

пир и экстрапир дегенер+ болезни обмена 14.ppt
- Количество слайдов: 135
 ОМСКАЯ ГОСУДАРСТВЕННАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ КАФЕДРА НЕВРОЛОГИИ И НЕЙРОХИРУРГИИ Наследственные болезни нервной системы. Пирамидные и экстрапирамидные дегенерации Наследственные болезни обмена д. м. н. Ларькин В. И.
ОМСКАЯ ГОСУДАРСТВЕННАЯ МЕДИЦИНСКАЯ АКАДЕМИЯ КАФЕДРА НЕВРОЛОГИИ И НЕЙРОХИРУРГИИ Наследственные болезни нервной системы. Пирамидные и экстрапирамидные дегенерации Наследственные болезни обмена д. м. н. Ларькин В. И.
 План лекции l Наследственные системные дегенерации нервной системы l l 1. ДЕГЕНЕРАЦИИ С ПРЕИМУЩЕСТВЕННЫМ ПОРАЖЕНИЕМ МОЗЖЕЧКА И ЕГО СВЯЗЕЙ Семейная атаксия Фридрейха. Болезнь Рефсума. 2. БОЛЕЗНИ С ПОРАЖЕНИЕМ ЭКСТРАПИРАМИДНОЙ НЕРВНОЙ СИСТЕМЫ. Хорея Гентингтона 3. БОЛЕЗНИ С ПОРАЖЕНИЕМ ПИРАМИДНОЙ СИСТЕМЫ. Спастический семейный паралич Штрюмпеля. 4. Синдромы, сопровождающиеся постепенным развитием нарушений позы и движений. болезнь Паркинсона. Торсионная дистония. Оливопонтоцеребеллярные дегенерации 5. Лейкодистрофии (лейкоэнцефалопатии) l 6. Наследственные болезни обмена. Патология аминокислот l Патология липидов. Муколипидозы. Патология углеводов. l l l
План лекции l Наследственные системные дегенерации нервной системы l l 1. ДЕГЕНЕРАЦИИ С ПРЕИМУЩЕСТВЕННЫМ ПОРАЖЕНИЕМ МОЗЖЕЧКА И ЕГО СВЯЗЕЙ Семейная атаксия Фридрейха. Болезнь Рефсума. 2. БОЛЕЗНИ С ПОРАЖЕНИЕМ ЭКСТРАПИРАМИДНОЙ НЕРВНОЙ СИСТЕМЫ. Хорея Гентингтона 3. БОЛЕЗНИ С ПОРАЖЕНИЕМ ПИРАМИДНОЙ СИСТЕМЫ. Спастический семейный паралич Штрюмпеля. 4. Синдромы, сопровождающиеся постепенным развитием нарушений позы и движений. болезнь Паркинсона. Торсионная дистония. Оливопонтоцеребеллярные дегенерации 5. Лейкодистрофии (лейкоэнцефалопатии) l 6. Наследственные болезни обмена. Патология аминокислот l Патология липидов. Муколипидозы. Патология углеводов. l l l
 НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ l l l l В основе наследственных заболеваний нервной системы лежат генные мутации, ведущие к нарушению синтеза определенного полипептида. нарушения усвоения отдельных веществ или их извращенный синтез, что приводит к дистрофическому процессу. повышенный распад ряда соединений в результате деструкции тканей или накопление веществ, отложение их в тканях, что способствует нарушению функционирования последних. характерны прогрессирующее течение, поражение определенной системы мозга, нервов, мышц. Дегенерация включает процессы деструкции, дистрофии, атрофии. Патология нервной системы может сочетаться с патологией внутренних органов.
НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ l l l l В основе наследственных заболеваний нервной системы лежат генные мутации, ведущие к нарушению синтеза определенного полипептида. нарушения усвоения отдельных веществ или их извращенный синтез, что приводит к дистрофическому процессу. повышенный распад ряда соединений в результате деструкции тканей или накопление веществ, отложение их в тканях, что способствует нарушению функционирования последних. характерны прогрессирующее течение, поражение определенной системы мозга, нервов, мышц. Дегенерация включает процессы деструкции, дистрофии, атрофии. Патология нервной системы может сочетаться с патологией внутренних органов.
 КЛАССИФИКАЦИЯ наследственных заболеваний l l l 1. Наследственные системные дегенерации нервной системы Дегенерации с преимущественным поражением мозжечка и его связей Сочетанные дегенерации мозжечковых путей и периферических нервов Болезни с поражением экстрапирамидной нервной системы Болезни с поражением пирамидной нервной системы l l l 2. Наследственные болезни обмена Патология аминокислот Патология липидов Муколипидозы Патология углеводов l 3. Болезни соединительной ткани l 4. Факоматозы l l 5. Нервно мышечные заболевания Прогрессирующие мышечные дистрофии миопатии Спинальные и невральные атрофии. Наследственные нервно мышечные заболевания с миотоническим синдромом Пароксизмальные миоплегии Миастения l l
КЛАССИФИКАЦИЯ наследственных заболеваний l l l 1. Наследственные системные дегенерации нервной системы Дегенерации с преимущественным поражением мозжечка и его связей Сочетанные дегенерации мозжечковых путей и периферических нервов Болезни с поражением экстрапирамидной нервной системы Болезни с поражением пирамидной нервной системы l l l 2. Наследственные болезни обмена Патология аминокислот Патология липидов Муколипидозы Патология углеводов l 3. Болезни соединительной ткани l 4. Факоматозы l l 5. Нервно мышечные заболевания Прогрессирующие мышечные дистрофии миопатии Спинальные и невральные атрофии. Наследственные нервно мышечные заболевания с миотоническим синдромом Пароксизмальные миоплегии Миастения l l
 ДЕГЕНЕРАЦИИ С ПРЕИМУЩЕСТВЕННЫМ ПОРАЖЕНИЕМ МОЗЖЕЧКА И ЕГО СВЯЗЕЙ
ДЕГЕНЕРАЦИИ С ПРЕИМУЩЕСТВЕННЫМ ПОРАЖЕНИЕМ МОЗЖЕЧКА И ЕГО СВЯЗЕЙ
 l Семейная атаксия Фридрейха. наследственное нейродегенеративное заболевание, начинающееся в детском или юношеском возрасте и характеризующееся прогрессирующей атаксией, деформациями скелета и кардиомиодистрофией. Тип наследования аутосомно рецессивный l Выявляются склероз задних и боковых канатиков спинного мозга, преимущественно пучков Голля, в меньшей степени — Бурдаха, Флексига, Говерса, волокон пирамидной системы, задних корешков, а также уменьшение клеток Пуркинье и нейронов зубчатых ядер мозжечка, подкорковых ганглиев, коры головного мозга, дистрофические изменения миокарда. Заболевание дебютирует в 8— 15 лет. Первым симптомом болезни является неустойчивая походка, которая была охарактеризована Шарко как табетически мозжечковая. l При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохокинез, дисметрия, скандированная речь, расстройства мышечно суставного чувства и вибрационной чувствительности. При исследовании глазного дна нередко выявляется атрофия соска зрительного нерва. Нарушается почерк. Ранними симптомами являются снижение мышечного тонуса и угасание глубоких рефлексов l l
l Семейная атаксия Фридрейха. наследственное нейродегенеративное заболевание, начинающееся в детском или юношеском возрасте и характеризующееся прогрессирующей атаксией, деформациями скелета и кардиомиодистрофией. Тип наследования аутосомно рецессивный l Выявляются склероз задних и боковых канатиков спинного мозга, преимущественно пучков Голля, в меньшей степени — Бурдаха, Флексига, Говерса, волокон пирамидной системы, задних корешков, а также уменьшение клеток Пуркинье и нейронов зубчатых ядер мозжечка, подкорковых ганглиев, коры головного мозга, дистрофические изменения миокарда. Заболевание дебютирует в 8— 15 лет. Первым симптомом болезни является неустойчивая походка, которая была охарактеризована Шарко как табетически мозжечковая. l При неврологическом обследовании выявляются крупноразмашистый нистагм, атаксия в руках и ногах, адиадохокинез, дисметрия, скандированная речь, расстройства мышечно суставного чувства и вибрационной чувствительности. При исследовании глазного дна нередко выявляется атрофия соска зрительного нерва. Нарушается почерк. Ранними симптомами являются снижение мышечного тонуса и угасание глубоких рефлексов l l
 В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей с патологическими пирамидными знаками, атрофиями мышц дистальных отделов конечностей. Интеллект снижен. Обнаруживают кифосколиоз, деформацию стоп, гипертрофию миокарда. l Заболевание медленно прогрессирует. Средняя продолжительность жизни 10— 15 лет с момента его развития. l Лечение. Применяют симптоматические средства: общеукрепляющие препараты (витамины, аминокислоты, ноотропные), лечебная физкультура, массаж l Прогноз. Кардиомиопатия приводит к застойной сердечной недостаточности. Продолжительность жизни больных редко превышает 30 лет. l
В более поздних стадиях болезни присоединяются афферентный парез нижних, а затем верхних конечностей с патологическими пирамидными знаками, атрофиями мышц дистальных отделов конечностей. Интеллект снижен. Обнаруживают кифосколиоз, деформацию стоп, гипертрофию миокарда. l Заболевание медленно прогрессирует. Средняя продолжительность жизни 10— 15 лет с момента его развития. l Лечение. Применяют симптоматические средства: общеукрепляющие препараты (витамины, аминокислоты, ноотропные), лечебная физкультура, массаж l Прогноз. Кардиомиопатия приводит к застойной сердечной недостаточности. Продолжительность жизни больных редко превышает 30 лет. l
 Наследственная мозжечковая атаксия Пьера Мари наследственное дегенеративное заболевание с преимущественным поражением мозжечка и его проводящих путей. Тип наследования аутосомно доминантный. l дегенеративные поражения клеток коры и ядер мозжечка, спиноцеребеллярных путей в боковых канатиках спинного мозга, в ядрах моста мозга и продолговатого мозга. l l
Наследственная мозжечковая атаксия Пьера Мари наследственное дегенеративное заболевание с преимущественным поражением мозжечка и его проводящих путей. Тип наследования аутосомно доминантный. l дегенеративные поражения клеток коры и ядер мозжечка, спиноцеребеллярных путей в боковых канатиках спинного мозга, в ядрах моста мозга и продолговатого мозга. l l
 Наблюдаются атаксия при выполнении координаторных проб, нарушение походки, скандированная речь, интенционное дрожание, нистагм. Мозжечковые симптомы сочетаются с умеренными или выраженными признаками пирамидной недостаточности со зрительными и глазодвигательными нарушениями, снижение интеллекта наличие зрительных и глазодвигательных расстройств l Рассеянный склероз в отличие от семейной атаксии Пьера Мари характеризуется ремиттирующим течением, большей выраженностью нижнего спастического парапареза, расстройством функций тазовых органов. l Лечение симптоматическое. l
Наблюдаются атаксия при выполнении координаторных проб, нарушение походки, скандированная речь, интенционное дрожание, нистагм. Мозжечковые симптомы сочетаются с умеренными или выраженными признаками пирамидной недостаточности со зрительными и глазодвигательными нарушениями, снижение интеллекта наличие зрительных и глазодвигательных расстройств l Рассеянный склероз в отличие от семейной атаксии Пьера Мари характеризуется ремиттирующим течением, большей выраженностью нижнего спастического парапареза, расстройством функций тазовых органов. l Лечение симптоматическое. l
 Сочетанные дегенерации мозжечковых путей и периферических нервов
Сочетанные дегенерации мозжечковых путей и периферических нервов
. l l l Тип наследования аутосомно рецессивный. Заболевание обычно начинается") Болезнь Рефсума (полиневритическая атаксия). l l l Тип наследования аутосомно рецессивный. Заболевание обычно начинается в раннем детстве. В основе патологии лежат демиелинизирующая полинейропатия, нарушения метаболизма липидов в виде жировой дегенерации нервов, клеток передних рогов спинного мозга, варолиевого моста, продолговатого мозга, мозжечка. В крови повышено содержание фитановой кислоты, меди, церуллоплазмина. определяются медленно прогрессирующие двигательные расстройства по периферическому типу, явления мозжечковой атаксии, расстройства чувствительности, снижение остроты зрения, слуха, обоняния. Иногда могут быть кардиомиопатия, деформация стоп, сколиоз, патология почек, снижение интеллекта. Лечение предусматривает диету с исключением продуктов, содержащих фитановую кислоту Болезнь Руси-Леви снижение скорости проведения возбуждения по нервам. l Гипертрофический интерстициальный неврит Дежерина. Скотта. является гипертрофия леммоцитов, разрастание элементов (швановской оболочки) l
Болезнь Рефсума (полиневритическая атаксия). l l l Тип наследования аутосомно рецессивный. Заболевание обычно начинается в раннем детстве. В основе патологии лежат демиелинизирующая полинейропатия, нарушения метаболизма липидов в виде жировой дегенерации нервов, клеток передних рогов спинного мозга, варолиевого моста, продолговатого мозга, мозжечка. В крови повышено содержание фитановой кислоты, меди, церуллоплазмина. определяются медленно прогрессирующие двигательные расстройства по периферическому типу, явления мозжечковой атаксии, расстройства чувствительности, снижение остроты зрения, слуха, обоняния. Иногда могут быть кардиомиопатия, деформация стоп, сколиоз, патология почек, снижение интеллекта. Лечение предусматривает диету с исключением продуктов, содержащих фитановую кислоту Болезнь Руси-Леви снижение скорости проведения возбуждения по нервам. l Гипертрофический интерстициальный неврит Дежерина. Скотта. является гипертрофия леммоцитов, разрастание элементов (швановской оболочки) l
 БОЛЕЗНИ С ПОРАЖЕНИЕМ ЭКСТРАПИРАМИДНОЙ НЕРВНОЙ СИСТЕМЫ
БОЛЕЗНИ С ПОРАЖЕНИЕМ ЭКСТРАПИРАМИДНОЙ НЕРВНОЙ СИСТЕМЫ
 хроническое прогрессирующее наследственно дегенеративное заболевание, характеризующееся сочетанным поражением") Гепатоцеребральная дистрофия (гепатолентикулярная дегенерация, болезнь Вестфаля—Вильсона—Коновалова) хроническое прогрессирующее наследственно дегенеративное заболевание, характеризующееся сочетанным поражением подкорковых узлов ЦНС и печени. l Описано в 1883 г. К. Вестфалем и в 1912 г. С. Вильсоном. l Термин «Гепатоцеребральная дистрофия» предложен Н. В. Коноваловым. l
Гепатоцеребральная дистрофия (гепатолентикулярная дегенерация, болезнь Вестфаля—Вильсона—Коновалова) хроническое прогрессирующее наследственно дегенеративное заболевание, характеризующееся сочетанным поражением подкорковых узлов ЦНС и печени. l Описано в 1883 г. К. Вестфалем и в 1912 г. С. Вильсоном. l Термин «Гепатоцеребральная дистрофия» предложен Н. В. Коноваловым. l
 Гепато церебральная дегенерация l l l наследственно дегенеративное заболевание, характеризующееся сочетанным поражением подкорковых узлов головного мозга и печени Тип наследования аутосомнорецессивный нарушение синтеза белка церулоплазмина, входящего в состав осглобулинов, транспортирующего медь. Вследствие этого создается высокая концентрация меди в крови и происходит ее отложение в органах и тканях, преимущественно в печени, мозге, роговице, а также в почках и других органах. дистрофические изменения нервных клеток с очаговыми размягчениями, образованием микрокист, разрастанием глии. изменения мелких сосудов мозговой ткани, кровоизлияния вокруг них, периваскулярный отек поступление меди в организм варьируется в пределах от 1 до 10 мг в сутки.
Гепато церебральная дегенерация l l l наследственно дегенеративное заболевание, характеризующееся сочетанным поражением подкорковых узлов головного мозга и печени Тип наследования аутосомнорецессивный нарушение синтеза белка церулоплазмина, входящего в состав осглобулинов, транспортирующего медь. Вследствие этого создается высокая концентрация меди в крови и происходит ее отложение в органах и тканях, преимущественно в печени, мозге, роговице, а также в почках и других органах. дистрофические изменения нервных клеток с очаговыми размягчениями, образованием микрокист, разрастанием глии. изменения мелких сосудов мозговой ткани, кровоизлияния вокруг них, периваскулярный отек поступление меди в организм варьируется в пределах от 1 до 10 мг в сутки.
 Складываются из симптомов поражения ЦНС и внутренних органов. l У больных появляются и нарастают мышечная ригидность, разнообразные гиперкинезы, псевдобульбарные симптомы, прогрессирующее снижение интеллекта, изменения со стороны печени и радужки l Клиническая картина.
Складываются из симптомов поражения ЦНС и внутренних органов. l У больных появляются и нарастают мышечная ригидность, разнообразные гиперкинезы, псевдобульбарные симптомы, прогрессирующее снижение интеллекта, изменения со стороны печени и радужки l Клиническая картина.
 выделяют четыре формы l 1. Ранняя ригидноаритмогиперкинетическая форма имеет наиболее злокачественное течение. Неврологические проявления развиваются в возрасте 7— 15 лет. Этому, как правило, предшествуют признаки поражения печени. В клинической картине преобладают мышечная ригидность и гиперкинезы. l 2. Дрожательно-ригидная и дрожательная формы, проявляющиеся в более позднем возрасте (17— 20 лет). Характеризуются одновременным появлением ригидности и дрожания, которое часто бывает первым признаком заболевания; постепенно нарастая, оно может становиться общим, захватывая мышцы туловища, конечностей, лица, челюстей, мягкого неба, надгортанника, голосовых связок, дыхательную мускулатуру, диафрагму. Нарушается глотание, речь становится скандированной. Часто отмечаются выраженные изменения психики.
