11 b, CD 11 c, характерна гетерогенность экспрессии CD 13, коэкспрессия лимфоидных маркеров CD 2, CD 9
 ОМЛ М 0 недифференцированный острый миелоидный лейкоз Миелопероксидаза и липиды отрицательные, PAS –реакция положительная в гранулярной форме в единичных бластах, CD 34+, CD 13+, CD 33+ Del 5, 7 t(9; 22) 3% ОНЛЛ
ОМЛ М 0 недифференцированный острый миелоидный лейкоз Миелопероксидаза и липиды отрицательные, PAS –реакция положительная в гранулярной форме в единичных бластах, CD 34+, CD 13+, CD 33+ Del 5, 7 t(9; 22) 3% ОНЛЛ
 ОМЛ M 1 без признаков созревания МПО + СD 33+, 13+, 65+, 117+ t(8; 21) 16% ОНЛЛ
ОМЛ M 1 без признаков созревания МПО + СD 33+, 13+, 65+, 117+ t(8; 21) 16% ОНЛЛ
 ОМЛ M 2 с признаками созревания. Бласты содержат большое количество гранул, ядро может иметь нуклеолы. Палочки Ауэра. Спленомегалия Хлоромы (миелоидная саркома мягких тканей) t(8; 22) 16% ОНЛЛ
ОМЛ M 2 с признаками созревания. Бласты содержат большое количество гранул, ядро может иметь нуклеолы. Палочки Ауэра. Спленомегалия Хлоромы (миелоидная саркома мягких тканей) t(8; 22) 16% ОНЛЛ
 ОМЛ М 3 промиелоцитарный лейкоз Гипергранулярные бласты МПО+ CD 13+, 33+, 9+, 11 в+ t (15; 17)
ОМЛ М 3 промиелоцитарный лейкоз Гипергранулярные бласты МПО+ CD 13+, 33+, 9+, 11 в+ t (15; 17)
 ОМЛ М 4 миеломонобластный лейкоз Биклональный МПО+ СD 13+, 14+, 15+ Гипертрофия десен t(8; 21) 17% ОНЛЛ
ОМЛ М 4 миеломонобластный лейкоз Биклональный МПО+ СD 13+, 14+, 15+ Гипертрофия десен t(8; 21) 17% ОНЛЛ
 М 4 Е 0 миеломонобластный лейкоз с эозинофилией • 1% ОНЛЛ бласты представлены миело и монобластами, число эозинофилов в к/м более 6% • > 90% - инверсия 16 хромосомы, t(16; 16) с точкой разрыва 16 q 22 • Хороший прогноз
М 4 Е 0 миеломонобластный лейкоз с эозинофилией • 1% ОНЛЛ бласты представлены миело и монобластами, число эозинофилов в к/м более 6% • > 90% - инверсия 16 хромосомы, t(16; 16) с точкой разрыва 16 q 22 • Хороший прогноз
 ОМЛ М 5 монобластный лейкоз • > 20% бластов имеют характерную моноцитоидную формую • МПО+, HLA-DR+, CD 33+, CD 14+, CD 15+ перестройка 11 q 23, t(9; 11), t(6; 11) – MLL ген Лейкемиды кожи, гипертрофия десен, высокий лейкоцитоз, плохой прогноз 14% ОНЛЛ
ОМЛ М 5 монобластный лейкоз • > 20% бластов имеют характерную моноцитоидную формую • МПО+, HLA-DR+, CD 33+, CD 14+, CD 15+ перестройка 11 q 23, t(9; 11), t(6; 11) – MLL ген Лейкемиды кожи, гипертрофия десен, высокий лейкоцитоз, плохой прогноз 14% ОНЛЛ
 ОМЛ M 6 эритробластный лейкоз • Бласты представлены 2 типами клеток: эритробластами и миелобластами (М 1), увеличено число эритроидных клеток с признаками морфологической и цитохимической дисплазии ГЛИКОФОРИН А+ , CD 34+, CD 38+, CD 71+, CD 33+ • Делеция 5 и 7 хромосом • Плохой прогноз, в анамнезе часто МДС • 5% всех ОМЛ
ОМЛ M 6 эритробластный лейкоз • Бласты представлены 2 типами клеток: эритробластами и миелобластами (М 1), увеличено число эритроидных клеток с признаками морфологической и цитохимической дисплазии ГЛИКОФОРИН А+ , CD 34+, CD 38+, CD 71+, CD 33+ • Делеция 5 и 7 хромосом • Плохой прогноз, в анамнезе часто МДС • 5% всех ОМЛ
 ОМЛ М 7 мегакариобластный лейкоз • Иммунофенотип: CD 41 а+, CD 61+ • Цитогенетика: трисомия 21, инверсии или транслокации 3, t(9; 22) • Может встречаться при синдроме Кланфельда XX • Плохой прогноз • 1% ОНЛЛ
ОМЛ М 7 мегакариобластный лейкоз • Иммунофенотип: CD 41 а+, CD 61+ • Цитогенетика: трисомия 21, инверсии или транслокации 3, t(9; 22) • Может встречаться при синдроме Кланфельда XX • Плохой прогноз • 1% ОНЛЛ
 Прогностические факторы ОМЛ • Giles R. Hematology, 1, 2002 Кариотип % % CR % EFS 5 -10 90 50 -70 5 -10 80 -90 70 40 -50 70 -80 20 -40 -5/-7 20 -30 40 5 -10 +8, FLT 3 мутации 10 60 10 -20 t(9; 11)q 23; 20 q-, др 10 -120 60 10 Благоприятный t(8; 21) Инверсии 16 t (15; 17) Промежуточный diploid Неблагоприятный
Прогностические факторы ОМЛ • Giles R. Hematology, 1, 2002 Кариотип % % CR % EFS 5 -10 90 50 -70 5 -10 80 -90 70 40 -50 70 -80 20 -40 -5/-7 20 -30 40 5 -10 +8, FLT 3 мутации 10 60 10 -20 t(9; 11)q 23; 20 q-, др 10 -120 60 10 Благоприятный t(8; 21) Инверсии 16 t (15; 17) Промежуточный diploid Неблагоприятный