выделяют четыре формы l 1. Ранняя ригидноаритмогиперкинетическая форма имеет наиболее злокачественное течение. Неврологические проявления развиваются в возрасте 7— 15 лет. Этому, как правило, предшествуют признаки поражения печени. В клинической картине преобладают мышечная ригидность и гиперкинезы. l 2. Дрожательно-ригидная и дрожательная формы, проявляющиеся в более позднем возрасте (17— 20 лет). Характеризуются одновременным появлением ригидности и дрожания, которое часто бывает первым признаком заболевания; постепенно нарастая, оно может становиться общим, захватывая мышцы туловища, конечностей, лица, челюстей, мягкого неба, надгортанника, голосовых связок, дыхательную мускулатуру, диафрагму. Нарушается глотание, речь становится скандированной. Часто отмечаются выраженные изменения психики.
 l l 3. Экстрапирамиднокорковая форма, выделенная Н. В. Коноваловым, отличается расстройством высших мозговых функций, наличием параличей, часто эпилептических припадков, грубым снижением интеллекта с изменениями личности. 4. Абдоминальная форма характеризуется преимущественным нарушением функции печени. Неврологические симптомы присоединяются в более поздних стадиях болезни.
l l 3. Экстрапирамиднокорковая форма, выделенная Н. В. Коноваловым, отличается расстройством высших мозговых функций, наличием параличей, часто эпилептических припадков, грубым снижением интеллекта с изменениями личности. 4. Абдоминальная форма характеризуется преимущественным нарушением функции печени. Неврологические симптомы присоединяются в более поздних стадиях болезни.
 Ведущим является синдром экстрапирамидных нарушений: ригидность мышц туловища, конечностей, лица, глотки и как следствие этого — нарушения походки, глотания, речи. Параллельно возникают гиперкинезы различного характера: тремор, атетоз, торсионная дистония интенционное дрожание, усиливающиеся при попытке выполнения произвольных движений. l Ранняя ригидно аритмогиперкинетическая форма, наиболее злокачественная по течению в возрасте 7— 15 лет. l
Ведущим является синдром экстрапирамидных нарушений: ригидность мышц туловища, конечностей, лица, глотки и как следствие этого — нарушения походки, глотания, речи. Параллельно возникают гиперкинезы различного характера: тремор, атетоз, торсионная дистония интенционное дрожание, усиливающиеся при попытке выполнения произвольных движений. l Ранняя ригидно аритмогиперкинетическая форма, наиболее злокачественная по течению в возрасте 7— 15 лет. l
, l") лабораторные исследования снижение содержания церулоплазмина (ниже 10 ЕД, при норме 25— 45 ЕД), l гипопротеинемия, гиперкупрумия (до 1000 мкг сут. и выше при норме 150 мкг/сут. ) и l Течение неуклонно прогрессирующее. Продолжительность жизни зависит от клинической формы заболевания, своевременности начатого лечения. l Прогноз. Средняя продолжительность жизни больных без лечения около 6 лет. l l гипераминоацидурия (до 1000 мг/сут. при норме 350 мг/сут. ). l повышение содержания аммиака в крови, изменение печеночных проб.
лабораторные исследования снижение содержания церулоплазмина (ниже 10 ЕД, при норме 25— 45 ЕД), l гипопротеинемия, гиперкупрумия (до 1000 мкг сут. и выше при норме 150 мкг/сут. ) и l Течение неуклонно прогрессирующее. Продолжительность жизни зависит от клинической формы заболевания, своевременности начатого лечения. l Прогноз. Средняя продолжительность жизни больных без лечения около 6 лет. l l гипераминоацидурия (до 1000 мг/сут. при норме 350 мг/сут. ). l повышение содержания аммиака в крови, изменение печеночных проб.
 Лечение l l l Основным методом лечения является выведение из организма избытка меди. Для этого используют тиоловые препараты, к которым относятся унитиол и D-neнцилламин. Дозы подбирают индивидуально: D пеницилламин в среднем назначают в дозе от 0, 45 до 2 г в сутки после еды Унитиол назначают повторными курсами по 5 мл 5% раствора внутримышечно ежедневно или через день (на курс 25 инъекций с перерывом между курсами 5— 6 мес). диета с ограничением продуктов, богатых медью, животных жиров, белков. Пища должна быть богата витаминами и углеводами. продолжительность жизни больных без лечения около 6 лет.
Лечение l l l Основным методом лечения является выведение из организма избытка меди. Для этого используют тиоловые препараты, к которым относятся унитиол и D-neнцилламин. Дозы подбирают индивидуально: D пеницилламин в среднем назначают в дозе от 0, 45 до 2 г в сутки после еды Унитиол назначают повторными курсами по 5 мл 5% раствора внутримышечно ежедневно или через день (на курс 25 инъекций с перерывом между курсами 5— 6 мес). диета с ограничением продуктов, богатых медью, животных жиров, белков. Пища должна быть богата витаминами и углеводами. продолжительность жизни больных без лечения около 6 лет.
 II. Синдромы прогрессирующей деменции, сочетающейся с другими выраженными неврологическими нарушениями Болезнь Гентингтона Это заболевание, характеризующееся сочетанием хореоатетоидных движений и прогрессирующей деменции, обычно начинается в среднем возрасте. Наследуется по аутосомно-доминантному типу. Как показали современные генетические исследования, детерминирующий его ген локализуется на коротком плече 4 -й хромосомы. l В связи с тем что болезнь неуклонно прогрессирует и неизбежно приводит к инвалидизации, а также затрагивает интересы всех членов семьи больного (риск развития заболевания у детей пораженного человека равен 50%), хорея Гентингтона в последние годы привлекает особое внимание. l Ложное впечатление о том, что семейный анамнез может быть негативным, складывается тогда, когда болезнь имеет поздний дебют и ее классифицируют как сенильную хорею, особенно если члены семьи умирали от других причин до манифестации заболевания. l
II. Синдромы прогрессирующей деменции, сочетающейся с другими выраженными неврологическими нарушениями Болезнь Гентингтона Это заболевание, характеризующееся сочетанием хореоатетоидных движений и прогрессирующей деменции, обычно начинается в среднем возрасте. Наследуется по аутосомно-доминантному типу. Как показали современные генетические исследования, детерминирующий его ген локализуется на коротком плече 4 -й хромосомы. l В связи с тем что болезнь неуклонно прогрессирует и неизбежно приводит к инвалидизации, а также затрагивает интересы всех членов семьи больного (риск развития заболевания у детей пораженного человека равен 50%), хорея Гентингтона в последние годы привлекает особое внимание. l Ложное впечатление о том, что семейный анамнез может быть негативным, складывается тогда, когда болезнь имеет поздний дебют и ее классифицируют как сенильную хорею, особенно если члены семьи умирали от других причин до манифестации заболевания. l
 Патогномоничными для болезни Гентингтона считают атрофию хвостатого ядра и, в меньшей степени, других структур базальных (подкорковых) узлов (скорлупа и бледный шар), выраженность которых не зависит от каких либо других изменений в мозге. Степень атрофии непосредственно коррелирует с тяжестью и длительностью течения заболевания. l На поздних стадиях в области хвостатого ядра, образующего в норме округлый выступ на поверхности боковой стенки бокового желудочка, формируется уплощение или углубление. В результате утраты ткани происходит соответствующее расширение желудочковой системы, особенно в области лобных рогов l происходит диффузная атрофия извилин, наиболее значительная на конвекситальной поверхности полушарий l потерю нейронов преимущественно в лобной коре l Патологические изменения
Патогномоничными для болезни Гентингтона считают атрофию хвостатого ядра и, в меньшей степени, других структур базальных (подкорковых) узлов (скорлупа и бледный шар), выраженность которых не зависит от каких либо других изменений в мозге. Степень атрофии непосредственно коррелирует с тяжестью и длительностью течения заболевания. l На поздних стадиях в области хвостатого ядра, образующего в норме округлый выступ на поверхности боковой стенки бокового желудочка, формируется уплощение или углубление. В результате утраты ткани происходит соответствующее расширение желудочковой системы, особенно в области лобных рогов l происходит диффузная атрофия извилин, наиболее значительная на конвекситальной поверхности полушарий l потерю нейронов преимущественно в лобной коре l Патологические изменения
 видно расширение боковых") МРТ l А. На фронтальном срезе (МРТ, Т 1 взвешенное изображение) видно расширение боковых желудочков (отмечено стрелками) вследствие типичной для болезни Гентингтона атрофии хвостатого ядра. l Б. На горизонтальном срезе (МРТ, Т 2 взвешенное изображение) видно увеличение интенсивности сигнала от скорлупы (отмечено стрелками).
МРТ l А. На фронтальном срезе (МРТ, Т 1 взвешенное изображение) видно расширение боковых желудочков (отмечено стрелками) вследствие типичной для болезни Гентингтона атрофии хвостатого ядра. l Б. На горизонтальном срезе (МРТ, Т 2 взвешенное изображение) видно увеличение интенсивности сигнала от скорлупы (отмечено стрелками).
 и участвующего") При нейрохимических исследованиях l l l резкое снижение уровня гамма-аминомасляной кислоты (ГАМК) и участвующего в ее синтезе фермента — декарбоксилазы глутаминовой кислоты (ДГК) в хвостатом ядре, скорлупе, бледном шаре, ретикулярной зоне черного вещества, а также некоторое уменьшение активности холинацетилтрансферазы (ХАТ) в хвостатом ядре. в полосатом теле и проекционно связанных с ним областях происходит истощение других нейромедиаторов повышено содержание пептидного нейромедиатора соматостатина в хвостатом ядре и скорлупе, существует селективная ранимость некоторых нейронов в определенных областях при сохранности других открыт патологический белок — гентингтин, который накапливается в нейронах.
При нейрохимических исследованиях l l l резкое снижение уровня гамма-аминомасляной кислоты (ГАМК) и участвующего в ее синтезе фермента — декарбоксилазы глутаминовой кислоты (ДГК) в хвостатом ядре, скорлупе, бледном шаре, ретикулярной зоне черного вещества, а также некоторое уменьшение активности холинацетилтрансферазы (ХАТ) в хвостатом ядре. в полосатом теле и проекционно связанных с ним областях происходит истощение других нейромедиаторов повышено содержание пептидного нейромедиатора соматостатина в хвостатом ядре и скорлупе, существует селективная ранимость некоторых нейронов в определенных областях при сохранности других открыт патологический белок — гентингтин, который накапливается в нейронах.
 Клинические аспекты l l l Двигательные нарушения обычно проявляются в молодом или среднем возрасте (35— 40 лет). Для данного заболевания характерно то, что более молодые пациенты, у которых первые симптомы заболевания возникли в возрасте 15— 40 лет, страдают более тяжелой его формой приводит к летальному исходу Начальные симптомы — типичный хореоатетоз или снижение интеллекта, на дальнейших стадиях имеются оба синдрома Атипичные движения, объединяющие элементы хореи, атетоза и дистонии, по внешнему виду как бы произвольные (как при двигательном беспокойстве), захватывают мышцы конечностей, туловища и головы. Насильственные движения (причудливые гримасы, нерегулярный ритм дыхания, нарушения артикуляции речи и нерегулярные, аритмичные, бессистемные движения конечностей, пританцовывающая походка) Эти движения мешают ходьбе и всем произвольным движениям.
Клинические аспекты l l l Двигательные нарушения обычно проявляются в молодом или среднем возрасте (35— 40 лет). Для данного заболевания характерно то, что более молодые пациенты, у которых первые симптомы заболевания возникли в возрасте 15— 40 лет, страдают более тяжелой его формой приводит к летальному исходу Начальные симптомы — типичный хореоатетоз или снижение интеллекта, на дальнейших стадиях имеются оба синдрома Атипичные движения, объединяющие элементы хореи, атетоза и дистонии, по внешнему виду как бы произвольные (как при двигательном беспокойстве), захватывают мышцы конечностей, туловища и головы. Насильственные движения (причудливые гримасы, нерегулярный ритм дыхания, нарушения артикуляции речи и нерегулярные, аритмичные, бессистемные движения конечностей, пританцовывающая походка) Эти движения мешают ходьбе и всем произвольным движениям.
 Обычно деменция и двигательные расстройства развиваются параллельно. Иногда деменция появляется до возникновения хореи или после этого; очень редко оба признака бывают слабо выраженными или отсутствуют. l Нейропсихические нарушения в виде депрессии, беспорядочного поведения и эмоциональных вспышек часто создают для больного значительные сложности еще до того, как деменция и двигательные расстройства станут глубокими. l Болезнь протекает медленно. Инвалидизация обусловлена усугублением как двигательных расстройств, так и изменений психики. l Смерть наступает спустя многие годы от интеркуррентной инфекции или, что нередко случается, в результате самоубийства. l
Обычно деменция и двигательные расстройства развиваются параллельно. Иногда деменция появляется до возникновения хореи или после этого; очень редко оба признака бывают слабо выраженными или отсутствуют. l Нейропсихические нарушения в виде депрессии, беспорядочного поведения и эмоциональных вспышек часто создают для больного значительные сложности еще до того, как деменция и двигательные расстройства станут глубокими. l Болезнь протекает медленно. Инвалидизация обусловлена усугублением как двигательных расстройств, так и изменений психики. l Смерть наступает спустя многие годы от интеркуррентной инфекции или, что нередко случается, в результате самоубийства. l
 l l l Специфическая терапия отсутствует. не существует ни одного метода лечения, который мог бы приостановить неуклонное прогрессирование болезни. Антагонисты дофаминовых рецепторов (бутирофеноны и фенотиазины) могут в какой-то степени уменьшить выраженность хореи У пациентов с болезнью Паркинсона при передозировке L дофа могут развиться распространенная хорея или хореоатетоз Галоперидол в дозах 2— 10 мг/сут уменьшает двигательные нарушения, но не влияет на прогноз заболевания.
l l l Специфическая терапия отсутствует. не существует ни одного метода лечения, который мог бы приостановить неуклонное прогрессирование болезни. Антагонисты дофаминовых рецепторов (бутирофеноны и фенотиазины) могут в какой-то степени уменьшить выраженность хореи У пациентов с болезнью Паркинсона при передозировке L дофа могут развиться распространенная хорея или хореоатетоз Галоперидол в дозах 2— 10 мг/сут уменьшает двигательные нарушения, но не влияет на прогноз заболевания.
 ДНК тестирование с исследованием тринуклеотидного ЦАГ участка гена Наиболее совершенным методом диагностики болезни Гентингтона (а также методом выявления пресимптомного носительства мутантного гена у клинически здоровых родственников) является ДНК тестирование с исследованием тринуклеотидного ЦАГ участка гена. l Обнаружение экспансии тринуклеотидных повторов в гене (свыше 36 копий ЦАГ-триплетов) позволяет с абсолютной точностью подтвердить наличие мутации. l Это имеет особое значение в атипичных и спорадических случаях БГ, а также в практике медико генетического консультирования при обследовании лиц из группы высокого риска. l
ДНК тестирование с исследованием тринуклеотидного ЦАГ участка гена Наиболее совершенным методом диагностики болезни Гентингтона (а также методом выявления пресимптомного носительства мутантного гена у клинически здоровых родственников) является ДНК тестирование с исследованием тринуклеотидного ЦАГ участка гена. l Обнаружение экспансии тринуклеотидных повторов в гене (свыше 36 копий ЦАГ-триплетов) позволяет с абсолютной точностью подтвердить наличие мутации. l Это имеет особое значение в атипичных и спорадических случаях БГ, а также в практике медико генетического консультирования при обследовании лиц из группы высокого риска. l
 Болезнь Галлервордена Шпатца l Первичный биохимический дефект при болезни Галлервордена Шпатца неизвестен. Принимая во внимание отложение железа в globus pallidus, substantia nigra и n. ruber, можно предположить, что развитие заболевания связано с нарушением метаболических процессов, протекающих с участием железосодержащих соединений. l Генетические данные. Болезнь Галлервордена Шпатца наследуется по аутосомно-рецессивному типу. Ген картирован на хромосоме 20 р. 12. 3 13.
Болезнь Галлервордена Шпатца l Первичный биохимический дефект при болезни Галлервордена Шпатца неизвестен. Принимая во внимание отложение железа в globus pallidus, substantia nigra и n. ruber, можно предположить, что развитие заболевания связано с нарушением метаболических процессов, протекающих с участием железосодержащих соединений. l Генетические данные. Болезнь Галлервордена Шпатца наследуется по аутосомно-рецессивному типу. Ген картирован на хромосоме 20 р. 12. 3 13.
 Критерии диагноза. Аутосомно рецессивный тип наследования. Дебют в детском или юношеском возрасте, но есть случаи и на 2 м, 3 м десятилетии жизни. l Раннее начало: генерализованная мышечная дистония, первично вовлекающая нижние конечности, пирамидный синдром, постуральные нарушения, снижение интеллекта, эпилептические припадки, пигментный ретинит или атрофия зрительного нерва. l Позднее начало: более медленное течение, преобладает паркинсонизм, характеризующийся акинезией, мышечной ригидностью, типичным тремором покоя, про и латеропульсией, постуральной неустойчивостью с частыми падениями, «маскообразным лицом» , орофасциальной (оромандибулярной) дистонией, дизартрией. l Прогрессирующая деменция. l l
Критерии диагноза. Аутосомно рецессивный тип наследования. Дебют в детском или юношеском возрасте, но есть случаи и на 2 м, 3 м десятилетии жизни. l Раннее начало: генерализованная мышечная дистония, первично вовлекающая нижние конечности, пирамидный синдром, постуральные нарушения, снижение интеллекта, эпилептические припадки, пигментный ретинит или атрофия зрительного нерва. l Позднее начало: более медленное течение, преобладает паркинсонизм, характеризующийся акинезией, мышечной ригидностью, типичным тремором покоя, про и латеропульсией, постуральной неустойчивостью с частыми падениями, «маскообразным лицом» , орофасциальной (оромандибулярной) дистонией, дизартрией. l Прогрессирующая деменция. l l
 С. «тигриных глаз» l Критерии диагноза l Характерная картина на МРТ (в режиме Т 2): в области бледного шара и черной субстанции – зоны резкого снижения интенсивности сигнала (отражение избыточного накопления железа в этих областях мозга). l Специфичностью является наличие зоны повышенного сигнала в переднемедиальной или центральной части бледного шара (как отражение гибели нейронов, глиоза и демиелинизации «глаз тигра» ).
С. «тигриных глаз» l Критерии диагноза l Характерная картина на МРТ (в режиме Т 2): в области бледного шара и черной субстанции – зоны резкого снижения интенсивности сигнала (отражение избыточного накопления железа в этих областях мозга). l Специфичностью является наличие зоны повышенного сигнала в переднемедиальной или центральной части бледного шара (как отражение гибели нейронов, глиоза и демиелинизации «глаз тигра» ).
 — характерная область") миндалевидый комплекс l l l Миндалевидное тело, миндалина (лат. corpus amygdaloideum) — характерная область мозга, имеющая форму миндалины, расположенной внутри височной доли (Lobus temporalis) головного мозга. Миндалина играет ключевую роль в формировании как отрицательных (страх), так и положительных эмоций (удовольствие). Её размер положительно коррелирует с агрессивным поведением. У людей это самая сексуально диморфная структура мозга — у мужчин после кастрации она сжимается более чем на 30 %. Предполагается, что такие состояния, как беспокойство, аутизм, депрессия, посттравматический шок и фобии, связаны с ненормальным функционированием миндалины. Миндалина — это фактически несколько отдельно функционирующих ядер, ключевыми являются: базально латеральный комплекс, центрально медиальные ядра и корково медиальные ядра. У пациентов, миндалевидное тело которых оказалось разрушено вследствие болезни Урбаха-Вите, наблюдается полное отсутствие страха.
миндалевидый комплекс l l l Миндалевидное тело, миндалина (лат. corpus amygdaloideum) — характерная область мозга, имеющая форму миндалины, расположенной внутри височной доли (Lobus temporalis) головного мозга. Миндалина играет ключевую роль в формировании как отрицательных (страх), так и положительных эмоций (удовольствие). Её размер положительно коррелирует с агрессивным поведением. У людей это самая сексуально диморфная структура мозга — у мужчин после кастрации она сжимается более чем на 30 %. Предполагается, что такие состояния, как беспокойство, аутизм, депрессия, посттравматический шок и фобии, связаны с ненормальным функционированием миндалины. Миндалина — это фактически несколько отдельно функционирующих ядер, ключевыми являются: базально латеральный комплекс, центрально медиальные ядра и корково медиальные ядра. У пациентов, миндалевидное тело которых оказалось разрушено вследствие болезни Урбаха-Вите, наблюдается полное отсутствие страха.
 III. Синдромы, сопровождающиеся постепенным развитием нарушений позы и движений. болезнь Паркинсона l Хроническое прогрессирующее дегенеративное заболевание головного мозга, преимущественно связанное с гибелью нейронов черной субстанции и других структур мозга и проявляющейся сочетание гипокинезии с ригидностью, тремором покоя, постуральной неустойчивостью, а также психическими и вегетативными расстройствами. l К моменту появления клинических симптомов заболевания происходит уменьшение количества дофаминэргических нейронов на 60 80%. l Дрожательный паралич — обычно встречается в виде спорадических случаев, но наблюдаются также и семейные случаи (частота последних составляет 1— 2%).
III. Синдромы, сопровождающиеся постепенным развитием нарушений позы и движений. болезнь Паркинсона l Хроническое прогрессирующее дегенеративное заболевание головного мозга, преимущественно связанное с гибелью нейронов черной субстанции и других структур мозга и проявляющейся сочетание гипокинезии с ригидностью, тремором покоя, постуральной неустойчивостью, а также психическими и вегетативными расстройствами. l К моменту появления клинических симптомов заболевания происходит уменьшение количества дофаминэргических нейронов на 60 80%. l Дрожательный паралич — обычно встречается в виде спорадических случаев, но наблюдаются также и семейные случаи (частота последних составляет 1— 2%).

 Патологические изменения Патологоанатомические изменения при дрожательном параличе изучены не полностью. l Чаще всего наблюдают скопления меланинсодержащих нервных клеток в стволе мозга (черное вещество, голубоватое место), где отмечаются различной степени выраженности гибель клеток и реактивный глиоз (особенно значительные в области черные вещества), а также l появляются характерные эозинофильные цитоплазматические включения (называемые тельцами Леви) l Результаты биохимических исследований, указывающие на снижение уровня дофамина в хвостатом ядре и скорлупе заболевания с преимущественным поражением нигростриарной дофаминергической системы. l
Патологические изменения Патологоанатомические изменения при дрожательном параличе изучены не полностью. l Чаще всего наблюдают скопления меланинсодержащих нервных клеток в стволе мозга (черное вещество, голубоватое место), где отмечаются различной степени выраженности гибель клеток и реактивный глиоз (особенно значительные в области черные вещества), а также l появляются характерные эозинофильные цитоплазматические включения (называемые тельцами Леви) l Результаты биохимических исследований, указывающие на снижение уровня дофамина в хвостатом ядре и скорлупе заболевания с преимущественным поражением нигростриарной дофаминергической системы. l
 Патоморфология болезни Паркинсона Норма Болезнь Паркинсона Substantia nigra: норма Substantia nigra: Депигментация Substantia nigra: норма Дегенерация клеток черной субстанции
Патоморфология болезни Паркинсона Норма Болезнь Паркинсона Substantia nigra: норма Substantia nigra: Депигментация Substantia nigra: норма Дегенерация клеток черной субстанции
 «У шимпанзе не было выявлено заметных изменений в структуре мозга, чего нельзя сказать о людях» , – пишут ученые, подводя итоги исследования. В мужских мозгах возрастные изменения объема структур мозга были меньшими по сравнению с женскими. l Ученые полагают, что возрастной деменцией человек расплачивается за годы, которые он проживает после утраты репродуктивных способностей. Мозг – самая уязвимая часть лишь потому, что он – «энергетический обжора» . l
«У шимпанзе не было выявлено заметных изменений в структуре мозга, чего нельзя сказать о людях» , – пишут ученые, подводя итоги исследования. В мужских мозгах возрастные изменения объема структур мозга были меньшими по сравнению с женскими. l Ученые полагают, что возрастной деменцией человек расплачивается за годы, которые он проживает после утраты репродуктивных способностей. Мозг – самая уязвимая часть лишь потому, что он – «энергетический обжора» . l
 мультисистемная дегенерация 6 стадий. l Стадия 1 поражение дорсального моторного ядра") Н. Braak (2002) мультисистемная дегенерация 6 стадий. l Стадия 1 поражение дорсального моторного ядра блуждающего нерва, а также обонятельной луковицы и прилегающей части переднего обонятельного ядра (аносмия и дисфункция желудочно кишечного тракта запоры). l Стадия 2 нарастание дегенеративных изменений в дорсальном моторном ядре и распространением дегенеративного процесса на ядра шва и магноцеллюлярные части ретикулярной формации, (тельца Леви впервые появляются в области голубого пятна). Клинически аффективные расстройства, а также сенсорные изменения, в частности болевые проявления.
Н. Braak (2002) мультисистемная дегенерация 6 стадий. l Стадия 1 поражение дорсального моторного ядра блуждающего нерва, а также обонятельной луковицы и прилегающей части переднего обонятельного ядра (аносмия и дисфункция желудочно кишечного тракта запоры). l Стадия 2 нарастание дегенеративных изменений в дорсальном моторном ядре и распространением дегенеративного процесса на ядра шва и магноцеллюлярные части ретикулярной формации, (тельца Леви впервые появляются в области голубого пятна). Клинически аффективные расстройства, а также сенсорные изменения, в частности болевые проявления.
 l Стадия 3 вовлечение компактной части черной субстанции (при этом не происходит снижения численности нейронов) и миндалины, а также педункулопонтинного ядра, орального ядра шва. Клинически могут развиваться нарушение сна и аффективные расстройства. Стадия 4 снижение количества нейронов компактной части черной субстанции, приводящим к развитию классических моторных проявлений БП. Вовлекаются переднемедиальные отделы височной доли, а также гиппокамп. l Появляется когнитивный дефицит, в том числе ослабление памяти, а также снижение личностной инициативы. l
l Стадия 3 вовлечение компактной части черной субстанции (при этом не происходит снижения численности нейронов) и миндалины, а также педункулопонтинного ядра, орального ядра шва. Клинически могут развиваться нарушение сна и аффективные расстройства. Стадия 4 снижение количества нейронов компактной части черной субстанции, приводящим к развитию классических моторных проявлений БП. Вовлекаются переднемедиальные отделы височной доли, а также гиппокамп. l Появляется когнитивный дефицит, в том числе ослабление памяти, а также снижение личностной инициативы. l
 l Стадия 5 критическая гибель нейронов компактной части черной субстанции. Соответственно клинически развиваются моторные флуктуации. Вовлекаются структуры неокортекса, прежде всего зоны в префронтальной, височной и теменной коре, что сопровождается нарастанием когнитивных нарушений и развитием психотических расстройств. l Стадия 6 усугубление изменений в ранее пораженных структурах с вовлечением первичных моторных и сенсорных зон коры с развитием выраженных когнитивных нарушений
l Стадия 5 критическая гибель нейронов компактной части черной субстанции. Соответственно клинически развиваются моторные флуктуации. Вовлекаются структуры неокортекса, прежде всего зоны в префронтальной, височной и теменной коре, что сопровождается нарастанием когнитивных нарушений и развитием психотических расстройств. l Стадия 6 усугубление изменений в ранее пораженных структурах с вовлечением первичных моторных и сенсорных зон коры с развитием выраженных когнитивных нарушений
 Симптомы паркинсонизма
Симптомы паркинсонизма
 l l l Ригидность — повышение мышечного тонуса, проявляются повышенным сопротивлением пассивным движениям. Повышенное сопротивление может быть: монотонным, часто нарастающим при повторном движении (феномен «восковая кукла» ); толчкообразно меняющимся (феномен «зубчатое колесо» ). У большинства больных с БП ригидность быстрее всего развивается в дистальных отделах рук и имеет четко асимметричный характер усиливается при движении. Относительно рано при БП развивается и ригидность мышц шеи, которая выявляется при пассивных движениях головой.
l l l Ригидность — повышение мышечного тонуса, проявляются повышенным сопротивлением пассивным движениям. Повышенное сопротивление может быть: монотонным, часто нарастающим при повторном движении (феномен «восковая кукла» ); толчкообразно меняющимся (феномен «зубчатое колесо» ). У большинства больных с БП ригидность быстрее всего развивается в дистальных отделах рук и имеет четко асимметричный характер усиливается при движении. Относительно рано при БП развивается и ригидность мышц шеи, которая выявляется при пассивных движениях головой.
 l Повышение тонуса мыщц антагонистов при пассивных движениях можно определить следующим приемом: если поднять голову лежащего, а потом резко отпустить руку, то голова не упадет на подушку, а опустится относительно плавно. Иногда голова в положении лежа несколько приподнята — феномен «воображаемой подушки» .
l Повышение тонуса мыщц антагонистов при пассивных движениях можно определить следующим приемом: если поднять голову лежащего, а потом резко отпустить руку, то голова не упадет на подушку, а опустится относительно плавно. Иногда голова в положении лежа несколько приподнята — феномен «воображаемой подушки» .
 Тремор — характерный, хотя и не обязательный для синдрома паркинсонизма симптом. l Это ритмичное, регулярное, непроизвольное дрожание конечностей, лицевой мускулатуры, головы, нижней челюсти, языка, более выраженное в покое, уменьшающееся при активных движениях. Частота колебаний 4— 8 в секунду. l Тремор усиливается при волнениях, практически исчезает во сне. l
Тремор — характерный, хотя и не обязательный для синдрома паркинсонизма симптом. l Это ритмичное, регулярное, непроизвольное дрожание конечностей, лицевой мускулатуры, головы, нижней челюсти, языка, более выраженное в покое, уменьшающееся при активных движениях. Частота колебаний 4— 8 в секунду. l Тремор усиливается при волнениях, практически исчезает во сне. l
 Тремор Для БП характерен тремор покоя, который проявляется в покоящейся конечности (чаще всего в дистальном отделе руки или ноги), уменьшается при ее движении, но усиливается при активных движениях другими конечностями. Выраженный тремор покоя в руке напоминает "скатывание пилюль" или "счет монет". Тремор покоя может быть начальным симптомом примерно у половины больных, а в течение заболевания развивается у 85% больных БП. l Постуральный тремор, возникающий при удержании позы (например, при удержании вытянутых вперед рук), или l кинетический тремор, возникающий при движении (например, тремор в руке при выполнении пальценосовой пробы). l Интенционный тремор, возникающий приближении к цели исключает БП. l
Тремор Для БП характерен тремор покоя, который проявляется в покоящейся конечности (чаще всего в дистальном отделе руки или ноги), уменьшается при ее движении, но усиливается при активных движениях другими конечностями. Выраженный тремор покоя в руке напоминает "скатывание пилюль" или "счет монет". Тремор покоя может быть начальным симптомом примерно у половины больных, а в течение заболевания развивается у 85% больных БП. l Постуральный тремор, возникающий при удержании позы (например, при удержании вытянутых вперед рук), или l кинетический тремор, возникающий при движении (например, тремор в руке при выполнении пальценосовой пробы). l Интенционный тремор, возникающий приближении к цели исключает БП. l
 Постуральные нарушения представлены изменением позы и постуральной неустойчивостью — нарушением способности удерживать равновесие при изменении положения или ходьбе. Постуральная неустойчивость —приводит к частым падениям. Изменение позы связано с преобладанием тонуса в сгибательной мускулатуре, что приводит к сгибательной позе ( «поза просителя» ). l В наиболее тяжелых случаях может развиваться выраженный наклон туловища вперед — камптокормия. l Падения чаще происходят вперед.
Постуральные нарушения представлены изменением позы и постуральной неустойчивостью — нарушением способности удерживать равновесие при изменении положения или ходьбе. Постуральная неустойчивость —приводит к частым падениям. Изменение позы связано с преобладанием тонуса в сгибательной мускулатуре, что приводит к сгибательной позе ( «поза просителя» ). l В наиболее тяжелых случаях может развиваться выраженный наклон туловища вперед — камптокормия. l Падения чаще происходят вперед.
 Появляется своеобразная сгибательная поза: голова и туловище наклонены вперед, руки полусогнуты в локтевых, лучезапястных и фаланговых суставах, нередко плотно приведены к боковым поверхностям грудной клетки, туловища, ноги полусогнуты в коленных суставах. Отмечается бедность мимики. l Темп произвольных движений с развитием заболевания постепенно замедляется, иногда довольно рано может наступить полная обездвиженность l
Появляется своеобразная сгибательная поза: голова и туловище наклонены вперед, руки полусогнуты в локтевых, лучезапястных и фаланговых суставах, нередко плотно приведены к боковым поверхностям грудной клетки, туловища, ноги полусогнуты в коленных суставах. Отмечается бедность мимики. l Темп произвольных движений с развитием заболевания постепенно замедляется, иногда довольно рано может наступить полная обездвиженность l
 Нарушение ходьбы l l l Нарушения ходьбы наблюдаются начиная с ранней стадии заболевания. Поначалу они бывают представлены уменьшением длины шага (микробазия), отсутствием содружественных движений рук при ходьбе (ахейрокинез), затруднением начала (инициации) ходьбы, шарканьем. Скорость ходьбы снижена, главным образом, за счет уменьшения длины шага, но не частоты шагов. При необходимости увеличить скорость ходьбы больные переходят на более быстрый шаг, но не могут увеличить длину шага — из за неспособности генерировать адекватное мышечное усилие, что преимущественно обусловлено гипокинезией.
Нарушение ходьбы l l l Нарушения ходьбы наблюдаются начиная с ранней стадии заболевания. Поначалу они бывают представлены уменьшением длины шага (микробазия), отсутствием содружественных движений рук при ходьбе (ахейрокинез), затруднением начала (инициации) ходьбы, шарканьем. Скорость ходьбы снижена, главным образом, за счет уменьшения длины шага, но не частоты шагов. При необходимости увеличить скорость ходьбы больные переходят на более быстрый шаг, но не могут увеличить длину шага — из за неспособности генерировать адекватное мышечное усилие, что преимущественно обусловлено гипокинезией.