 Общая выживаемость в разных группах риска Благоприятный Промежуточный Неблагоприятный
Общая выживаемость в разных группах риска Благоприятный Промежуточный Неблагоприятный
 Встречаемость мутаций без нарушения кариотипа
Встречаемость мутаций без нарушения кариотипа
(q 22; q 22);") Прогноз при различных мутациях и транслокациях Группа Аномалия Благоприятная t(8; 21)(q 22; q 22); RUNX 1 -RUNX 1 T 1 inv(16)(p 13. 1 q 22) or t(16; 16)(p 13. 1; q 22); CBFB-MYH 11 Мутация NPM 1 без FLT 3 -ITD (normal karyotype) Мутация CEBPA (normal karyotype) Промежуточная-1 Мутация NPM 1 and FLT 3 -ITD (normal karyotype) Wild-type NPM 1 without FLT 3 -ITD (normal karyotype Промежуточная-2 t(9; 11)(p 22; q 23); MLLT 3 -MLL + неклассифицируемые аномалии Неблагоприятная inv(3)(q 21 q 26. 2) or t(3; 3)(q 21; q 26. 2); RPN 1 -EVI 1 t(6; 9)(p 23; q 34); DEK-NUP 214 t(v; 11)(v; q 23); complex karyotype
Прогноз при различных мутациях и транслокациях Группа Аномалия Благоприятная t(8; 21)(q 22; q 22); RUNX 1 -RUNX 1 T 1 inv(16)(p 13. 1 q 22) or t(16; 16)(p 13. 1; q 22); CBFB-MYH 11 Мутация NPM 1 без FLT 3 -ITD (normal karyotype) Мутация CEBPA (normal karyotype) Промежуточная-1 Мутация NPM 1 and FLT 3 -ITD (normal karyotype) Wild-type NPM 1 without FLT 3 -ITD (normal karyotype Промежуточная-2 t(9; 11)(p 22; q 23); MLLT 3 -MLL + неклассифицируемые аномалии Неблагоприятная inv(3)(q 21 q 26. 2) or t(3; 3)(q 21; q 26. 2); RPN 1 -EVI 1 t(6; 9)(p 23; q 34); DEK-NUP 214 t(v; 11)(v; q 23); complex karyotype
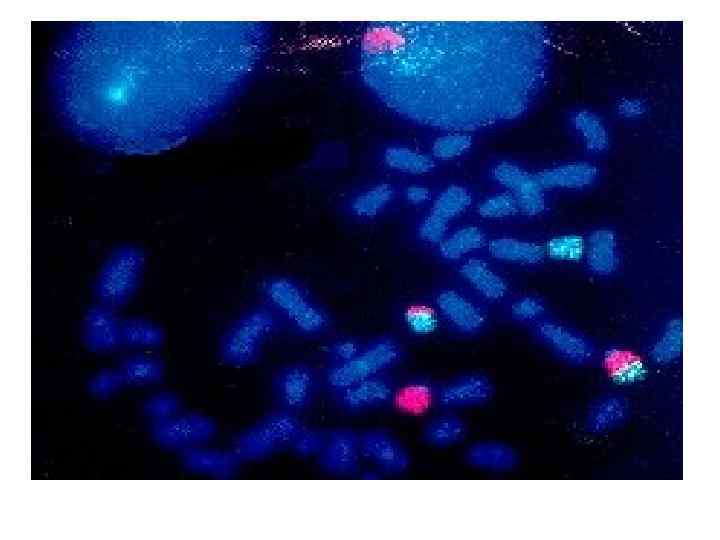
(q 26; q 21), ассоциированная с повреждениями гена EVI-1") Транслокация (3; 3)(q 26; q 21), ассоциированная с повреждениями гена EVI-1
Транслокация (3; 3)(q 26; q 21), ассоциированная с повреждениями гена EVI-1
(q 21 p 32)x 2,") Больной с EVI-1+ ОНЛЛ G-banding M-FISH 72 -75, XY, inv(1)(q 21 p 32)x 2, +3 x 2, +4 x 2, +del(5)(q 13)x 2, +6 x 2, +8, +del(8)(q 11 q 23), +10, +11, +12, +der(13)t(1; 13)(q 21; q 34), , +del(13)(q 11), +14, +15 x 2, +1`6, +17, +18, +1`9 x 2, +20 x 2, +21 x 2, +22 (Т. Л. Гиндина, 2011)
Больной с EVI-1+ ОНЛЛ G-banding M-FISH 72 -75, XY, inv(1)(q 21 p 32)x 2, +3 x 2, +4 x 2, +del(5)(q 13)x 2, +6 x 2, +8, +del(8)(q 11 q 23), +10, +11, +12, +der(13)t(1; 13)(q 21; q 34), , +del(13)(q 11), +14, +15 x 2, +1`6, +17, +18, +1`9 x 2, +20 x 2, +21 x 2, +22 (Т. Л. Гиндина, 2011)
 Экспрессия EVI-1 ассоциируется с плохим прогнозом при ОМЛ Kaplan Meier analysis : A -ОВ B -БСВ, C -БРВ N=574 пациента (Lugthart S. , Blood. 2008; 111: 4329)
Экспрессия EVI-1 ассоциируется с плохим прогнозом при ОМЛ Kaplan Meier analysis : A -ОВ B -БСВ, C -БРВ N=574 пациента (Lugthart S. , Blood. 2008; 111: 4329)
 7+3+VP-16 TAD (тиогуанин, цитарабин,") Лечение ОМЛ • • Индукционная терапия 7+3 (цитарабин + даунорубицин) 7+3+VP-16 TAD (тиогуанин, цитарабин, даунорубицин) HDAC (высокие дозы цитарабина) Консолидация ремиссии • Цитарабин в средних/высоких дозах • Цитарабин в высоких дозах+митоксантрон (HAM)
Лечение ОМЛ • • Индукционная терапия 7+3 (цитарабин + даунорубицин) 7+3+VP-16 TAD (тиогуанин, цитарабин, даунорубицин) HDAC (высокие дозы цитарабина) Консолидация ремиссии • Цитарабин в средних/высоких дозах • Цитарабин в высоких дозах+митоксантрон (HAM)
 Индукция ремиссии ОМЛ • « 7+3» Полная ремиссия достигается в 58 - 64%. ( Bishop J. , 1997) • Цитарабин 100 мг/м 2 х 2/день (д 1 -7) • Даунорубицин 45 мг/м 2 1/день (д 1 -3)
Индукция ремиссии ОМЛ • « 7+3» Полная ремиссия достигается в 58 - 64%. ( Bishop J. , 1997) • Цитарабин 100 мг/м 2 х 2/день (д 1 -7) • Даунорубицин 45 мг/м 2 1/день (д 1 -3)
 Протокол лечения ОМЛ Индукция ремиссии Консолидация Благоприятный прогноз: 1. Стандартная поддерживающая ХТ Промежуточный прогноз: 1. алло ТКМ (sibling donor) Неблагоприятный прогноз: 1. алло ТКМ (sibling, MUD, Haplo) 2. ауто ТКМ 2. алло ТКМ (MUD)
Протокол лечения ОМЛ Индукция ремиссии Консолидация Благоприятный прогноз: 1. Стандартная поддерживающая ХТ Промежуточный прогноз: 1. алло ТКМ (sibling donor) Неблагоприятный прогноз: 1. алло ТКМ (sibling, MUD, Haplo) 2. ауто ТКМ 2. алло ТКМ (MUD)
 Программа 2 -й линии ПХТ FLAG Флюдарабин 30 мг/м 2/д Д 2 -6 30 мин инфузия Цитарабин 2 г/м 2/ д Д 2 -6 ч/з 4 часа после флюдара Г-КСФ 400 мкг С д 1 до нейтрофи лов>1. 0
Программа 2 -й линии ПХТ FLAG Флюдарабин 30 мг/м 2/д Д 2 -6 30 мин инфузия Цитарабин 2 г/м 2/ д Д 2 -6 ч/з 4 часа после флюдара Г-КСФ 400 мкг С д 1 до нейтрофи лов>1. 0
 Основные понятия и критерии • Полная клинико-гематологическая ремиссия. В КМ ≤ 5% бластов при нормальном соотношении всех ростков гемопоэза, нейтрофилы в ПК >1, 5 Х 10 9/л, тромбоциты > 100 Х 10 9/л. Указанные показатели должны сохраняться более 1 месяца. • Цитогенетическая ремиссия. Полная клинико-гематологическая ремиссия, при которой методами стандартной цитогенетики не выявляются исходные аномалии кариотипа. • Молекулярная ремиссия. Полная клинико-гематологическая ремиссия при отсутствии определявшихся ранее маркеров ОЛ (ПЦР).
Основные понятия и критерии • Полная клинико-гематологическая ремиссия. В КМ ≤ 5% бластов при нормальном соотношении всех ростков гемопоэза, нейтрофилы в ПК >1, 5 Х 10 9/л, тромбоциты > 100 Х 10 9/л. Указанные показатели должны сохраняться более 1 месяца. • Цитогенетическая ремиссия. Полная клинико-гематологическая ремиссия, при которой методами стандартной цитогенетики не выявляются исходные аномалии кариотипа. • Молекулярная ремиссия. Полная клинико-гематологическая ремиссия при отсутствии определявшихся ранее маркеров ОЛ (ПЦР).
 Основные понятия и критерии • Резистентная форма ОЛ. Отсутствие полной ремиссии после проведения 2 -х курсов индукционной терапии. • Рецидив. >5% бластов в КМ ± нейролейкемия, спленомегалия, лимфоаденопатия и т. д, ). • Ранний рецидив. Рецидив в течение 1 года после достижения ремиссии. • Нейролейкемия. Цитоз ≥ 5/мкл. полной
Основные понятия и критерии • Резистентная форма ОЛ. Отсутствие полной ремиссии после проведения 2 -х курсов индукционной терапии. • Рецидив. >5% бластов в КМ ± нейролейкемия, спленомегалия, лимфоаденопатия и т. д, ). • Ранний рецидив. Рецидив в течение 1 года после достижения ремиссии. • Нейролейкемия. Цитоз ≥ 5/мкл. полной
 Безрецидивная выживаемость пациентов с ОМЛ •
Безрецидивная выживаемость пациентов с ОМЛ •
 ТКМ в первой ремиссии ОМЛ
ТКМ в первой ремиссии ОМЛ
 Характеристика пациентов ОМЛ после алогенной ТКМ 2000 -2012 гг. ИДГи. Т им. Р. М. Горбачевой СПб. ГМУ им. И. П. Павлова • • • • Количество пациентов 104 Женщин 57 (55%) Мужчин 47 (45%) Возраст 15 -68 лет Медиана возраста 29 лет FAB классификация: М 0 – 15 (14%) М 1 – 19 (18%) М 2 – 31 (30%) М 4 – 22 (21%) М 5 – 12 (12%) М 6 – 4 (4%) М 7 – 1 (1%) • • De novo – 91 (88%) Вторичный ОМЛ – 13 (12%) • • Родственная ТКМ – 41 (39%) Неродственная ТКМ – 63 (61%) • • • Ремиссия 1 – 33 (32%) Ремиссия 2 -3 – 31 (30%) Вне ремиссии – 40 (38%)
Характеристика пациентов ОМЛ после алогенной ТКМ 2000 -2012 гг. ИДГи. Т им. Р. М. Горбачевой СПб. ГМУ им. И. П. Павлова • • • • Количество пациентов 104 Женщин 57 (55%) Мужчин 47 (45%) Возраст 15 -68 лет Медиана возраста 29 лет FAB классификация: М 0 – 15 (14%) М 1 – 19 (18%) М 2 – 31 (30%) М 4 – 22 (21%) М 5 – 12 (12%) М 6 – 4 (4%) М 7 – 1 (1%) • • De novo – 91 (88%) Вторичный ОМЛ – 13 (12%) • • Родственная ТКМ – 41 (39%) Неродственная ТКМ – 63 (61%) • • • Ремиссия 1 – 33 (32%) Ремиссия 2 -3 – 31 (30%) Вне ремиссии – 40 (38%)
: влияние ремиссионного статуса на момент ТКМ") Общая выживаемость больных ОМЛ после аллогенной ТКМ (n=104): влияние ремиссионного статуса на момент ТКМ Ремиссия 1, n=33, 56 % Ремиссия 2 -3, n=31, 24 % Вне ремиссии, n=40, 10 % p=0. 0002
Общая выживаемость больных ОМЛ после аллогенной ТКМ (n=104): влияние ремиссионного статуса на момент ТКМ Ремиссия 1, n=33, 56 % Ремиссия 2 -3, n=31, 24 % Вне ремиссии, n=40, 10 % p=0. 0002
: значение ремиссионного статуса на момент") Безрецидивная выживаемость больных ОМЛ после аллогенной неродственной ТКМ (n=52): значение ремиссионного статуса на момент ТКМ Ремиссия 1, 64 % Ремиссия 2 -3, 8 % Вне ремиссии, 10 % p=0. 01962
Безрецидивная выживаемость больных ОМЛ после аллогенной неродственной ТКМ (n=52): значение ремиссионного статуса на момент ТКМ Ремиссия 1, 64 % Ремиссия 2 -3, 8 % Вне ремиссии, 10 % p=0. 01962
, AML") Пациент А. возраст: 21 год Диагноз: Острый миелобластный лейкоз М 1, t(8; 21), AML 1/ETO, нейролейкоз (03. 2011 г. ).