 парадоксальная кинезия: больной, не способный самостоятельно идти по ровной поверхности, легко взбирается и спускается по лестнице или идет по нарисованной линии. l Значительно чаще на поздних стадиях развивается так называемая парадоксальная акинезия, или застывания (freezing) — внезапная блокада осуществляемого движения, в результате которой ноги пациента буквально «прирастают» к полу. l
парадоксальная кинезия: больной, не способный самостоятельно идти по ровной поверхности, легко взбирается и спускается по лестнице или идет по нарисованной линии. l Значительно чаще на поздних стадиях развивается так называемая парадоксальная акинезия, или застывания (freezing) — внезапная блокада осуществляемого движения, в результате которой ноги пациента буквально «прирастают» к полу. l
. Если толкнуть больного вперед, он бежит,") Нередко наблюдается склонность к непроизвольному бегу вперед (пропульсии). Если толкнуть больного вперед, он бежит, чтобы не упасть, как бы «догоняя свой центр тяжести» . Часто толчок в грудь ведет к бегу назад (ретро пульсии), в сторону (латеропульсии). l Часто при резко выраженном синдроме позы больного напоминают каталептические. l Акинез и пластическая гипертония особенно резко проявляются в мускулатуре лица, жевательных и затылочных мышцах, мышцах конечностей. l Речь тихая, монотонная, без модуляций, с наклонностью к затуханию в конце фразы l
Нередко наблюдается склонность к непроизвольному бегу вперед (пропульсии). Если толкнуть больного вперед, он бежит, чтобы не упасть, как бы «догоняя свой центр тяжести» . Часто толчок в грудь ведет к бегу назад (ретро пульсии), в сторону (латеропульсии). l Часто при резко выраженном синдроме позы больного напоминают каталептические. l Акинез и пластическая гипертония особенно резко проявляются в мускулатуре лица, жевательных и затылочных мышцах, мышцах конечностей. l Речь тихая, монотонная, без модуляций, с наклонностью к затуханию в конце фразы l
 Психические нарушения l Психические нарушения проявляются утратой инициативы, активности, сужением кругозора и интересов, резким понижением различных эмоциональных реакций и аффектов, а также некоторой поверхностью и медлительностью мышления (брадифрения). Наблюдаются брадипсихия — трудное активное переключение с одной мысли на другую, акайрия — прилипчивость, вязкость, эгоцентризм. Иногда возникают пароксизмы психического возбуждения.
Психические нарушения l Психические нарушения проявляются утратой инициативы, активности, сужением кругозора и интересов, резким понижением различных эмоциональных реакций и аффектов, а также некоторой поверхностью и медлительностью мышления (брадифрения). Наблюдаются брадипсихия — трудное активное переключение с одной мысли на другую, акайрия — прилипчивость, вязкость, эгоцентризм. Иногда возникают пароксизмы психического возбуждения.
 Немоторные проявления БП
Немоторные проявления БП
 клинические формы l Ригиднобрадикинетическая форма характеризуется повышением тонуса мышц по пластическому типу, прогрессирующим замедлением активных движений вплоть до обездвиженности; появляются мышечные контрактуры, флексорная поза больных ригидно брадикинетическую
клинические формы l Ригиднобрадикинетическая форма характеризуется повышением тонуса мышц по пластическому типу, прогрессирующим замедлением активных движений вплоть до обездвиженности; появляются мышечные контрактуры, флексорная поза больных ригидно брадикинетическую
 l Дрожательно-ригидная форма характеризуется тремором конечностей, преимущественно их дистальных отделов, к которому с развитием заболевания присоединяется скованность произвольных движений
l Дрожательно-ригидная форма характеризуется тремором конечностей, преимущественно их дистальных отделов, к которому с развитием заболевания присоединяется скованность произвольных движений
 l Для дрожательной формы паркинсонизма характерно наличие постоянного или почти постоянного средне и крупноамплитудного тремора конечностей, языка, головы, нижней челюсти. Тонус мышц нормальный или несколько повышен. Темп произвольных движений сохранен.
l Для дрожательной формы паркинсонизма характерно наличие постоянного или почти постоянного средне и крупноамплитудного тремора конечностей, языка, головы, нижней челюсти. Тонус мышц нормальный или несколько повышен. Темп произвольных движений сохранен.
 Словарь l моторные флюктуации колебание активности двигательной период «включения» период двигательной активности пациентов l период «выключения» период снижения двигательной активности пациентов l феномен «истощения» конца дозы или акинезия конца дозы l дискинезии избыточная двигательная активность l Гипокинезия проявляющееся замедленностью и обеднением рисунка движений l Ригидность пластическое повышение тонуса мышц, l постуральная неустойчивость при изменении позы , l
Словарь l моторные флюктуации колебание активности двигательной период «включения» период двигательной активности пациентов l период «выключения» период снижения двигательной активности пациентов l феномен «истощения» конца дозы или акинезия конца дозы l дискинезии избыточная двигательная активность l Гипокинезия проявляющееся замедленностью и обеднением рисунка движений l Ригидность пластическое повышение тонуса мышц, l постуральная неустойчивость при изменении позы , l
 Стадия 1. 0 — только односторонние") Классификация l l l l Шкала Хен—Яра (1967) Стадия 1. 0 — только односторонние проявления. Стадия 1. 5 — односторонние проявления с вовлечением аксиальной мускулатуры. Стадия 2. 0 — двухсторонние проявления без признаков нарушения равновесия. Стадия 2. 5 — мягкие двухсторонние проявления. Сохранена способность преодолевать вызванную ретропульсию. Стадия 3. 0 — умеренные или средней тяжести двухсторонние проявления. Небольшая постуральная неустойчивость. Но больной не нуждается в посторонней помощи. Стадия 4. 0 — тяжёлая обездвиженность; однако ещё может ходить или стоять без поддержки. Стадия 5. 0 — без посторонней помощи прикован к креслу или кровати.
Классификация l l l l Шкала Хен—Яра (1967) Стадия 1. 0 — только односторонние проявления. Стадия 1. 5 — односторонние проявления с вовлечением аксиальной мускулатуры. Стадия 2. 0 — двухсторонние проявления без признаков нарушения равновесия. Стадия 2. 5 — мягкие двухсторонние проявления. Сохранена способность преодолевать вызванную ретропульсию. Стадия 3. 0 — умеренные или средней тяжести двухсторонние проявления. Небольшая постуральная неустойчивость. Но больной не нуждается в посторонней помощи. Стадия 4. 0 — тяжёлая обездвиженность; однако ещё может ходить или стоять без поддержки. Стадия 5. 0 — без посторонней помощи прикован к креслу или кровати.
 Темп прогрессирования Быстрый темп прогрессирования, при котором смена стадий заболевания (первая —> вторая или вторая —> третья) происходит в течение 2 или менее лет. l Умеренный темп прогрессирования, при котором смена стадий происходит более чем за 2 года, но не более чем за 5 l Медленный темп прогрессирования со сменой стадий более чем через 5 лет. l
Темп прогрессирования Быстрый темп прогрессирования, при котором смена стадий заболевания (первая —> вторая или вторая —> третья) происходит в течение 2 или менее лет. l Умеренный темп прогрессирования, при котором смена стадий происходит более чем за 2 года, но не более чем за 5 l Медленный темп прогрессирования со сменой стадий более чем через 5 лет. l
 Ювенильный паркинсонизм l вариант первичного паркинсонизма с дебютом на 2 3 десятилетии жизни, наследуется по аутосомно рецессивному типу, связан с мутациями на 6 хромосоме - в гене, кодирующем белок паркин. При этом варианте происходит дегенерация нигростриарных дофаминергических нейронов без образования телец Леви. Клинически характерно медленно прогрессирующее течение, наличие постурального тремора и дистонического гиперкинеза, отсутствие выраженных когнитивных нарушений, раннее развитие моторных флуктуации и дискинезий.
Ювенильный паркинсонизм l вариант первичного паркинсонизма с дебютом на 2 3 десятилетии жизни, наследуется по аутосомно рецессивному типу, связан с мутациями на 6 хромосоме - в гене, кодирующем белок паркин. При этом варианте происходит дегенерация нигростриарных дофаминергических нейронов без образования телец Леви. Клинически характерно медленно прогрессирующее течение, наличие постурального тремора и дистонического гиперкинеза, отсутствие выраженных когнитивных нарушений, раннее развитие моторных флуктуации и дискинезий.
 Посттравматический паркинсонизм l редок, развивается в результате острой тяжелой черепно мозговой травмы или повторяющихся легких черепно мозговых травм «энцефалопатия боксёров» . Помимо проявлений паркинсонизма характерны пирамидные, псевдобульбарные, мозжечковые проявления, медленно прогрессирующая деменция, малая эффективность препаратов леводопы.
Посттравматический паркинсонизм l редок, развивается в результате острой тяжелой черепно мозговой травмы или повторяющихся легких черепно мозговых травм «энцефалопатия боксёров» . Помимо проявлений паркинсонизма характерны пирамидные, псевдобульбарные, мозжечковые проявления, медленно прогрессирующая деменция, малая эффективность препаратов леводопы.
 Сосудистый паркинсонизм l l синдром, встречающийся примерно в 10 раз реже, чем болезнь Паркинсона, развивается на фоне хронической ишемии головного мозга, либо вследствие инсульта. Периоды прогрессирования симптомов чередуются с периодами стабилизации. l Клинически характерно преимущественное вовлечение нижних конечностей, выраженные нарушения ходьбы и равновесия, отсутствие тремора покоя, неэффективность дофаминергических средств, частое сочетание с псевдобульбарными, пирамидными, мозжечковыми, тазовыми нарушениями, раннее развитие деменции. Диагноз уточняют с помощью КТ или МРТ, лечение направлено на предупреждение дальнейшего сосудистого повреждения мозга.
Сосудистый паркинсонизм l l синдром, встречающийся примерно в 10 раз реже, чем болезнь Паркинсона, развивается на фоне хронической ишемии головного мозга, либо вследствие инсульта. Периоды прогрессирования симптомов чередуются с периодами стабилизации. l Клинически характерно преимущественное вовлечение нижних конечностей, выраженные нарушения ходьбы и равновесия, отсутствие тремора покоя, неэффективность дофаминергических средств, частое сочетание с псевдобульбарными, пирамидными, мозжечковыми, тазовыми нарушениями, раннее развитие деменции. Диагноз уточняют с помощью КТ или МРТ, лечение направлено на предупреждение дальнейшего сосудистого повреждения мозга.
 Изменения МРТ при сосудистом паркинсонизме
Изменения МРТ при сосудистом паркинсонизме
 Лекарственный паркинсонизм редок, встречается у больных психиатрических лечебниц, отличается быстрым развитием, симметричностью симптоматики, редкостью типичного тремора покоя, но частой встречаемостью грубого постурального тремора и акинезии. l Вызывается нейролептиками, метилдофой, резерпином, амиодароном, антагонистами кальция (циннаризин, флунаризин, дилгиазем), индометацином, циклоспорином, вальпроатом натрия, препаратами лития. l Обычно симптоматика исчезает после отмены препарата в течение 4 8 недель, l Прогрессирование симптомов паркинсонизма после отмены препарата указывает на наличие у больного нейродегенеративного заболевания l
Лекарственный паркинсонизм редок, встречается у больных психиатрических лечебниц, отличается быстрым развитием, симметричностью симптоматики, редкостью типичного тремора покоя, но частой встречаемостью грубого постурального тремора и акинезии. l Вызывается нейролептиками, метилдофой, резерпином, амиодароном, антагонистами кальция (циннаризин, флунаризин, дилгиазем), индометацином, циклоспорином, вальпроатом натрия, препаратами лития. l Обычно симптоматика исчезает после отмены препарата в течение 4 8 недель, l Прогрессирование симптомов паркинсонизма после отмены препарата указывает на наличие у больного нейродегенеративного заболевания l
 Постэнцефалитический паркинсонизм редок, является осложнением вирусного энцефалита, обычно имеет регрессирующее течение. l Прогрессирующий надъядерный паралич спорадическое дегенеративное заболевание, возникает после 60 70 лет, характеризуется сочетанием паркинсонизма с надъядерной офтальмоплегией (парез вертикального взора, преимущественно вниз). l Проявления паркинсонизма развиваются симметрично, выражена гипомимия, тремор покоя обычно отсутствует, гипокинезия и ригидность более выражены в проксимальных отделах конечностей, быстро нарастает деменция. l МРТ выявляет атрофию среднего мозга. l
Постэнцефалитический паркинсонизм редок, является осложнением вирусного энцефалита, обычно имеет регрессирующее течение. l Прогрессирующий надъядерный паралич спорадическое дегенеративное заболевание, возникает после 60 70 лет, характеризуется сочетанием паркинсонизма с надъядерной офтальмоплегией (парез вертикального взора, преимущественно вниз). l Проявления паркинсонизма развиваются симметрично, выражена гипомимия, тремор покоя обычно отсутствует, гипокинезия и ригидность более выражены в проксимальных отделах конечностей, быстро нарастает деменция. l МРТ выявляет атрофию среднего мозга. l
 l Лечение Нейропротекторная терапия призвана замедлять утрату дофаминэргических нейронов, и тем самым тормозить прогрессирование заболевания. В настоящее время отсутствуют нейропротекторные средств с доказанной эффективностью. В качестве возможных нейропротекторных препаратов рассматриваются ингибиторы МАО В ( селегилин ( Юмекс ), расагилин), агонисты дофаминовых рецепторов ( прамипексол ( Мирапекс ), ропинирол) и амантадин ( Мидантан , ПК-Мерц ). Леводопа является наиболее эффективным препаратом для лечения двигательных нарушений при болезни Паркинсона, она обеспечивает хороший контроль над симптомами заболевания в течение 4 – 6 лет. l При преобладании в клинической картине больного тремора покоя могут быть назначены антихолинергические препараты (циклодол, акинетон) пациентам сравнительно молодого возраста на ранних этапах заболевания, или больным старше 60 лет с дрожательной формой болезни Паркинсона (при недостаточной эффективности леводопы) l
l Лечение Нейропротекторная терапия призвана замедлять утрату дофаминэргических нейронов, и тем самым тормозить прогрессирование заболевания. В настоящее время отсутствуют нейропротекторные средств с доказанной эффективностью. В качестве возможных нейропротекторных препаратов рассматриваются ингибиторы МАО В ( селегилин ( Юмекс ), расагилин), агонисты дофаминовых рецепторов ( прамипексол ( Мирапекс ), ропинирол) и амантадин ( Мидантан , ПК-Мерц ). Леводопа является наиболее эффективным препаратом для лечения двигательных нарушений при болезни Паркинсона, она обеспечивает хороший контроль над симптомами заболевания в течение 4 – 6 лет. l При преобладании в клинической картине больного тремора покоя могут быть назначены антихолинергические препараты (циклодол, акинетон) пациентам сравнительно молодого возраста на ранних этапах заболевания, или больным старше 60 лет с дрожательной формой болезни Паркинсона (при недостаточной эффективности леводопы) l
 Контрольная МРТ головного мозга пациента после двухсторонней стимуляции субталамического ядра.
Контрольная МРТ головного мозга пациента после двухсторонней стимуляции субталамического ядра.
 l МРТ пациента после выполнения правосторонней таламотомии. l МРТ пациента после выполнения левосторонней паллидотомии.
l МРТ пациента после выполнения правосторонней таламотомии. l МРТ пациента после выполнения левосторонней паллидотомии.
 БОЛЕЗНИ С ПОРАЖЕНИЕМ ПИРАМИДНОЙ СИСТЕМЫ
БОЛЕЗНИ С ПОРАЖЕНИЕМ ПИРАМИДНОЙ СИСТЕМЫ
 Спастический семейный паралич Щтрюмпеля. Тип наследования аутосомно рецессивный. Морфологически у больных отмечаются дегенеративные изменения в боковых (реже в передних) столбах спинного мозга, продолговатом мозге, мозжечковых проводящих путях, иногда корковых отделах лобных долей. l Заболевание начинается в конце первого десятилетия жизни. l Клинически определяются спастические парезы в ногах с наличием гипертонуса, гиперрефлексии, появлением патологических рефлексов. Позже в процесс вовлекаются верхние конечности. Могут иметь место бульбарные симптомы. Походка затрудняется, появляются клонусы стон. Чувствительных расстройств и патологии тазовых органов обычно не бывает l Лечение заключается в назначении препаратов, снижающих мышечный тонус (мидокалм, баклофен, сирдалуд), стероидных гормонов, общеукрепляющих средств. l l
Спастический семейный паралич Щтрюмпеля. Тип наследования аутосомно рецессивный. Морфологически у больных отмечаются дегенеративные изменения в боковых (реже в передних) столбах спинного мозга, продолговатом мозге, мозжечковых проводящих путях, иногда корковых отделах лобных долей. l Заболевание начинается в конце первого десятилетия жизни. l Клинически определяются спастические парезы в ногах с наличием гипертонуса, гиперрефлексии, появлением патологических рефлексов. Позже в процесс вовлекаются верхние конечности. Могут иметь место бульбарные симптомы. Походка затрудняется, появляются клонусы стон. Чувствительных расстройств и патологии тазовых органов обычно не бывает l Лечение заключается в назначении препаратов, снижающих мышечный тонус (мидокалм, баклофен, сирдалуд), стероидных гормонов, общеукрепляющих средств. l l
 Органические гиперкинезы l Атетоз l Гемибаллизм l Дрожание l Миоклонии l Тики l Хореический или реперкуссивный гиперкинез
Органические гиперкинезы l Атетоз l Гемибаллизм l Дрожание l Миоклонии l Тики l Хореический или реперкуссивный гиперкинез
 – подвижный Изменчивый гиперкинез, медленный тонический спазм червеобразного типа") Атетоз l l Athetosis (греческий) – подвижный Изменчивый гиперкинез, медленный тонический спазм червеобразного типа мышц дистальных отделов конечностей, иногда лица и тела. Попеременное повышение и снижение мышечного тонуса агонистов и антагонистов ( «щупальца спрута» ), наблюдается нерегулярное повышение мышечного напряжения в агонистах и антагонистах. l Атетоз обычно вызывается повреждением полосатого тела. Поражаются клетки скорлупы и хвостатого тела
Атетоз l l Athetosis (греческий) – подвижный Изменчивый гиперкинез, медленный тонический спазм червеобразного типа мышц дистальных отделов конечностей, иногда лица и тела. Попеременное повышение и снижение мышечного тонуса агонистов и антагонистов ( «щупальца спрута» ), наблюдается нерегулярное повышение мышечного напряжения в агонистах и антагонистах. l Атетоз обычно вызывается повреждением полосатого тела. Поражаются клетки скорлупы и хвостатого тела
 Лицевой параспазм l локальный гиперкинез, проявляющийся тоническими симметричными сокращениями мимических мышц, мускулатуры языка, век. l Блефароспазм l Провоцируется разговором, едой, улыбкой, усиливается при волнении, ярком освещении и исчезает во сне. характеризуется спонтанным форсированным зажмуриванием глаз, что может привести к функциональной слепоте. Эти движения могут отмечаться у пациентов с паркинсонизмом и торсионной дистонией, но бывают и результатом побочного эффекта нейролептической терапии. Блефароспазм описан при поражении дорсомедиальных отделов покрышки моста или верхней части ствола.