Пациент А. возраст: 21 год Диагноз: Острый миелобластный лейкоз М 1, t(8; 21), AML 1/ETO, нейролейкоз (03. 2011 г. ).
 Лихорадка 39 С Изменения в гемограмме Обследован в РКБ: Нарастающая слабость Июнь … Миелограмма: бласты 69% МПО+ Диагноз: острый нелимфобластный лейкоз Октябрь Ноябрь 2010 г. 7+3 Doxo
Лихорадка 39 С Изменения в гемограмме Обследован в РКБ: Нарастающая слабость Июнь … Миелограмма: бласты 69% МПО+ Диагноз: острый нелимфобластный лейкоз Октябрь Ноябрь 2010 г. 7+3 Doxo
 Обследован в ГКБ им. Боткина Контрольная миелограмма Бласты 5% Миелограмма Бласты 74% МПО+ PAS+ Ликвор: Цитоз 11/3 Бласты 11% Контрольная миелограмма Бласты 2, 5% Диагноз: ОМЛ М 1, Первичная химиорезистентность Нейролейкоз Ноябрь … Март Первая Клиникогематологическая ремиссия Апрель Май 2011 г. 7+3 Doxo 7+3 Dauno +цитостатики интротекально HAI HA
Обследован в ГКБ им. Боткина Контрольная миелограмма Бласты 5% Миелограмма Бласты 74% МПО+ PAS+ Ликвор: Цитоз 11/3 Бласты 11% Контрольная миелограмма Бласты 2, 5% Диагноз: ОМЛ М 1, Первичная химиорезистентность Нейролейкоз Ноябрь … Март Первая Клиникогематологическая ремиссия Апрель Май 2011 г. 7+3 Doxo 7+3 Dauno +цитостатики интротекально HAI HA
 По данным HLA типирования имеется 2 родственных донора 14. 07. 2011 выполнена родственная алло-ТГСК Приживление: Leu на д+21 Neu на д +25 Tr на д+12 Консультирован В ИДГи. Т, рекомендована родственная алло. ТГСК Май Июнь Июль 2011 г. HA Bu+Cp Август Tac + MMF
По данным HLA типирования имеется 2 родственных донора 14. 07. 2011 выполнена родственная алло-ТГСК Приживление: Leu на д+21 Neu на д +25 Tr на д+12 Консультирован В ИДГи. Т, рекомендована родственная алло. ТГСК Май Июнь Июль 2011 г. HA Bu+Cp Август Tac + MMF
. • Донор – сестра, 1976 г. р.") Протокол трансплантации костного мозга (14. 07. 2011). • Донор – сестра, 1976 г. р. , гемотрансфузий 0, беременностей 3. • СD 34+/кг веса реципиента = 3, 7 х10^6. NC/кг веса реципиента = 3, 7 х10^9. • CD 34+ = 1, 0%, CD 3+ = 3, 5%.
Протокол трансплантации костного мозга (14. 07. 2011). • Донор – сестра, 1976 г. р. , гемотрансфузий 0, беременностей 3. • СD 34+/кг веса реципиента = 3, 7 х10^6. NC/кг веса реципиента = 3, 7 х10^9. • CD 34+ = 1, 0%, CD 3+ = 3, 5%.
 • Химеризм 90 -95%") • Приживление трансплантата на день +21 (04. 08. 2011) • Химеризм 90 -95% • Признаков о. РТПХ нет
• Приживление трансплантата на день +21 (04. 08. 2011) • Химеризм 90 -95% • Признаков о. РТПХ нет
 • Базисная терапия – такролимус 2 мг/сут. • Сохраняется полная ремиссия заболевания. • С 15. 11. 2011 начинается отмена ИСТ. • Полная отмена ИСТ в начале декабря 2011 г. • Декабрь 2011 года: появление болей в спине • Нарастающая слабость нижних конечностей • Парапарез, нарушение функции тазовых органов
• Базисная терапия – такролимус 2 мг/сут. • Сохраняется полная ремиссия заболевания. • С 15. 11. 2011 начинается отмена ИСТ. • Полная отмена ИСТ в начале декабря 2011 г. • Декабрь 2011 года: появление болей в спине • Нарастающая слабость нижних конечностей • Парапарез, нарушение функции тазовых органов
 – Эр 3, 9 х10^12, Гем 119 г/л,") • ОАК (07. 12. 2011) – Эр 3, 9 х10^12, Гем 119 г/л, Л 4, 5 х10^9, бласты 75%. • Ликвор (06. 12. 2011): бесцветный, прозрачный, цитоз 8/3, белок 4, 56 г/л. • МРТ грудного отдела позвоночника (18. 12. 2011): интрадуральные объемные образования на уровне тел Th 6 -7, Th 10 -L 1, объемные образования паравертебральных мягких тканей, ребернопозвоночных углов на уровнях Th 6 -10, S 1 -4. • Миелограмма – бласты 89, 6%. • Химеризм (07. 12. 2011) 40 -50% клеток имеют донорское происхождение. • Ds: комбинированный (костномозговой, экстрамедуллярный) рецидив ОМЛ.