Лицевой параспазм l локальный гиперкинез, проявляющийся тоническими симметричными сокращениями мимических мышц, мускулатуры языка, век. l Блефароспазм l Провоцируется разговором, едой, улыбкой, усиливается при волнении, ярком освещении и исчезает во сне. характеризуется спонтанным форсированным зажмуриванием глаз, что может привести к функциональной слепоте. Эти движения могут отмечаться у пациентов с паркинсонизмом и торсионной дистонией, но бывают и результатом побочного эффекта нейролептической терапии. Блефароспазм описан при поражении дорсомедиальных отделов покрышки моста или верхней части ствола.
 Спастическая кривошея l l l наиболее частые формы мышечной дистонии. При обоих заболеваниях обычно поражаются скорлупа и центромедианное ядре таламуса, а также другие экстрапирамидные ядра (бледный шар, черное вещество и др. ). Спастическая кривошея — тоническое расстройство, выражающееся в спастических сокращениях мышц шейной области, приводящих к медленным, непроизвольным поворотам и наклонам головы. Больные часто используют компенсаторные приемы для уменьшения гиперкинеза, в частности поддерживают голову рукой. Помимо других мышц шеи, особенно часто вовлекаются ; процесс грудинно ключично сосцевидная и трапециевидная мышцы. Спастическая кривошея может представлять собой локальную форм, торсионной дистонии или
Спастическая кривошея l l l наиболее частые формы мышечной дистонии. При обоих заболеваниях обычно поражаются скорлупа и центромедианное ядре таламуса, а также другие экстрапирамидные ядра (бледный шар, черное вещество и др. ). Спастическая кривошея — тоническое расстройство, выражающееся в спастических сокращениях мышц шейной области, приводящих к медленным, непроизвольным поворотам и наклонам головы. Больные часто используют компенсаторные приемы для уменьшения гиперкинеза, в частности поддерживают голову рукой. Помимо других мышц шеи, особенно часто вовлекаются ; процесс грудинно ключично сосцевидная и трапециевидная мышцы. Спастическая кривошея может представлять собой локальную форм, торсионной дистонии или
 Торсионная дистония — вовлечение в патологический процесс мускулатуры туловища, грудной клетки с вращательными движениями туловища и проксимальных сегментов конечностей. Они могут быть настолько выраженными, что без поддержки больной не может ни стоять, ни ходить. l Возможна идиопатическая торсионная дистония или дистония как проявление энцефалита, хореи Гентингтона, болезни Галлевердена—Шпатца, гепатоцеребральной дистрофии. l
Торсионная дистония — вовлечение в патологический процесс мускулатуры туловища, грудной клетки с вращательными движениями туловища и проксимальных сегментов конечностей. Они могут быть настолько выраженными, что без поддержки больной не может ни стоять, ни ходить. l Возможна идиопатическая торсионная дистония или дистония как проявление энцефалита, хореи Гентингтона, болезни Галлевердена—Шпатца, гепатоцеребральной дистрофии. l
 торсионную и симптоматическую дистонию. l Тип наследования при") Этиология и патогенез. Различают идиопатическую (семейную) торсионную и симптоматическую дистонию. l Тип наследования при идиопатической торсионной дистонии как аутосомнодоминантный, так и аутосомнорецессивный. l Симптоматическая торсионная дистония встречается при гепатоцеребральной дистрофии, хорее Гентингтона, опухолях мозга, эпидемическом энцефалите, детском церебральном параличе. Имеются указания, что в патогенезе наследственной торсионной дистонии имеет значение нарушение допаминового обмена. При обследовании у этих больных обнаруживается повышение содержания допамингидроксилазы в сыворотке крови. l
Этиология и патогенез. Различают идиопатическую (семейную) торсионную и симптоматическую дистонию. l Тип наследования при идиопатической торсионной дистонии как аутосомнодоминантный, так и аутосомнорецессивный. l Симптоматическая торсионная дистония встречается при гепатоцеребральной дистрофии, хорее Гентингтона, опухолях мозга, эпидемическом энцефалите, детском церебральном параличе. Имеются указания, что в патогенезе наследственной торсионной дистонии имеет значение нарушение допаминового обмена. При обследовании у этих больных обнаруживается повышение содержания допамингидроксилазы в сыворотке крови. l
 Клинические проявления l Развивается заболевание постепенно, в 2/3 случаев в возрасте до 15 лет. В детском возрасте первыми симптомами болезни могут быть нарушение походки, спастическая кривошея; у взрослых чаще встречаются первично генерализованные формы. В результате нарушения соотношения функции мышц синергистов и антагонистов возникают насильственные длительные тонические сокращения мышц туловища, головы, тазового пояса, конечностей, обычно ротаторного характера, сочетающиеся с атетоидными движениями в пальцах. Создается впечатление, что мышцы постоянно сокращаются для преодоления действия антагонистов. Возникающие позы, даже самые неудобные, сохраняются в течение длительного времени. Гиперкинезы усиливаются при волнении, активных движениях, во сне исчезают. Постепенно, по мере прогрессирования заболевания, поза пациента становится постоянно дистонической, с усиленным поясничным лордозом, флексией бедер, медиальной ротацией рук и ног.
Клинические проявления l Развивается заболевание постепенно, в 2/3 случаев в возрасте до 15 лет. В детском возрасте первыми симптомами болезни могут быть нарушение походки, спастическая кривошея; у взрослых чаще встречаются первично генерализованные формы. В результате нарушения соотношения функции мышц синергистов и антагонистов возникают насильственные длительные тонические сокращения мышц туловища, головы, тазового пояса, конечностей, обычно ротаторного характера, сочетающиеся с атетоидными движениями в пальцах. Создается впечатление, что мышцы постоянно сокращаются для преодоления действия антагонистов. Возникающие позы, даже самые неудобные, сохраняются в течение длительного времени. Гиперкинезы усиливаются при волнении, активных движениях, во сне исчезают. Постепенно, по мере прогрессирования заболевания, поза пациента становится постоянно дистонической, с усиленным поясничным лордозом, флексией бедер, медиальной ротацией рук и ног.
 Клинические проявления В зависимости от распространенности дистонических явлений выделяют локальную и генерализованную формы заболевания. l При локальных дистонических симптомах возникает тоническое сокращение отдельных мышечных групп, нарушаются произвольные движения и возникает аномальная поза. К таким симптомам относятся спастическая кривошея, писчий спазм, оромандибулярная дистония (открывание и закрывание рта и непроизвольные движения языка), блефароспазм, щечно лицевая, щечно язычная дистония, хореоатетоз. l
Клинические проявления В зависимости от распространенности дистонических явлений выделяют локальную и генерализованную формы заболевания. l При локальных дистонических симптомах возникает тоническое сокращение отдельных мышечных групп, нарушаются произвольные движения и возникает аномальная поза. К таким симптомам относятся спастическая кривошея, писчий спазм, оромандибулярная дистония (открывание и закрывание рта и непроизвольные движения языка), блефароспазм, щечно лицевая, щечно язычная дистония, хореоатетоз. l
 Лечение. Длительное, симптоматическое. Применяют комбинации холинолитиков и седативных препаратов, в некоторых случаях эффективно использование леводопы. Назначается также галоперидол или резерпин. l Очень редко прибегают к стереотаксическим операциям на подкорковых ядрах. l
Лечение. Длительное, симптоматическое. Применяют комбинации холинолитиков и седативных препаратов, в некоторых случаях эффективно использование леводопы. Назначается также галоперидол или резерпин. l Очень редко прибегают к стереотаксическим операциям на подкорковых ядрах. l
 Оливопонтоцеребеллярные дегенерации l Группа наследственных заболеваний нервной системы, характеризующихся дегенеративными изменениями нейронов мозжечка, ядер нижних олив и моста мозга, в ряде случаев – ядер черепных нервов каудальной группы, в меньшей степени – поражением проводящих путей и клеток передних рогов спинного мозга, базальных ганглиев. Заболевания отличаются типом наследования и различным сочетанием клинических симптомов.
Оливопонтоцеребеллярные дегенерации l Группа наследственных заболеваний нервной системы, характеризующихся дегенеративными изменениями нейронов мозжечка, ядер нижних олив и моста мозга, в ряде случаев – ядер черепных нервов каудальной группы, в меньшей степени – поражением проводящих путей и клеток передних рогов спинного мозга, базальных ганглиев. Заболевания отличаются типом наследования и различным сочетанием клинических симптомов.
 По классификации Кенигсмарка и Вайнера Тип I – оливопонтоцеребеллярная дегенерация Менделя. l Наследуется по аутосомно доминантному типу. Течение медленно прогрессирующее. Проявляться может в возрасте от 11 до 60 лет. Клиническая картина складывается из симптомов поражения мозжечка (атаксия, мышечная гипотония, скандированная речь с элементами дизартрии, интенционное дрожание), ядер каудальных черепных нервов (дизартрия, дисфагия), подкорковых ганглиев (гиперкинезы); реже выявляются пирамидные и глазодвигательные симптомы. l Тип II – оливопонтоцеребеллярная дегенерация Фиклера— Винклера. l Наследуется по аутосомно рецессивному типу. Проявляется в возрасте от 20 до 80 лет симптомами поражения мозжечка, преимущественно атаксией в конечностях. Чувствительность и сухожильные рефлексы не изменены. Парезов не наблюдается. l
По классификации Кенигсмарка и Вайнера Тип I – оливопонтоцеребеллярная дегенерация Менделя. l Наследуется по аутосомно доминантному типу. Течение медленно прогрессирующее. Проявляться может в возрасте от 11 до 60 лет. Клиническая картина складывается из симптомов поражения мозжечка (атаксия, мышечная гипотония, скандированная речь с элементами дизартрии, интенционное дрожание), ядер каудальных черепных нервов (дизартрия, дисфагия), подкорковых ганглиев (гиперкинезы); реже выявляются пирамидные и глазодвигательные симптомы. l Тип II – оливопонтоцеребеллярная дегенерация Фиклера— Винклера. l Наследуется по аутосомно рецессивному типу. Проявляется в возрасте от 20 до 80 лет симптомами поражения мозжечка, преимущественно атаксией в конечностях. Чувствительность и сухожильные рефлексы не изменены. Парезов не наблюдается. l
 По классификации Кенигсмарка и Вайнера Тип III – оливопонтоцеребеллярная дегенерация с ретинальной дегенерацией. l Наследуется по аутосомно доминантному типу. Возникает в молодом возрасте. Наряду с мозжечковыми и экстрапирамидными симптомами определяется прогрессирующее снижение остроты зрения вследствие пигментной дегенерации ганглиозных клеток сетчатки. l Тип IV – оливопонтоцеребеллярная дегенерация Шута—Хайкмана. l Наследуется по аутосомно доминантному типу. Проявляется в детском и молодом возрасте. Кроме мозжечковых симптомов, выявляется поражение ядер VII, IX, Х и XII пар черепных нервов (паралич лицевого нерва, бульбарные симптомы) и задних канатиков спинного мозга (расстройства мышечно суставного чувства и вибрационной чувствительности). l
По классификации Кенигсмарка и Вайнера Тип III – оливопонтоцеребеллярная дегенерация с ретинальной дегенерацией. l Наследуется по аутосомно доминантному типу. Возникает в молодом возрасте. Наряду с мозжечковыми и экстрапирамидными симптомами определяется прогрессирующее снижение остроты зрения вследствие пигментной дегенерации ганглиозных клеток сетчатки. l Тип IV – оливопонтоцеребеллярная дегенерация Шута—Хайкмана. l Наследуется по аутосомно доминантному типу. Проявляется в детском и молодом возрасте. Кроме мозжечковых симптомов, выявляется поражение ядер VII, IX, Х и XII пар черепных нервов (паралич лицевого нерва, бульбарные симптомы) и задних канатиков спинного мозга (расстройства мышечно суставного чувства и вибрационной чувствительности). l
 Тип V – оливопонтоцеребеллярная дегенерация с деменцией, офтальмоплегией и экстрапирамидными нарушениями. l Тип наследования аутосомно доминантный. Развивается в среднем возрасте. Характеризуется деменцией, прогрессирующей офтальмоплегией, экстрапирамидными и мозжечковыми симптомами l
Тип V – оливопонтоцеребеллярная дегенерация с деменцией, офтальмоплегией и экстрапирамидными нарушениями. l Тип наследования аутосомно доминантный. Развивается в среднем возрасте. Характеризуется деменцией, прогрессирующей офтальмоплегией, экстрапирамидными и мозжечковыми симптомами l
 Лечение. Симптоматическое l Проводят курсы неспецифического общеукрепляющего лечения, массаж, лечебную физкультуру. l
Лечение. Симптоматическое l Проводят курсы неспецифического общеукрепляющего лечения, массаж, лечебную физкультуру. l
 Болезнь Лафора. Ген болезни Лафора локализован на длинном плече хромосомы 6 (6 q 23— 25). Он кодирует белок лафорин, который функционирует как тирозинфосфатаза. Состояние наследуется аутосомно рецессивно. Множество концентрических телец (тельца Лафора) обнаруживаются в ганглионарных клетках на всем их протяжении, особенно в зубчатом ядре, черном веществе, ретикулярной формации и гиппокампе. Сходное амилоидное вещество можно найти в сердце и печени l Дебют возраст от 11 до 18 лет, когда возникают эпилептические приступы (миоклонические и тонико клонические). l Терминальная стадия, для которой типичны деменция, спастический тетрапарез и почти постоянные миоклонии, наступает через 4— 10 лет после появления первых симптомов. l
Болезнь Лафора. Ген болезни Лафора локализован на длинном плече хромосомы 6 (6 q 23— 25). Он кодирует белок лафорин, который функционирует как тирозинфосфатаза. Состояние наследуется аутосомно рецессивно. Множество концентрических телец (тельца Лафора) обнаруживаются в ганглионарных клетках на всем их протяжении, особенно в зубчатом ядре, черном веществе, ретикулярной формации и гиппокампе. Сходное амилоидное вещество можно найти в сердце и печени l Дебют возраст от 11 до 18 лет, когда возникают эпилептические приступы (миоклонические и тонико клонические). l Терминальная стадия, для которой типичны деменция, спастический тетрапарез и почти постоянные миоклонии, наступает через 4— 10 лет после появления первых симптомов. l
 перерыв
перерыв
 характеризуется") IV. Синдромы, сопровождающиеся прогрессирующей атаксией Атаксия Фридрейха l Спиноцеребеллярные дегенерации (наследственная атаксия Фридрейха) характеризуется дегенерацией систем восходящих и нисходящих волокон спинного мозга, включая спиноцеребеллярные пути, с сопутствующей дегенерацией периферических аксонов и миелиновых оболочек по типу хронической полиневропатии. Причины болезни аутосомно-рецессивно наследуемая мутация в гене FXN, кодирующем белок фратаксин. Фратаксин играет важную роль в работе митохондрий, в частности, в выведении железа из около митохондриального пространства. l В отсутствие фратаксина избыток железа вызывает образование свободных радикалов и повреждения. l
IV. Синдромы, сопровождающиеся прогрессирующей атаксией Атаксия Фридрейха l Спиноцеребеллярные дегенерации (наследственная атаксия Фридрейха) характеризуется дегенерацией систем восходящих и нисходящих волокон спинного мозга, включая спиноцеребеллярные пути, с сопутствующей дегенерацией периферических аксонов и миелиновых оболочек по типу хронической полиневропатии. Причины болезни аутосомно-рецессивно наследуемая мутация в гене FXN, кодирующем белок фратаксин. Фратаксин играет важную роль в работе митохондрий, в частности, в выведении железа из около митохондриального пространства. l В отсутствие фратаксина избыток железа вызывает образование свободных радикалов и повреждения. l
 Патологические изменения l Заболевание характеризуется синдромом поражения задних и боковых канатиков спинного мозга, чаще в люмбосакральных сегментах, гибелью клеток столбов Кларка и дорсальных спиноцеребеллярных трактов и периферических нервов. l Пучки Голля поражаются в большей степени, чем пучки Бурдаха. В большинстве случаев дегенерация охватывает также кортико спинальные тракты. Степень поражения мозжечка различна. l На поздних стадиях характерна дегенерация ядер черепных нервов, зубчатого ядра, ножки мозжечка, несколько реже страдают клетки больших полушарий мозга. l l Помимо перечисленных нейропатологических изменений, у некоторых пациентов развивается типичная форма дегенерации миокарда, приводящая к гибели волокон и фиброзу.
Патологические изменения l Заболевание характеризуется синдромом поражения задних и боковых канатиков спинного мозга, чаще в люмбосакральных сегментах, гибелью клеток столбов Кларка и дорсальных спиноцеребеллярных трактов и периферических нервов. l Пучки Голля поражаются в большей степени, чем пучки Бурдаха. В большинстве случаев дегенерация охватывает также кортико спинальные тракты. Степень поражения мозжечка различна. l На поздних стадиях характерна дегенерация ядер черепных нервов, зубчатого ядра, ножки мозжечка, несколько реже страдают клетки больших полушарий мозга. l l Помимо перечисленных нейропатологических изменений, у некоторых пациентов развивается типичная форма дегенерации миокарда, приводящая к гибели волокон и фиброзу.
 Клиническая картина Первые симптомы обычно появляются на первом, втором десятилетии жизни, однако первые проявления могут быть и на третьем и четвертом десятилетии. l Проявляется атаксия при ходьбе, а также нарушением почерка, дизартрией, слабостью в ногах, нарушением или потерей слуха. Нарушается глубокая чувствительность, постепенно нарастает мышечная атрофия, на начальных этапах более выражена на нижних конечностях, с течением болезни захватывает и верхние. Атрофируется зрительный нерв, развивается катаракта, что ведет к слепоте, нарушается функция тазовых органов, развивается деменция. l l Возникают скелетные деформации — искривление позвоночника, «стопа Фридрейха» .
Клиническая картина Первые симптомы обычно появляются на первом, втором десятилетии жизни, однако первые проявления могут быть и на третьем и четвертом десятилетии. l Проявляется атаксия при ходьбе, а также нарушением почерка, дизартрией, слабостью в ногах, нарушением или потерей слуха. Нарушается глубокая чувствительность, постепенно нарастает мышечная атрофия, на начальных этапах более выражена на нижних конечностях, с течением болезни захватывает и верхние. Атрофируется зрительный нерв, развивается катаракта, что ведет к слепоте, нарушается функция тазовых органов, развивается деменция. l l Возникают скелетные деформации — искривление позвоночника, «стопа Фридрейха» .
 Неврологические проявления l l l l • Заболевание манифестирует обычно появлением неловкости, неуверенности при ходьбе, особенно в темноте, больные начинают пошатываться, часто спотыкаются. Вскоре к атаксии при ходьбе присоединяются дискоординация в руках, изменение почерка, слабость в йогах. Уже в самом начале заболевания может отмечаться дизартрия. • Ранним признаком является исчезновение сухожильных и надкостничных рефлексов. Угнетение рефлексов (в первую очередь ахилловых и коленных) может на несколько лет опережать манифестацию других симптомов болезни • В развернутой стадии заболевания у больных обычно наблюдается тотальная арефлексия. • Типичным неврологическим проявлением болезни Фридрейха является нарушение глубокой (суставно-мышечной и вибрационной) чувствительности. • Довольно рано у больных при неврологическом осмотре может быть обнаружен симптом Бабинского, мышечная гипотония. • По мере прогрессирования заболевания постепенно нарастают мозжечковая и сенситивная атаксия, слабость и атрофия мышц ног. • В поздней стадии болезни часты, амиотрофии и расстройства глубокой чувствительности, которые распространяются на руки. Больные перестают самостоятельно ходить и обслуживать себя из за глубокого распада моторных функций. • В ряде случаев наблюдается нистагм, снижение слуха, атрофия зрительных нервов; при длительном течении болезни отмечается нарушение функции тазовых органов, деменция.
Неврологические проявления l l l l • Заболевание манифестирует обычно появлением неловкости, неуверенности при ходьбе, особенно в темноте, больные начинают пошатываться, часто спотыкаются. Вскоре к атаксии при ходьбе присоединяются дискоординация в руках, изменение почерка, слабость в йогах. Уже в самом начале заболевания может отмечаться дизартрия. • Ранним признаком является исчезновение сухожильных и надкостничных рефлексов. Угнетение рефлексов (в первую очередь ахилловых и коленных) может на несколько лет опережать манифестацию других симптомов болезни • В развернутой стадии заболевания у больных обычно наблюдается тотальная арефлексия. • Типичным неврологическим проявлением болезни Фридрейха является нарушение глубокой (суставно-мышечной и вибрационной) чувствительности. • Довольно рано у больных при неврологическом осмотре может быть обнаружен симптом Бабинского, мышечная гипотония. • По мере прогрессирования заболевания постепенно нарастают мозжечковая и сенситивная атаксия, слабость и атрофия мышц ног. • В поздней стадии болезни часты, амиотрофии и расстройства глубокой чувствительности, которые распространяются на руки. Больные перестают самостоятельно ходить и обслуживать себя из за глубокого распада моторных функций. • В ряде случаев наблюдается нистагм, снижение слуха, атрофия зрительных нервов; при длительном течении болезни отмечается нарушение функции тазовых органов, деменция.
") Экстраневральные проявления l l l Поражение сердца (встречается более чем у 90 % больных) Характерным является развитие типичной прогрессирующей кардиомиопатии. Кардиомиопатия преимущественно носит характер гипертрофической, но в отдельных случаях возможно развитие дилатационной кардиомиопатии. Кардиомиопатия проявляется: • болями в области сердца • сердцебиением • одышкой при физической нагрузке • систолическим шумом и другими симптомами. Более чем у половины больных кардиомиопатия является непосредственной причиной смерти. Соответствующие изменения обычно обнаруживаются: • на ЭКГ нарушение ритма, инверсия зубца. Т, изменения проводимости В ряде случаев клинические и электрокардиографические симптомы поражения сердца иногда на несколько лет опережают появление неврологических нарушений. Скелетные деформации: • сколиоз • “стопа Фридрейха” - высокий вогнутый свод стопы с переразгибанием пальцев в основных фалангах и сгибанием в дистальных • деформация пальцев рук и ног и др. Эти нарушения также могут появляться задолго до развития первых неврологических симптомов. Эндокринные расстройства: • сахарный диабет • гипогонадизм • инфантилизм • дисфункция яичников Катаракта
Экстраневральные проявления l l l Поражение сердца (встречается более чем у 90 % больных) Характерным является развитие типичной прогрессирующей кардиомиопатии. Кардиомиопатия преимущественно носит характер гипертрофической, но в отдельных случаях возможно развитие дилатационной кардиомиопатии. Кардиомиопатия проявляется: • болями в области сердца • сердцебиением • одышкой при физической нагрузке • систолическим шумом и другими симптомами. Более чем у половины больных кардиомиопатия является непосредственной причиной смерти. Соответствующие изменения обычно обнаруживаются: • на ЭКГ нарушение ритма, инверсия зубца. Т, изменения проводимости В ряде случаев клинические и электрокардиографические симптомы поражения сердца иногда на несколько лет опережают появление неврологических нарушений. Скелетные деформации: • сколиоз • “стопа Фридрейха” - высокий вогнутый свод стопы с переразгибанием пальцев в основных фалангах и сгибанием в дистальных • деформация пальцев рук и ног и др. Эти нарушения также могут появляться задолго до развития первых неврологических симптомов. Эндокринные расстройства: • сахарный диабет • гипогонадизм • инфантилизм • дисфункция яичников Катаракта
 Клинические проявления Как и при других прогрессирующих атаксиях, нарушения сначала появляются в нижних конечностях. Дебют заболевания приходится на поздний детский возраст. Больной, бывший до этого совершенно здоровым, начинает ходить неровно, пошатываться при ходьбе, ощущает неустойчивость в положении стоя. Позднее возникают неловкость движений и мозжечковый тремор в руках и кистях, а также дизартрия с патологическим ритмом речи (скандирование). l Эти симптомы обусловливаются изменениями со стороны межпозвоночных узлов, спиноцеребеллярных путей и мозжечка; однако связать атаксию с анатомическим распределением патологических изменений затруднительно. l В конечностях, помимо атаксии, обычно обнаруживают также значительную слабость. При осмотре, как правило, можно выявить нистагм и скелетные аномалии: кифосколиоз, причина которого непонятна, а также типичную деформацию стоп (pes cavus, полая стопа) с подъемом (разгибание) пальцев, которую объясняют атрофией и контрактурами мышц стоп в то время, пока ее кости остаются податливыми. l
Клинические проявления Как и при других прогрессирующих атаксиях, нарушения сначала появляются в нижних конечностях. Дебют заболевания приходится на поздний детский возраст. Больной, бывший до этого совершенно здоровым, начинает ходить неровно, пошатываться при ходьбе, ощущает неустойчивость в положении стоя. Позднее возникают неловкость движений и мозжечковый тремор в руках и кистях, а также дизартрия с патологическим ритмом речи (скандирование). l Эти симптомы обусловливаются изменениями со стороны межпозвоночных узлов, спиноцеребеллярных путей и мозжечка; однако связать атаксию с анатомическим распределением патологических изменений затруднительно. l В конечностях, помимо атаксии, обычно обнаруживают также значительную слабость. При осмотре, как правило, можно выявить нистагм и скелетные аномалии: кифосколиоз, причина которого непонятна, а также типичную деформацию стоп (pes cavus, полая стопа) с подъемом (разгибание) пальцев, которую объясняют атрофией и контрактурами мышц стоп в то время, пока ее кости остаются податливыми. l
.") Характерно необычное сочетание полного выпадения сухожильных рефлексов с присутствием патологических разгибательных рефлексов (симптом Бабинского). l Выражены расстройства чувства положения и вибрационной чувствительности в конечностях. У некоторых больных в связи с аксононейропатией, поражающей мелкие нервные волокна, симметрично снижена болевая, температурная и тактильная чувствительность в дистальных отделах конечностей. l Интеллект обычно сохранен, хотя у незначительного числа больных происходит его снижение с развитием деменции на поздних стадиях заболевания. До взрослого возраста больные доживают редко. l Смерть чаще наступает в результате сопутствующей кардиомиопатии. l
Характерно необычное сочетание полного выпадения сухожильных рефлексов с присутствием патологических разгибательных рефлексов (симптом Бабинского). l Выражены расстройства чувства положения и вибрационной чувствительности в конечностях. У некоторых больных в связи с аксононейропатией, поражающей мелкие нервные волокна, симметрично снижена болевая, температурная и тактильная чувствительность в дистальных отделах конечностей. l Интеллект обычно сохранен, хотя у незначительного числа больных происходит его снижение с развитием деменции на поздних стадиях заболевания. До взрослого возраста больные доживают редко. l Смерть чаще наступает в результате сопутствующей кардиомиопатии. l
 абортивная форма может протекать в легкой или абортивной форме (например, наличие pes cavus, выпадение или повышение сухожильных рефлексов) и приводить к незначительной инвалидизации или вообще не сопровождаться таковой. l Подобные аномалии можно обнаружить среди родственников пациентов, страдающих развернутой клинической формой заболевания. l Эффективных методов лечения к настоящему времени не найдено. l
абортивная форма может протекать в легкой или абортивной форме (например, наличие pes cavus, выпадение или повышение сухожильных рефлексов) и приводить к незначительной инвалидизации или вообще не сопровождаться таковой. l Подобные аномалии можно обнаружить среди родственников пациентов, страдающих развернутой клинической формой заболевания. l Эффективных методов лечения к настоящему времени не найдено. l
 Лечение атаксии Фридрейха l l l l Лечения, преводящего к полному выздоровлению нет. Используются препараты так называемого митохондриального ряда, антиоксиданты и соединения, способствующие уменьшению аккумуляции железа в митохондриях. Среди антиоксидантов широко используется витамины А и Е, а также препарат идебенон (нобен), являющийся синтетическим аналогом коэнзима Q 10. Препарат обладает мощным антиоксидантным и цитопротективным действием, что помогает «затормозить» нейродегенративный процесс. Кроме того, органом мишенью идебенона является миокард, таким образом, препарат замедляет развитие гипертрофической кардиомиопатии. Необходимо наблюдение эндокринолога, ортопедическая коррекция стоп (стопа Фридрейха). Большое значение имеет также лечебная физкультура и физиотерапия.
Лечение атаксии Фридрейха l l l l Лечения, преводящего к полному выздоровлению нет. Используются препараты так называемого митохондриального ряда, антиоксиданты и соединения, способствующие уменьшению аккумуляции железа в митохондриях. Среди антиоксидантов широко используется витамины А и Е, а также препарат идебенон (нобен), являющийся синтетическим аналогом коэнзима Q 10. Препарат обладает мощным антиоксидантным и цитопротективным действием, что помогает «затормозить» нейродегенративный процесс. Кроме того, органом мишенью идебенона является миокард, таким образом, препарат замедляет развитие гипертрофической кардиомиопатии. Необходимо наблюдение эндокринолога, ортопедическая коррекция стоп (стопа Фридрейха). Большое значение имеет также лечебная физкультура и физиотерапия.
") Лейкодистрофии (лейкоэнцефалопатии)
Лейкодистрофии (лейкоэнцефалопатии)
 l Дегенеративные заболевания мозга с преимущественным поражением белого вещества) — наследственные прогрессирующие") Лейкодистрофии (лейкоэнцефалопатии) l Дегенеративные заболевания мозга с преимущественным поражением белого вещества) — наследственные прогрессирующие заболевания нервной системы, сопровождающиеся преимущественным поражением белого вещества ЦНС, а иногда и периферической нервной системы l Спонгиозная дегенерация белого вещества (болезнь Канавана-ван-Богарта-Бертрана). КТ: симметричные участки понижения плотности во внутренних и наружных капсулах, белом веществе больших полушарий, г — МРТв Т 1 и Т 2 (д) режимах: диффузное изменение сигнала белого вещества больших полушарий, субкортикальных структур, внутренних (стрелки) и наружных капсул. l l
Лейкодистрофии (лейкоэнцефалопатии) l Дегенеративные заболевания мозга с преимущественным поражением белого вещества) — наследственные прогрессирующие заболевания нервной системы, сопровождающиеся преимущественным поражением белого вещества ЦНС, а иногда и периферической нервной системы l Спонгиозная дегенерация белого вещества (болезнь Канавана-ван-Богарта-Бертрана). КТ: симметричные участки понижения плотности во внутренних и наружных капсулах, белом веществе больших полушарий, г — МРТв Т 1 и Т 2 (д) режимах: диффузное изменение сигнала белого вещества больших полушарий, субкортикальных структур, внутренних (стрелки) и наружных капсул. l l
 Общие признаки лейкодистрофии — наличие симметричных дистрофических поражений белого вещества больших полушарий мозга, мозжечка и спинного мозга, сливающихся очагов демиелинизации проводящих путей и ассоциативных волокон. Демиелинизирующие процессы сочетаются со спонгиозом (множеством овальных вакуолей) l В белом веществе уменьшается содержание липидов, образуются глиозные рубцы. l Серое вещество страдает меньше, в нем обнаруживаются участки гибели и перерождения нейронов. l Глобоидно-клеточная лейкодистрофия. а — КТ: множество мелких кальцинатов в зрительных буграх. б — МРТ в режиме Т 2: повышение интенсивности сигнала в белом веществе теменных областей
Общие признаки лейкодистрофии — наличие симметричных дистрофических поражений белого вещества больших полушарий мозга, мозжечка и спинного мозга, сливающихся очагов демиелинизации проводящих путей и ассоциативных волокон. Демиелинизирующие процессы сочетаются со спонгиозом (множеством овальных вакуолей) l В белом веществе уменьшается содержание липидов, образуются глиозные рубцы. l Серое вещество страдает меньше, в нем обнаруживаются участки гибели и перерождения нейронов. l Глобоидно-клеточная лейкодистрофия. а — КТ: множество мелких кальцинатов в зрительных буграх. б — МРТ в режиме Т 2: повышение интенсивности сигнала в белом веществе теменных областей
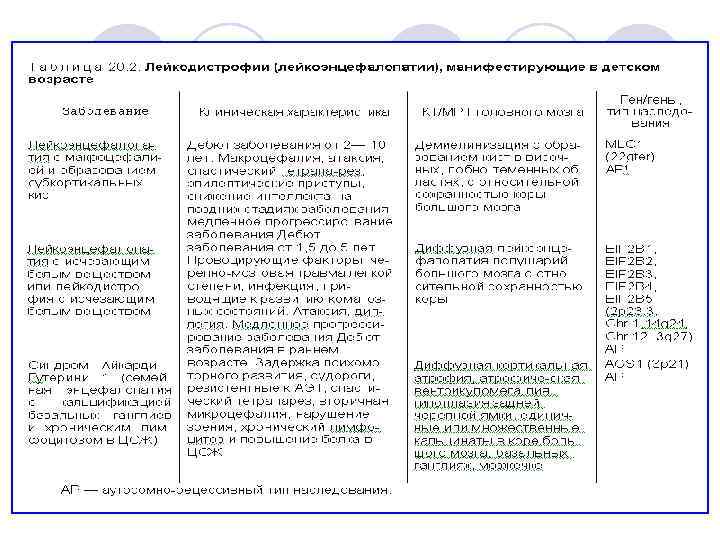
 2. Наследственные болезни обмена Патология аминокислот Патология липидов Муколипидозы Патология углеводов
2. Наследственные болезни обмена Патология аминокислот Патология липидов Муколипидозы Патология углеводов
 КЛАССИФИКАЦИЯ наследственных заболеваний l 1. Наследственные системные дегенерации нервной системы l Дегенерации с преимущественным поражением мозжечка и его связей Сочетанные дегенерации мозжечковых путей и периферических нервов Болезни с поражением экстрапирамидной нервной системы Болезни с поражением пирамидной нервной системы l l l l 2. Наследственные болезни обмена Патология аминокислот Патология липидов Муколипидозы Патология углеводов l l 3. Болезни соединительной ткани 4. Факоматозы 5. Нервно мышечные заболевания Прогрессирующие мышечные дистрофии миопатии Спинальные и невральные атрофии. Наследственные нервно мышечные заболевания с миотоническим синдромом Пароксизмальные миоплегии Миастения
КЛАССИФИКАЦИЯ наследственных заболеваний l 1. Наследственные системные дегенерации нервной системы l Дегенерации с преимущественным поражением мозжечка и его связей Сочетанные дегенерации мозжечковых путей и периферических нервов Болезни с поражением экстрапирамидной нервной системы Болезни с поражением пирамидной нервной системы l l l l 2. Наследственные болезни обмена Патология аминокислот Патология липидов Муколипидозы Патология углеводов l l 3. Болезни соединительной ткани 4. Факоматозы 5. Нервно мышечные заболевания Прогрессирующие мышечные дистрофии миопатии Спинальные и невральные атрофии. Наследственные нервно мышечные заболевания с миотоническим синдромом Пароксизмальные миоплегии Миастения
 НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ОБМЕНА ВЕЩЕСТВ Это обширный класс моногенных наследственных заболеваний, обусловленных мутациями генов, кодирующих ферменты, транспортные или сигнальные белки. С момента открытия первых форм НБО прошло уже более 100 лет, на сегодняшний день к НБО относят около 500 форм, и это число постоянно увеличивается. l В подавляющем большинстве случаев НБО не имеют отличительных фенотипических признаков, а в раннем неонатальном периоде симптомы заболевания, как правило, неспецифичны: задержка физического развития, нарушения вскармливания, эпизоды сонливости и вялости, что ведет к установлению неправильного диагноза. l
НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ОБМЕНА ВЕЩЕСТВ Это обширный класс моногенных наследственных заболеваний, обусловленных мутациями генов, кодирующих ферменты, транспортные или сигнальные белки. С момента открытия первых форм НБО прошло уже более 100 лет, на сегодняшний день к НБО относят около 500 форм, и это число постоянно увеличивается. l В подавляющем большинстве случаев НБО не имеют отличительных фенотипических признаков, а в раннем неонатальном периоде симптомы заболевания, как правило, неспецифичны: задержка физического развития, нарушения вскармливания, эпизоды сонливости и вялости, что ведет к установлению неправильного диагноза. l
 Метаболические сдвиги при мутационной блокаде трансформации одного вещества в другое
Метаболические сдвиги при мутационной блокаде трансформации одного вещества в другое
 В зависимости от пораженного метаболического пути выделяют 22 подкласса НБО l митохондриальные болезни; l лизосомные болезни накопления; l пероксисомные болезни; l нарушения обмена аминокислот/ органических кислот; l нарушения обмена углеводов; l эндокринопатии.