• ОАК (07. 12. 2011) – Эр 3, 9 х10^12, Гем 119 г/л, Л 4, 5 х10^9, бласты 75%. • Ликвор (06. 12. 2011): бесцветный, прозрачный, цитоз 8/3, белок 4, 56 г/л. • МРТ грудного отдела позвоночника (18. 12. 2011): интрадуральные объемные образования на уровне тел Th 6 -7, Th 10 -L 1, объемные образования паравертебральных мягких тканей, ребернопозвоночных углов на уровнях Th 6 -10, S 1 -4. • Миелограмма – бласты 89, 6%. • Химеризм (07. 12. 2011) 40 -50% клеток имеют донорское происхождение. • Ds: комбинированный (костномозговой, экстрамедуллярный) рецидив ОМЛ.
 • • Проточная цитометрия – в костном мозге бластные клетки с миелоидным фенотипом (CD 13+/CD 15+/CD 33+/CD 34+/CD 117+/HLADR/MPO+) Цитогенетическое исследование (06. 12. 2011): 45, Х, -Y t(8; 21) • Молекулярное исследование (06. 12. 2011): обнаружен AML 1 -ETO • С 07. 12. 2011 – 12. 2011 проведен курс химиотерапии по схеме FLAG • 20. 12. 2011 введение донорских лимфоцитов № 1, в дозе 1 х10^6 на кг, начата терапия реафероном + ГМ-КСФ
• • Проточная цитометрия – в костном мозге бластные клетки с миелоидным фенотипом (CD 13+/CD 15+/CD 33+/CD 34+/CD 117+/HLADR/MPO+) Цитогенетическое исследование (06. 12. 2011): 45, Х, -Y t(8; 21) • Молекулярное исследование (06. 12. 2011): обнаружен AML 1 -ETO • С 07. 12. 2011 – 12. 2011 проведен курс химиотерапии по схеме FLAG • 20. 12. 2011 введение донорских лимфоцитов № 1, в дозе 1 х10^6 на кг, начата терапия реафероном + ГМ-КСФ
 – бласты 2, 2%, химеризм: 80 -90% клеток") • Миелограмма (11. 01. 2012) – бласты 2, 2%, химеризм: 80 -90% клеток имеют донорское происхождение • Цитогенетическое исследование (11. 01. 2012): 46 ХХ • Молекулярное исследование (11. 01. 2012): обнаружена транслокация AML 1 -ETO • МРТ грудного отдела позвоночника (13. 01. 2012): положительная динамика по сравнению с исследованием от 06. 12. 2011
• Миелограмма (11. 01. 2012) – бласты 2, 2%, химеризм: 80 -90% клеток имеют донорское происхождение • Цитогенетическое исследование (11. 01. 2012): 46 ХХ • Молекулярное исследование (11. 01. 2012): обнаружена транслокация AML 1 -ETO • МРТ грудного отдела позвоночника (13. 01. 2012): положительная динамика по сравнению с исследованием от 06. 12. 2011
 • С 18. 01. 2012 начат второй курс химиотерапии HD ARA-C • 25. 01. 2012 введение донорских лимфоцитов № 2 в дозе 4 x 10^6 на кг, реаферон + ГМ-КСФ • 24. 02. 2012 введение донорских лимфоцитов № 3 в дозе 6 x 10^6 на кг, реаферон + ГМ-КСФ • 25. 04. 2012 оперативное удаление экстрамедуллярных очагов с целью декомпрессии спинного мозга • 05 – 08. 2012 продолжение полихимиотерапии и иммуноадоптивной терапии, достижение молекулярной ремиссии
• С 18. 01. 2012 начат второй курс химиотерапии HD ARA-C • 25. 01. 2012 введение донорских лимфоцитов № 2 в дозе 4 x 10^6 на кг, реаферон + ГМ-КСФ • 24. 02. 2012 введение донорских лимфоцитов № 3 в дозе 6 x 10^6 на кг, реаферон + ГМ-КСФ • 25. 04. 2012 оперативное удаление экстрамедуллярных очагов с целью декомпрессии спинного мозга • 05 – 08. 2012 продолжение полихимиотерапии и иммуноадоптивной терапии, достижение молекулярной ремиссии
 • Иматиниб,") Новые препараты • Клофорабин • Миелотарг (gemtuzumab-ozogamicin –anti CD 33 + calcheamicin) • Иматиниб, дазатиниб (CD 117+, KIT-RTK мутации) • Децитабин, азацитидин (гипермитиллирование ДНК) • Тандутиниб, сунитиниб (ингибитор FLT 3 – FMS –like tyrosine kinase 3) • Типифарниб (ингибитор фарнезилтрансферазы)
Новые препараты • Клофорабин • Миелотарг (gemtuzumab-ozogamicin –anti CD 33 + calcheamicin) • Иматиниб, дазатиниб (CD 117+, KIT-RTK мутации) • Децитабин, азацитидин (гипермитиллирование ДНК) • Тандутиниб, сунитиниб (ингибитор FLT 3 – FMS –like tyrosine kinase 3) • Типифарниб (ингибитор фарнезилтрансферазы)
 • 10% всех ОМЛ • Выделяют два морфологических варианта") Острый промиелоцитарный лейкоз (М 3) • 10% всех ОМЛ • Выделяют два морфологических варианта ОПЛ: 1. Типичный ОПЛ (75 -85%): характерная морфология бластных клеток. 2. Атипичный М 3 v (variant) с нетипичной морфологией опухолевых клеток. • Для типичного ОПЛ характерна лейкопения ( L<5. 0, нередко < 1. 0) • Для М 3 v характерен лейкоцитоз (L 20. 0 – 200. 0) • Экстрамедулярные очаги не характерны для ОПЛ, однако в последнее время возросло количество сообщений о поражении ЦНС.