В зависимости от пораженного метаболического пути выделяют 22 подкласса НБО l митохондриальные болезни; l лизосомные болезни накопления; l пероксисомные болезни; l нарушения обмена аминокислот/ органических кислот; l нарушения обмена углеводов; l эндокринопатии.
 обусловлены нарушениями пяти ферментных комплексов дыхательной цепи") Митохондриальные болезни Болезни дыхательной цепи митохондрий (БДЦМ) обусловлены нарушениями пяти ферментных комплексов дыхательной цепи митохондрий (КДЦМ), которые обеспечивают заключительный этап клеточного дыхания и синтез аденозинтрифосфорной кислоты (АТФ). l В состав комплекса дыхательной цепи митохондрий (КДЦМ) входит более чем 80 различных полипептидов, подавляющее большинство которых кодируется ядерной ДНК (я. ДНК). l
Митохондриальные болезни Болезни дыхательной цепи митохондрий (БДЦМ) обусловлены нарушениями пяти ферментных комплексов дыхательной цепи митохондрий (КДЦМ), которые обеспечивают заключительный этап клеточного дыхания и синтез аденозинтрифосфорной кислоты (АТФ). l В состав комплекса дыхательной цепи митохондрий (КДЦМ) входит более чем 80 различных полипептидов, подавляющее большинство которых кодируется ядерной ДНК (я. ДНК). l
 Молекулярно-генетическая классификация митохондриальных болезней l l l I. Дефекты митохондриальной ДНК Точечные мутации. Единичные делеции. Дупликации или дупликации/ делеции. II. Дефекты ядерной ДНК 1. Дефекты в генах я. ДНК, кодирующих КДЦМ. 2. Дефекты в генах я. ДНК, ответственных за сборку КДЦМ ( комплекса дыхательной цепи митохондрий) III. Дефекты митохондриальной ДНК, вызванные нарушениями ядерной ДНК Тканеспецифичные делении и дупликации митохондриальной ДНК. Истощение мт. ДНК.
Молекулярно-генетическая классификация митохондриальных болезней l l l I. Дефекты митохондриальной ДНК Точечные мутации. Единичные делеции. Дупликации или дупликации/ делеции. II. Дефекты ядерной ДНК 1. Дефекты в генах я. ДНК, кодирующих КДЦМ. 2. Дефекты в генах я. ДНК, ответственных за сборку КДЦМ ( комплекса дыхательной цепи митохондрий) III. Дефекты митохондриальной ДНК, вызванные нарушениями ядерной ДНК Тканеспецифичные делении и дупликации митохондриальной ДНК. Истощение мт. ДНК.
 Расположение основных мутаций мт. ДНК l Яйцеклетка содержит сотни митохондрий, в то время как сперматозоид содержит 5— 10 митохондрий. В литературе описаны единичные случаи передачи небольшого количества мт. ДНК от отца, однако все описанные патогенные мутации мт. ДНК были унаследованы от матери [Di. Mauro S. et al. , 2003].
Расположение основных мутаций мт. ДНК l Яйцеклетка содержит сотни митохондрий, в то время как сперматозоид содержит 5— 10 митохондрий. В литературе описаны единичные случаи передачи небольшого количества мт. ДНК от отца, однако все описанные патогенные мутации мт. ДНК были унаследованы от матери [Di. Mauro S. et al. , 2003].
 Болезни дыхательной цепи митохондрий, обусловленные точковыми мутациями мт. ДНК. l l l l В настоящее время известно 5 клинических симптомокомплексов, связанных с точковыми мутациями мт. ДНК и наследуемых по материнской линии. Названия синдромов — акроним основных клинических составляющих. Это синдромы LHON (атрофия зрительных нервов Лебера), NARP (невропатия, атаксия, пигментная дегенерация сетчатки), MELAS (митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные состояния), MERRF (мио клонус эпилепсия, наличие «рваных красных волокон» в мышечном биоптате) и ММС (митохондриальная миокардиопатия, наследуемая по материнской линии). Все они, кроме синдрома ММС, сопровождаются неврологическими нарушениями МРТ головного мозга при синдроме ME LAS.
Болезни дыхательной цепи митохондрий, обусловленные точковыми мутациями мт. ДНК. l l l l В настоящее время известно 5 клинических симптомокомплексов, связанных с точковыми мутациями мт. ДНК и наследуемых по материнской линии. Названия синдромов — акроним основных клинических составляющих. Это синдромы LHON (атрофия зрительных нервов Лебера), NARP (невропатия, атаксия, пигментная дегенерация сетчатки), MELAS (митохондриальная энцефаломиопатия, лактатацидоз, инсультоподобные состояния), MERRF (мио клонус эпилепсия, наличие «рваных красных волокон» в мышечном биоптате) и ММС (митохондриальная миокардиопатия, наследуемая по материнской линии). Все они, кроме синдрома ММС, сопровождаются неврологическими нарушениями МРТ головного мозга при синдроме ME LAS.
, обусловленные крупными перестройками митохондриальной ДНК. l Синдром Кернса—Сейра Двусторонний") Болезни дыхательной цепи митохондрий (БДЦМ), обусловленные крупными перестройками митохондриальной ДНК. l Синдром Кернса—Сейра Двусторонний птоз, низкий рост, атаксия, задержка развития. Самыми распространенными являются синдром Кернса—Сейра и прогрессирующая наружная офтальмоплегия
Болезни дыхательной цепи митохондрий (БДЦМ), обусловленные крупными перестройками митохондриальной ДНК. l Синдром Кернса—Сейра Двусторонний птоз, низкий рост, атаксия, задержка развития. Самыми распространенными являются синдром Кернса—Сейра и прогрессирующая наружная офтальмоплегия
, обусловленные нарушением межгеномного взаимодействия. l К нарушениям межгеномного взаимодействия") Болезни дыхательной цепи митохондрий (БДЦМ), обусловленные нарушением межгеномного взаимодействия. l К нарушениям межгеномного взаимодействия относятся истощения и ¡ множественные делеции мт. ДНК (синдром Ли) [Краснопольская К. Д. , 2005; Di. Mauro S. et al. , 2003]. ¡ МРТ при синдроме Ли повышение интенсивности сигнала в перивентрикулярном белом веществе, в области базальных ганглиев и таламусе l
Болезни дыхательной цепи митохондрий (БДЦМ), обусловленные нарушением межгеномного взаимодействия. l К нарушениям межгеномного взаимодействия относятся истощения и ¡ множественные делеции мт. ДНК (синдром Ли) [Краснопольская К. Д. , 2005; Di. Mauro S. et al. , 2003]. ¡ МРТ при синдроме Ли повышение интенсивности сигнала в перивентрикулярном белом веществе, в области базальных ганглиев и таламусе l
 Синдром Ли — один из самых генетически гетерогенных и частых синдромов обусловлено мутациями мт. ДНК (гены, кодирующие субъединицы АТФазы или т. РНК), мутациями ядерных генов, кодирующих полипептиды КДЦМ l Дебют заболевания — в первые месяцы жизни. У этих больных с рождения могут отмечаться нарушения вскармливания (слабое сосание, нарушения глотания, снижение аппетита и повторные рвоты), признаки легочной недостаточности, миоклонические судороги, мышечная диффузная гипотония или спастическая диплегия, отсутствует фиксация взгляда, резко снижена реакция на яркий свет и звуковые раздражители. Течение заболевания быстро прогрессирующее с быстрым фатальным исходом. l При манифестации болезни в первые 3 мес жизни наблюдаются эпизоды угнетения сознания вплоть до комы, тонические и миоклонические судороги, нарушения вскармливания, снижение остроты зрения и прогрессирующая мышечная слабость. l При начале заболевания в старшем и подростковом возрасте ведущими симптомами являются экстрапирамидно мозжечковые расстройства, в дальнейшем присоединяются пирамидные нарушения, судороги l
Синдром Ли — один из самых генетически гетерогенных и частых синдромов обусловлено мутациями мт. ДНК (гены, кодирующие субъединицы АТФазы или т. РНК), мутациями ядерных генов, кодирующих полипептиды КДЦМ l Дебют заболевания — в первые месяцы жизни. У этих больных с рождения могут отмечаться нарушения вскармливания (слабое сосание, нарушения глотания, снижение аппетита и повторные рвоты), признаки легочной недостаточности, миоклонические судороги, мышечная диффузная гипотония или спастическая диплегия, отсутствует фиксация взгляда, резко снижена реакция на яркий свет и звуковые раздражители. Течение заболевания быстро прогрессирующее с быстрым фатальным исходом. l При манифестации болезни в первые 3 мес жизни наблюдаются эпизоды угнетения сознания вплоть до комы, тонические и миоклонические судороги, нарушения вскармливания, снижение остроты зрения и прогрессирующая мышечная слабость. l При начале заболевания в старшем и подростковом возрасте ведущими симптомами являются экстрапирамидно мозжечковые расстройства, в дальнейшем присоединяются пирамидные нарушения, судороги l
 Дебютные симптомы нарушений дыхательной цепи митохондрий.
Дебютные симптомы нарушений дыхательной цепи митохондрий.
 критерии клинической оценки необъяснимое сочетание неродственных симптомов у больного; l раннее начало и прогрессирующее течение заболевания; l вовлечение органов и тканей различного эмбрионального происхождения и различных биологических функций l l l l Принципы диагностики клинико генеалогический анализ; анализ неврологической симптоматики с использованием параклинических методов; биохимические методы; морфологические методы; молекулярно генетические методы.
критерии клинической оценки необъяснимое сочетание неродственных симптомов у больного; l раннее начало и прогрессирующее течение заболевания; l вовлечение органов и тканей различного эмбрионального происхождения и различных биологических функций l l l l Принципы диагностики клинико генеалогический анализ; анализ неврологической симптоматики с использованием параклинических методов; биохимические методы; морфологические методы; молекулярно генетические методы.
 Лизосомные болезни накопления - нарушение расщепления макромолекул в лизосомах. Лизосомы — это органеллы, окруженные одной мембраной и содержащие около 50 различных ферментов. При мутациях в генах, кодирующих лизосомные ферменты или транспортные белки, происходит накопление нерасщепленных субстратов, что вызывает нарушение работы лизосом и приводит к гибели клеток. В зависимости от природы накапливаемых в лизосомах макромолекул выделяют мукополисахаридозы, гликопротеинозы, сфинголипидозы, нейрональные цероидные липофусцинозы. l Частота ЛБН составляет 1 на 7000— 8000 живых новорожденных l Болезнь Краббе (БКр) (глобоидноклеточная лейкодистрофия, недостаточность галактоцереброзидазы l
Лизосомные болезни накопления - нарушение расщепления макромолекул в лизосомах. Лизосомы — это органеллы, окруженные одной мембраной и содержащие около 50 различных ферментов. При мутациях в генах, кодирующих лизосомные ферменты или транспортные белки, происходит накопление нерасщепленных субстратов, что вызывает нарушение работы лизосом и приводит к гибели клеток. В зависимости от природы накапливаемых в лизосомах макромолекул выделяют мукополисахаридозы, гликопротеинозы, сфинголипидозы, нейрональные цероидные липофусцинозы. l Частота ЛБН составляет 1 на 7000— 8000 живых новорожденных l Болезнь Краббе (БКр) (глобоидноклеточная лейкодистрофия, недостаточность галактоцереброзидазы l
 (сульфатидный липидоз, диффузный склероз мозга)") Метахроматическая лейкодистрофия (МЛД) (сульфатидный липидоз, диффузный склероз мозга)
Метахроматическая лейкодистрофия (МЛД) (сульфатидный липидоз, диффузный склероз мозга)
 Мукополисахариды представляют собой сложные соединения, молекулы которых состоят из белкового компонента и углеводных цепей, содержащих дисахаридные звенья из гексуроновых кислот и аминосахаров. Мукополисахариды входят в состав межклеточного вещества соединительной ткани, кожи, костей, синовиальной жидкости, суставов, хрящей, роговицы, сосудов и ряда органов. l Наследственные нарушения обмена последних приводит к накоплению их в лизосомах клеток и развитию мукополисахаридозов. Данная группа заболеваний связана с мутациями в разных генах, кодирующих ферменты одного метаболического пути распада молекул гликозоаминогликанов. l Явление генетической гетерогенности по критерию некоторой схожести клинических проявлений позволяет объединить заболевания в одну группу. Всего известно 7 клинико биохимических типов патологии. l
Мукополисахариды представляют собой сложные соединения, молекулы которых состоят из белкового компонента и углеводных цепей, содержащих дисахаридные звенья из гексуроновых кислот и аминосахаров. Мукополисахариды входят в состав межклеточного вещества соединительной ткани, кожи, костей, синовиальной жидкости, суставов, хрящей, роговицы, сосудов и ряда органов. l Наследственные нарушения обмена последних приводит к накоплению их в лизосомах клеток и развитию мукополисахаридозов. Данная группа заболеваний связана с мутациями в разных генах, кодирующих ферменты одного метаболического пути распада молекул гликозоаминогликанов. l Явление генетической гетерогенности по критерию некоторой схожести клинических проявлений позволяет объединить заболевания в одну группу. Всего известно 7 клинико биохимических типов патологии. l
 группа ЛБН, обусловленная нарушениями обмена гликозаминогликанов. Выделяют 10 форм мукополисахаридозов, различающихся по") Мукополисахаридозы (МПС) группа ЛБН, обусловленная нарушениями обмена гликозаминогликанов. Выделяют 10 форм мукополисахаридозов, различающихся по типу накапливаемых гликозаминогликанов, типу наследования и дефекту ферментов. l Клиническими особенностями этой группы являются сочетания поражения нервной системы, скелета и внутренних органов l l l. Мукополисахаридоз lпервого типа l (синдром Гурлера) (изменение черт лица по типу «гаргоилизма» ); б когтистая кисть у того же больного.
Мукополисахаридозы (МПС) группа ЛБН, обусловленная нарушениями обмена гликозаминогликанов. Выделяют 10 форм мукополисахаридозов, различающихся по типу накапливаемых гликозаминогликанов, типу наследования и дефекту ферментов. l Клиническими особенностями этой группы являются сочетания поражения нервной системы, скелета и внутренних органов l l l. Мукополисахаридоз lпервого типа l (синдром Гурлера) (изменение черт лица по типу «гаргоилизма» ); б когтистая кисть у того же больного.