Острый промиелоцитарный лейкоз (М 3) • 10% всех ОМЛ • Выделяют два морфологических варианта ОПЛ: 1. Типичный ОПЛ (75 -85%): характерная морфология бластных клеток. 2. Атипичный М 3 v (variant) с нетипичной морфологией опухолевых клеток. • Для типичного ОПЛ характерна лейкопения ( L<5. 0, нередко < 1. 0) • Для М 3 v характерен лейкоцитоз (L 20. 0 – 200. 0) • Экстрамедулярные очаги не характерны для ОПЛ, однако в последнее время возросло количество сообщений о поражении ЦНС.
 Особенности ОПЛ • Тяжелый геморрагический синдром, гематомный тип кровоточивости • ДВС-синдром • Молодой возраст больных Иммунофенотип ОПЛ: CD 13+, CD 34 - , HLA-DRХромосомные аберрации: t (15; 17)(q 22; q 12): химерный ген PML / RARα t(11; 17)(q 13, q 21): химерный ген NUMA / RARα
Особенности ОПЛ • Тяжелый геморрагический синдром, гематомный тип кровоточивости • ДВС-синдром • Молодой возраст больных Иммунофенотип ОПЛ: CD 13+, CD 34 - , HLA-DRХромосомные аберрации: t (15; 17)(q 22; q 12): химерный ген PML / RARα t(11; 17)(q 13, q 21): химерный ген NUMA / RARα
 Генетические аспекты ОПЛ • Продукт химерного гена PML/RARα -патологический белок, сохраняющий активные функциональные домены как белка RARα, так и PML белка. • При ОПЛ белок PML/RARα накапливается в цитоплазме и ядре миелоидных клеток, что приводит к блоку дифференцировки на стадии промиелоцитов. • PML/RARα подавляет апоптоз опухолевых клеток. • ТРАНС-РЕТИНОЕВАЯ КИСЛОТА (ATRA, весаноид) в высокой концентрации эффективна при ОПЛ с t(15; 17), t(11; 17)(q 13; 21) • Снимает блок дифференцировки, стимулируя активность нормального RARα гена.
Генетические аспекты ОПЛ • Продукт химерного гена PML/RARα -патологический белок, сохраняющий активные функциональные домены как белка RARα, так и PML белка. • При ОПЛ белок PML/RARα накапливается в цитоплазме и ядре миелоидных клеток, что приводит к блоку дифференцировки на стадии промиелоцитов. • PML/RARα подавляет апоптоз опухолевых клеток. • ТРАНС-РЕТИНОЕВАЯ КИСЛОТА (ATRA, весаноид) в высокой концентрации эффективна при ОПЛ с t(15; 17), t(11; 17)(q 13; 21) • Снимает блок дифференцировки, стимулируя активность нормального RARα гена.
 Терапия ОПЛ • ATRA без ХТ- 95 % полных ремиссий. Терапия одной ATRA всегда заканчивается рецидивом ОПЛ, длительность ремиссии- 3, 5 месяца. • 7+3+ ATRA в качестве индукции/консолидации (4 курса) Даунорубицин Цитарабин Поддерживающая терапия по программе POMP ( метотрексат, 6 меркаптопурин- постоянно, винкристин, преднизолон - блоки) в течение 2 -х лет. • Результаты -71% полных ремиссий, общая выживаемость - 80%, 8 летняя безрецидивная выживаемость - 70%.
Терапия ОПЛ • ATRA без ХТ- 95 % полных ремиссий. Терапия одной ATRA всегда заканчивается рецидивом ОПЛ, длительность ремиссии- 3, 5 месяца. • 7+3+ ATRA в качестве индукции/консолидации (4 курса) Даунорубицин Цитарабин Поддерживающая терапия по программе POMP ( метотрексат, 6 меркаптопурин- постоянно, винкристин, преднизолон - блоки) в течение 2 -х лет. • Результаты -71% полных ремиссий, общая выживаемость - 80%, 8 летняя безрецидивная выживаемость - 70%.
 Показания: индукция ремиссии и консолидация ремиссии у взрослых") ТЕРАПИЯ БОЛЬНЫХ ОПЛ ТРИСЕНОКСОМ (arsenic trioxide) Показания: индукция ремиссии и консолидация ремиссии у взрослых пациентов с резистентным течением или рецидивом ОПЛ t (15; 17)PML/RARα)
ТЕРАПИЯ БОЛЬНЫХ ОПЛ ТРИСЕНОКСОМ (arsenic trioxide) Показания: индукция ремиссии и консолидация ремиссии у взрослых пациентов с резистентным течением или рецидивом ОПЛ t (15; 17)PML/RARα)
 Выживаемость на терапии трисеноксом
Выживаемость на терапии трисеноксом
 Острые лимфобластные лейкозы
Острые лимфобластные лейкозы
 ОЛЛ Распространенность: 30 случаев на 1 000 Пик заболевания: 2 -5 и 50 -60 лет. У взрослых - 1% всех злокачественных заболеваний ОЛЛ - самые частые злокачественные опухоли у детей (30% всех опухолей) Факторы риска: рентгеновское облучение в пренатальном периоде, Дети с синдромом Дауна (+21), анемией Фанкони, синдромом Кляйтфельтера , нейрофиброматозом, телеангиоматозом
ОЛЛ Распространенность: 30 случаев на 1 000 Пик заболевания: 2 -5 и 50 -60 лет. У взрослых - 1% всех злокачественных заболеваний ОЛЛ - самые частые злокачественные опухоли у детей (30% всех опухолей) Факторы риска: рентгеновское облучение в пренатальном периоде, Дети с синдромом Дауна (+21), анемией Фанкони, синдромом Кляйтфельтера , нейрофиброматозом, телеангиоматозом
 Морфологическая классификация острых лимфобластных лейкозов • FAB • L 1 -микроформы бластов • L 2 - гетерогенные бласты • L 3 - беркитоподобные бласты Иммунологические варианты • Т-лейкозы • В-лейкозы • B-лейкозы 85 -90 % среди всех ОЛЛ
Морфологическая классификация острых лимфобластных лейкозов • FAB • L 1 -микроформы бластов • L 2 - гетерогенные бласты • L 3 - беркитоподобные бласты Иммунологические варианты • Т-лейкозы • В-лейкозы • B-лейкозы 85 -90 % среди всех ОЛЛ
 Иммунологическая классификация ОЛЛ Вариант Антигены Частота % Прогноз Цитогенетика Ранний пре-В CD 10 -, CD 19+ c. Ig-, SIg- 5 -10 Плохой ** t (4; 11) Commonолл CD 10+CD 19+ 40 -45 средний t (9; 22) Пре-В CD 10+CD 19+ 20 средний t(9; 22) t(4; 11) t(1; 19) 4 -5 Плохой*** t (8; 14) t (8; 22) t (2; 8) 5 -6 20 Плохой хороший 14 q 11 7 q 34 c. Ig- SIg- c. Ig+ SIg. B CD 10+CD 19+ c. Ig- SIg+ Пре-Т T CD 7, c. CD 3, CD 1, CD 3, CD 4 CD 7 CD 8
Иммунологическая классификация ОЛЛ Вариант Антигены Частота % Прогноз Цитогенетика Ранний пре-В CD 10 -, CD 19+ c. Ig-, SIg- 5 -10 Плохой ** t (4; 11) Commonолл CD 10+CD 19+ 40 -45 средний t (9; 22) Пре-В CD 10+CD 19+ 20 средний t(9; 22) t(4; 11) t(1; 19) 4 -5 Плохой*** t (8; 14) t (8; 22) t (2; 8) 5 -6 20 Плохой хороший 14 q 11 7 q 34 c. Ig- SIg- c. Ig+ SIg. B CD 10+CD 19+ c. Ig- SIg+ Пре-Т T CD 7, c. CD 3, CD 1, CD 3, CD 4 CD 7 CD 8
 Иммунофенотипирование костного мозга методом проточной цитометрии Острый лимфобластный лейкоз из В-клеток предшественников Бласты характеризуются снижением экспрессии панлейкоцитарного антигена CD 45, яркой экспрессией Вклеточных антигенов CD 19, CD 22, CD 79 a, CD 10 и экспрессией линейно неограниченного маркера CD 34, отражающего ранние стадии клеточного созревания
Иммунофенотипирование костного мозга методом проточной цитометрии Острый лимфобластный лейкоз из В-клеток предшественников Бласты характеризуются снижением экспрессии панлейкоцитарного антигена CD 45, яркой экспрессией Вклеточных антигенов CD 19, CD 22, CD 79 a, CD 10 и экспрессией линейно неограниченного маркера CD 34, отражающего ранние стадии клеточного созревания
 Иммунофенотипирование костного мозга методом проточной цитометрии Острый Т-клеточный лимфобластный лейкоз Бласты характеризуются снижением экспрессии панлейкоцитарного антигена CD 45, экспрессией Тклеточных антигенов CD 3, цитоплазматический CD 3, CD 7, CD 5, CD 2, CD 1 a и экспрессией линейно неограниченного маркера CD 34, отражающего аберрантность клеточного иммунофенотипа
Иммунофенотипирование костного мозга методом проточной цитометрии Острый Т-клеточный лимфобластный лейкоз Бласты характеризуются снижением экспрессии панлейкоцитарного антигена CD 45, экспрессией Тклеточных антигенов CD 3, цитоплазматический CD 3, CD 7, CD 5, CD 2, CD 1 a и экспрессией линейно неограниченного маркера CD 34, отражающего аберрантность клеточного иммунофенотипа
(q 34;") Цитогенетические аномалии при ОЛЛ Цитогенетические аберрации Вовлеченные гены Частота встречаемости % t(9; 22)(q 34; q 11. 2) BCR/ABL 11 -34 t(4; 11)(q 21; q 23) MLL/AF 4 3 -7% Del (9 p) или t (9 p) CDKN 2 A и CDKN 2 B 5 -15 Del (12 p)или t (9 p) ETV 6 4 -5 t(14 q 11 -q 13) TCR α , δ 4 -6 t(14 q 32) IGH, BCL 11 A 5 t(8; 14)(q 24; q 32) TCL-1 BCL 11 B 5 Del 6 q ? 2 -6 t(1; 19) E 2 A/PBX 1 3 + 9 q NUP 214/ABL 4
Цитогенетические аномалии при ОЛЛ Цитогенетические аберрации Вовлеченные гены Частота встречаемости % t(9; 22)(q 34; q 11. 2) BCR/ABL 11 -34 t(4; 11)(q 21; q 23) MLL/AF 4 3 -7% Del (9 p) или t (9 p) CDKN 2 A и CDKN 2 B 5 -15 Del (12 p)или t (9 p) ETV 6 4 -5 t(14 q 11 -q 13) TCR α , δ 4 -6 t(14 q 32) IGH, BCL 11 A 5 t(8; 14)(q 24; q 32) TCL-1 BCL 11 B 5 Del 6 q ? 2 -6 t(1; 19) E 2 A/PBX 1 3 + 9 q NUP 214/ABL 4
 Плохой прогноз:") Прогностически значимые цитогенетические изменения при ОЛЛ Благоприятный прогноз: гипердиплоидия >50 t(12; 21) Плохой прогноз: t(9; 22) t(4; 11) t(1; 19)
Прогностически значимые цитогенетические изменения при ОЛЛ Благоприятный прогноз: гипердиплоидия >50 t(12; 21) Плохой прогноз: t(9; 22) t(4; 11) t(1; 19)
 (q 34; q 11)- bcr/abl • У") Ph + «Филадельфийская хромосома» t (9; 22) (q 34; q 11)- bcr/abl • У детей и 50% взрослых с ОЛЛ продуцируется белок р190 kd • При ХМЛ и 50% ОЛЛ взрослых продуцируется белок p 210 kd • Оба белка активируют тирозин-киназу
Ph + «Филадельфийская хромосома» t (9; 22) (q 34; q 11)- bcr/abl • У детей и 50% взрослых с ОЛЛ продуцируется белок р190 kd • При ХМЛ и 50% ОЛЛ взрослых продуцируется белок p 210 kd • Оба белка активируют тирозин-киназу
 Ph+ ОЛЛ • 20 -30% всех ОЛЛ взрослых, 5% ОЛЛ у детей • 50 % пре-В – ОЛЛ • 5 -DFS: 45 % при L < 50, 0 • 5 -DFS: 20% при L > 100, 0 Гиперлейкоцитоз, спленомегалия, Показание к аллогенной ТКМ лимфоаденопатия
Ph+ ОЛЛ • 20 -30% всех ОЛЛ взрослых, 5% ОЛЛ у детей • 50 % пре-В – ОЛЛ • 5 -DFS: 45 % при L < 50, 0 • 5 -DFS: 20% при L > 100, 0 Гиперлейкоцитоз, спленомегалия, Показание к аллогенной ТКМ лимфоаденопатия
 Терапия ОЛЛ Протокол D. Hoeltzer • Индукция ремиссии: винкристин, глюкокортикоиды, антрациклины, циклофосфан • Консолидация и интенсификация ремиссии: модификации индукционных схем, ротационная консолидация, ТКМ • Поддерживающая терапия: винкристин, преднизолон, 6 МП, метотрексат • Профилактика и лечение нейролейкемии • Резистентные ОЛЛ: FLAG, Blinatumomab (CD 3 CD 19 bispecific AB)
Терапия ОЛЛ Протокол D. Hoeltzer • Индукция ремиссии: винкристин, глюкокортикоиды, антрациклины, циклофосфан • Консолидация и интенсификация ремиссии: модификации индукционных схем, ротационная консолидация, ТКМ • Поддерживающая терапия: винкристин, преднизолон, 6 МП, метотрексат • Профилактика и лечение нейролейкемии • Резистентные ОЛЛ: FLAG, Blinatumomab (CD 3 CD 19 bispecific AB)
 80, 0% 4, 0% 0,") Элиминация патологического клона на фоне лечения (минимальная остаточная болезнь) 80, 0% 4, 0% 0, 04% 80, 0% День 0 4, 0% День +15 0, 04% День +28
Элиминация патологического клона на фоне лечения (минимальная остаточная болезнь) 80, 0% 4, 0% 0, 04% 80, 0% День 0 4, 0% День +15 0, 04% День +28
 группа высокого риска рецидива Выводы: аллогенная ТКМ повышает выживаемость в группе высокого риска У больных стандартного риска аллогенная ТКМ не улучшает исхода заболевания по сравнению со стандартной ПХТ или ауто. ТКМ
группа высокого риска рецидива Выводы: аллогенная ТКМ повышает выживаемость в группе высокого риска У больных стандартного риска аллогенная ТКМ не улучшает исхода заболевания по сравнению со стандартной ПХТ или ауто. ТКМ
 Характеристика пациентов ОЛЛ после аллогенной ТКМ 2000 -2012 гг. (СПб. ГМУ им. акад. И. П. Павлова, ИДГи. Т им. Р. М. Горбачевой) Количество пациентов 103 Женщин 39 (38%) Мужчин 64 (62%) Возраст 14 -57 лет Медиана возраста 21 год Родственная ТКМ – 32 (31%) Неродственная ТКМ – 71 (69%) Ремиссия 1 – 21 (20%) t(9; 22) – 8 (38%) t(4; 11) – 4 (19%) Т-кл. – 5 (24%) Ремиссия 2 – 33 (32%) Вне ремиссии – 49 (48%) Классификация: В-кл. Ph(-) – 46 (45%) prо-B – 12 (26%) common-B – 21 (46%) pre-B – 11 (24%) mature-B – 1 (2%) B-lin – 1 (2%) В-кл. Ph(+) – 23 (22%) prо-B – 2 (9%) common-B – 11 (48%) pre-B – 3 (13%) B-lin – 1 (4%) н/данных – 6 (26%) Т-кл. – 24 (23%) prо-Т – 4 (17%) pre-Т – 5 (21%) cort-T – 3 (12%) Т-lin – 12 (50%) нет данных – 10 (10%)
Характеристика пациентов ОЛЛ после аллогенной ТКМ 2000 -2012 гг. (СПб. ГМУ им. акад. И. П. Павлова, ИДГи. Т им. Р. М. Горбачевой) Количество пациентов 103 Женщин 39 (38%) Мужчин 64 (62%) Возраст 14 -57 лет Медиана возраста 21 год Родственная ТКМ – 32 (31%) Неродственная ТКМ – 71 (69%) Ремиссия 1 – 21 (20%) t(9; 22) – 8 (38%) t(4; 11) – 4 (19%) Т-кл. – 5 (24%) Ремиссия 2 – 33 (32%) Вне ремиссии – 49 (48%) Классификация: В-кл. Ph(-) – 46 (45%) prо-B – 12 (26%) common-B – 21 (46%) pre-B – 11 (24%) mature-B – 1 (2%) B-lin – 1 (2%) В-кл. Ph(+) – 23 (22%) prо-B – 2 (9%) common-B – 11 (48%) pre-B – 3 (13%) B-lin – 1 (4%) н/данных – 6 (26%) Т-кл. – 24 (23%) prо-Т – 4 (17%) pre-Т – 5 (21%) cort-T – 3 (12%) Т-lin – 12 (50%) нет данных – 10 (10%)
 Безрецидивная выживаемость больных ОЛЛ в зависимости от статуса заболевания на момент аллогенной ТКМ (n 91) р=0, 00079 n=21, 57% n=33, 49% n=37, 29%
Безрецидивная выживаемость больных ОЛЛ в зависимости от статуса заболевания на момент аллогенной ТКМ (n 91) р=0, 00079 n=21, 57% n=33, 49% n=37, 29%
 Аллогенная ТКМ – единственный радикальный способ лечения многих вариантов ОЛ
Аллогенная ТКМ – единственный радикальный способ лечения многих вариантов ОЛ
 ВОПРОСЫ
ВОПРОСЫ