 Частота заболевания 1 на 144 000 Гликозоаминогликаны накапливаются повсеместно") Мукополисахаридоз первого типа (синдром Гурлера) Частота заболевания 1 на 144 000 Гликозоаминогликаны накапливаются повсеместно в хрящах, сухожилиях, надкостнице эндокарде и сосудистой стенке, печени, селезенке и нервной ткани. Отек мягкой мозговой оболочки вызывав частичную окклюзию субарахноидальных пространств, что приводит к прогрессирующей внутренней и наружной гидроцефалии. Нейроны раздуты и вакуолизированы. Поражаются клетки коры большого мозга, таламуса, ствола, передних рогов. l Тугоподвижность суставов является результатом деформации метафизов; утолщение суставной капсулы вторично к отложению в ней гликозоаминогликанов и фиброза. Обструкция дыхательных путей является следствием сужения трахеи, утолщения голосовых связок, избыточности отечных тканей в верхних дыхательных путях [Краснопольская К. Д. , 2005; Scriver С. R etal. , 2001]. l l
Мукополисахаридоз первого типа (синдром Гурлера) Частота заболевания 1 на 144 000 Гликозоаминогликаны накапливаются повсеместно в хрящах, сухожилиях, надкостнице эндокарде и сосудистой стенке, печени, селезенке и нервной ткани. Отек мягкой мозговой оболочки вызывав частичную окклюзию субарахноидальных пространств, что приводит к прогрессирующей внутренней и наружной гидроцефалии. Нейроны раздуты и вакуолизированы. Поражаются клетки коры большого мозга, таламуса, ствола, передних рогов. l Тугоподвижность суставов является результатом деформации метафизов; утолщение суставной капсулы вторично к отложению в ней гликозоаминогликанов и фиброза. Обструкция дыхательных путей является следствием сужения трахеи, утолщения голосовых связок, избыточности отечных тканей в верхних дыхательных путях [Краснопольская К. Д. , 2005; Scriver С. R etal. , 2001]. l l
 Гипоплазия подвздошных костей при мукополисахаридозе первого типа
Гипоплазия подвздошных костей при мукополисахаридозе первого типа
") Мукополисахаридоз четвертого типа (синдром Моркио)
Мукополисахаридоз четвертого типа (синдром Моркио)
 Муколипидозы снижения активности лизосомной N ацетилглюкозаминил 1 фосфотрансферазы, нарушение транспорта лизосомных ферментов внутрь лизосом l Ген картирован на хромосоме 4 q 21—q 23. Частота заболева ния 1 на 60 000 l низкие показатели роста и массы тела, задержка физического развития, грыжи (пупочная, паховая, белой линии живота или комбинированные); могут наблюдаться врожденные переломы костей, врожденный вывих головки бедра и/или плеча, врожденные пороки сердца. Ха рактерны изменения черт лица по типу «гаргоилизма» : макроцефалия, брахицефальная, акроцефальная или квадратная форма черепа, выступающие лобные бугры, гипертелоризм, эпикант, отечные веки, запавшаяширокая переносица, короткий нос с открытыми вперед ноздрями, носовые ходы расширены, большой и открытый рот, гиперплазия десен, макроглоссия, толстые губы. Грубые деформации скелета, множественный дизостоз: короткие шея и грудная клетка (в виде колокола), кифосколиоз, кифоз, прогрессирующая тугоподвижность большинства суставов, «когтистая кисть» . l У всех больных выявляются гепатоспленомегалия, пороки сердца, помутнение роговицы и прогрессирующая задержка психомоторного развития l l .
Муколипидозы снижения активности лизосомной N ацетилглюкозаминил 1 фосфотрансферазы, нарушение транспорта лизосомных ферментов внутрь лизосом l Ген картирован на хромосоме 4 q 21—q 23. Частота заболева ния 1 на 60 000 l низкие показатели роста и массы тела, задержка физического развития, грыжи (пупочная, паховая, белой линии живота или комбинированные); могут наблюдаться врожденные переломы костей, врожденный вывих головки бедра и/или плеча, врожденные пороки сердца. Ха рактерны изменения черт лица по типу «гаргоилизма» : макроцефалия, брахицефальная, акроцефальная или квадратная форма черепа, выступающие лобные бугры, гипертелоризм, эпикант, отечные веки, запавшаяширокая переносица, короткий нос с открытыми вперед ноздрями, носовые ходы расширены, большой и открытый рот, гиперплазия десен, макроглоссия, толстые губы. Грубые деформации скелета, множественный дизостоз: короткие шея и грудная клетка (в виде колокола), кифосколиоз, кифоз, прогрессирующая тугоподвижность большинства суставов, «когтистая кисть» . l У всех больных выявляются гепатоспленомегалия, пороки сердца, помутнение роговицы и прогрессирующая задержка психомоторного развития l l .
 Сиалидоз l l l снижения активности лизосомной а нейраминидазы, которая участвует в метаболизме сиалилсодержащих гликопротеинов При нарушении распада сиалилсодержащих гликопротеинов происходит их накопление в клетках, что приводит к нарушению функцио нирования и гибели клеток. Ген картирован на хромосоме q 23 прогрессирующее снижение зрения вплоть до слепоты, миоклонические приступы и нарушение походки. развитие тяжелых миоклонических и генерализованных тоникоклонических приступов, резистентных к антиэпилептической терапии. У большинства больных отмечаются мозжечковые расстройства (атаксия, интенционный тремор, дизартрия) и полиневропатический синдром симптоматическая терапия.
Сиалидоз l l l снижения активности лизосомной а нейраминидазы, которая участвует в метаболизме сиалилсодержащих гликопротеинов При нарушении распада сиалилсодержащих гликопротеинов происходит их накопление в клетках, что приводит к нарушению функцио нирования и гибели клеток. Ген картирован на хромосоме q 23 прогрессирующее снижение зрения вплоть до слепоты, миоклонические приступы и нарушение походки. развитие тяжелых миоклонических и генерализованных тоникоклонических приступов, резистентных к антиэпилептической терапии. У большинства больных отмечаются мозжечковые расстройства (атаксия, интенционный тремор, дизартрия) и полиневропатический синдром симптоматическая терапия.
 В результате дефекта глюкоцереброзидазы в клетках ретикулоэндотелиальной,") Болезнь Гоше ( А/р, 1 q 21) В результате дефекта глюкоцереброзидазы в клетках ретикулоэндотелиальной, нервной системы накапливаются глюцереброзиды и сфинголипиды. l тип I, или хроническая форма, без поражения нервной системы (ненейронопатическая), l тип II — острая нейронопатическая, l тип III — подострая нейронопатическая формы. l
Болезнь Гоше ( А/р, 1 q 21) В результате дефекта глюкоцереброзидазы в клетках ретикулоэндотелиальной, нервной системы накапливаются глюцереброзиды и сфинголипиды. l тип I, или хроническая форма, без поражения нервной системы (ненейронопатическая), l тип II — острая нейронопатическая, l тип III — подострая нейронопатическая формы. l
 Клетки Гоше в пунктате селезенки l Число и размер лизосом резко увеличиваются, приводя к гибели клеток. Гистохимически это выявляется как «пенистые» клетки (клетки Гоше) в ретикулоэндотелиальной системе селезенки, костного мозга, лимфатических узлов, печени, плаценты, мозга (в паренхиме мозга находятся периваскулярно).
Клетки Гоше в пунктате селезенки l Число и размер лизосом резко увеличиваются, приводя к гибели клеток. Гистохимически это выявляется как «пенистые» клетки (клетки Гоше) в ретикулоэндотелиальной системе селезенки, костного мозга, лимфатических узлов, печени, плаценты, мозга (в паренхиме мозга находятся периваскулярно).
 Болезнь Ниманна—Пика Аутосомно рецессивное заболевание, связанное с дефицитом активности фермента сфингомиелиназы, что приводит к накоплению сфингомиелина в клетках ретикулоэндотелиальной и нервной систем. l Ген сфингомиелиназы картирован на хромосоме 11 р15. 4— 15. 1. При микроскопии во всех органах обнаруживаются «пенистые» клетки — так называемые клетки Ниманна— Пика l гепатоспленомегалия, задержка психомоторного развития, нарушения вскармливания, рвота, диарея, гипертермия. l У 50 % больных выявляется дегенерация макулы по типу «вишневой косточки» l
Болезнь Ниманна—Пика Аутосомно рецессивное заболевание, связанное с дефицитом активности фермента сфингомиелиназы, что приводит к накоплению сфингомиелина в клетках ретикулоэндотелиальной и нервной систем. l Ген сфингомиелиназы картирован на хромосоме 11 р15. 4— 15. 1. При микроскопии во всех органах обнаруживаются «пенистые» клетки — так называемые клетки Ниманна— Пика l гепатоспленомегалия, задержка психомоторного развития, нарушения вскармливания, рвота, диарея, гипертермия. l У 50 % больных выявляется дегенерация макулы по типу «вишневой косточки» l
 Пероксисомные болезни Пероксисомы — небольшие овальные или круглые органеллы, окруженные одной мембраной. l Большинство ферментов пероксисом являются оксидазами, поэтому одной из главных функций пероксисом является защита клетки от агрессивных форм кислорода, образующихся в ее матриксе. Две большие группы: l 1 я —нарушение биогенеза пероксисом (их полное отсутствие или нарушение их функциональной активности). Указанная группа включает синдром Целлвегера, неонатальную адреногейкодистрофию, инфантильную болезнь Рефсума; l 2 я — нарушение единичных белков пероксисом — акаталаземию, Х сцепленную адренолейкодистрофию. l l
Пероксисомные болезни Пероксисомы — небольшие овальные или круглые органеллы, окруженные одной мембраной. l Большинство ферментов пероксисом являются оксидазами, поэтому одной из главных функций пероксисом является защита клетки от агрессивных форм кислорода, образующихся в ее матриксе. Две большие группы: l 1 я —нарушение биогенеза пероксисом (их полное отсутствие или нарушение их функциональной активности). Указанная группа включает синдром Целлвегера, неонатальную адреногейкодистрофию, инфантильную болезнь Рефсума; l 2 я — нарушение единичных белков пероксисом — акаталаземию, Х сцепленную адренолейкодистрофию. l l
, концентрическое сужение полей") Болезнь Рефсума. Основные клинические симптомы у пациентов включали ночную слепоту (гемералопию), концентрическое сужение полей зрения с атипичной пигментной дегенерацией, полиневропатию и атаксию. l Основная тетрада клинических симптомов включает пигментную дегенерацию сетчатки, периферическую полиневропатию, церебральную атаксию и повышение концентрации белка в ЦСЖ. Заболевание дебютирует в подростковом возрасте и позднее l Пигментная дегенерация сетчатки, поражение сердца (кардиомиопатия и нарушения сердечной проводимости), нейросенсорная тугоухость, изменения кожи (сухость, ихтиоз) и деформации скелета, аносмия, катаракта. l Диета с ограничением продуктов, богатых фитановой кислотой (тунец, треска, морской окунь, баранина и говядина, белый хлеб, белый рис, яичные желтки, сливочное масло), предупреждает появление неврологической симптоматики l
Болезнь Рефсума. Основные клинические симптомы у пациентов включали ночную слепоту (гемералопию), концентрическое сужение полей зрения с атипичной пигментной дегенерацией, полиневропатию и атаксию. l Основная тетрада клинических симптомов включает пигментную дегенерацию сетчатки, периферическую полиневропатию, церебральную атаксию и повышение концентрации белка в ЦСЖ. Заболевание дебютирует в подростковом возрасте и позднее l Пигментная дегенерация сетчатки, поражение сердца (кардиомиопатия и нарушения сердечной проводимости), нейросенсорная тугоухость, изменения кожи (сухость, ихтиоз) и деформации скелета, аносмия, катаракта. l Диета с ограничением продуктов, богатых фитановой кислотой (тунец, треска, морской окунь, баранина и говядина, белый хлеб, белый рис, яичные желтки, сливочное масло), предупреждает появление неврологической симптоматики l
 Нарушение обмена органических кислот/ аминокислот l l l Большинство болезней этой группы манифестируют в неонатальном периоде, характеризуются острым течением и ранним летальным исходом. Однако некоторые формы НБО могут проявляться позднее, на 2— 5 м месяцах жизни. К таким заболеваниям относятся Глутаровая ацидурия, тип I, фенилкетонурия, болезнь с запахом «кленового сиропа мочи» , множественная карбоксилазная недостаточность, нарушения обмена цикла мочевины и др. [Scriver С. R. et al. , 2001].
Нарушение обмена органических кислот/ аминокислот l l l Большинство болезней этой группы манифестируют в неонатальном периоде, характеризуются острым течением и ранним летальным исходом. Однако некоторые формы НБО могут проявляться позднее, на 2— 5 м месяцах жизни. К таким заболеваниям относятся Глутаровая ацидурия, тип I, фенилкетонурия, болезнь с запахом «кленового сиропа мочи» , множественная карбоксилазная недостаточность, нарушения обмена цикла мочевины и др. [Scriver С. R. et al. , 2001].
 l MPT головного мозга при глутаровой ацидурии типа I. Симметричное расширение сильвиевых щелей с формированием «эффекта надкушенного яблока l МРТ головного мозга при болезни Канавана. Диффузная дегенерация белого вещества головного мозга.
l MPT головного мозга при глутаровой ацидурии типа I. Симметричное расширение сильвиевых щелей с формированием «эффекта надкушенного яблока l МРТ головного мозга при болезни Канавана. Диффузная дегенерация белого вещества головного мозга.
 Фенилкетонурия. объединяют несколько клинически сходных, но генетически гетерогенных заболеваний: классическую фенилкетонурию ФКУ I и атипичные формы, связанные с недостаточностью птеринового кофактора: ФКУ II, III и некоторые другие варианты [Темин П. А. , 2001; Mc. Kusick V. , 1992]. l ФКУ I типа — аутосомно рецессивное заболевание, связанное с дефектом фенилаланингидроксилазы, ген которой картирован и локализован на длинном плече хромосомы 12 (12 q 22—q 24). l Частота заболевания в мире в среднем составляет 1 на 10 000 l
Фенилкетонурия. объединяют несколько клинически сходных, но генетически гетерогенных заболеваний: классическую фенилкетонурию ФКУ I и атипичные формы, связанные с недостаточностью птеринового кофактора: ФКУ II, III и некоторые другие варианты [Темин П. А. , 2001; Mc. Kusick V. , 1992]. l ФКУ I типа — аутосомно рецессивное заболевание, связанное с дефектом фенилаланингидроксилазы, ген которой картирован и локализован на длинном плече хромосомы 12 (12 q 22—q 24). l Частота заболевания в мире в среднем составляет 1 на 10 000 l
 A/p 1: 20000, 12 q 22 -24 l Слабая пигментации кожи, радужной оболочки глаз, волос, умеренная степень олигофреннии Фенилаланингидроксилаза превращает фенилаланин в незаменимую аминокислоту тирозин. В результате блока фермента происходит накопление поступающего с пищей фенилаланина и продуктов его распада — фенилпировиноградной, фенилмолочной, фенилуксусной кислот и других веществ. l прямое токсическое воздействие фенилаланина и его дериватов, l
A/p 1: 20000, 12 q 22 -24 l Слабая пигментации кожи, радужной оболочки глаз, волос, умеренная степень олигофреннии Фенилаланингидроксилаза превращает фенилаланин в незаменимую аминокислоту тирозин. В результате блока фермента происходит накопление поступающего с пищей фенилаланина и продуктов его распада — фенилпировиноградной, фенилмолочной, фенилуксусной кислот и других веществ. l прямое токсическое воздействие фенилаланина и его дериватов, l
 Дебют заболевания в первые 2 мес жизни. Манифестными симптомами являются частые срыгивания, рвота, беспокойство и мышечная гипотония. С рождения отмечаются характерные фенотипические особенности: светлые волосы, голубые глаза и правильные черты лица. В дальнейшем появляются нарушение психомоторного развития и необычный «мышиный» запах тела и мочи. l На 2 м году жизни присоединяются судороги по типу инфантильных спазмов, тонико клонические, тяжелая степень умственной отсталости (IQ менее 50). l
Дебют заболевания в первые 2 мес жизни. Манифестными симптомами являются частые срыгивания, рвота, беспокойство и мышечная гипотония. С рождения отмечаются характерные фенотипические особенности: светлые волосы, голубые глаза и правильные черты лица. В дальнейшем появляются нарушение психомоторного развития и необычный «мышиный» запах тела и мочи. l На 2 м году жизни присоединяются судороги по типу инфантильных спазмов, тонико клонические, тяжелая степень умственной отсталости (IQ менее 50). l
 повышение уровня фенилаланина в биологических жидкостях: его содержание 900 — 1200 мкмоль/л (норма 80— 120 мкмоль/л). l Скрининговые тесты проводят всем детям на 3 — 4 е сутки в специализированных лабораториях. l диетотерапия с пониженным содержанием фенилаланина. В пищевой рацион включают овощи, фрукты, соки, малобелковые продукты (саго, хлеб, вермишель на крахмальной основе), белковые гидролизаты, лишенные фенилаланина (берлофен, нофелан, апонти). Дополнительно назначают витамины (особенно группы В и микроэлементы. l
повышение уровня фенилаланина в биологических жидкостях: его содержание 900 — 1200 мкмоль/л (норма 80— 120 мкмоль/л). l Скрининговые тесты проводят всем детям на 3 — 4 е сутки в специализированных лабораториях. l диетотерапия с пониженным содержанием фенилаланина. В пищевой рацион включают овощи, фрукты, соки, малобелковые продукты (саго, хлеб, вермишель на крахмальной основе), белковые гидролизаты, лишенные фенилаланина (берлофен, нофелан, апонти). Дополнительно назначают витамины (особенно группы В и микроэлементы. l
,") Нарушения обмена углеводов Галактоземия I типа, или классическая галактоземия (недостаточность галактозо 1 фосфат уридилтрансферазы), относится к группе наследственных заболеваний, обусловленных недостаточностью ферментов, участвующих в метаболизме галактозы. Также известны две другие формы галактоземии: l галактоземия II типа (недостаточность галактокиназы) и l галактоземия III типа (недостаточность галактозо 4 эпимеразы). l
Нарушения обмена углеводов Галактоземия I типа, или классическая галактоземия (недостаточность галактозо 1 фосфат уридилтрансферазы), относится к группе наследственных заболеваний, обусловленных недостаточностью ферментов, участвующих в метаболизме галактозы. Также известны две другие формы галактоземии: l галактоземия II типа (недостаточность галактокиназы) и l галактоземия III типа (недостаточность галактозо 4 эпимеразы). l
 Галактоземия I типа l l l l 1 случай на 40 000— 70 000, описано более 180 различных мутаций, Следствием недостаточности фермента является накопление галактозы, галактозо 1 фосфата. Метаболиты токсически действуют на метаболизм многих тканей — мозг, печень, почки, кишечник. Первыми симптомами заболевания являются рвота, плохая прибавка массы тела, диарея. К 2 недельному возрасту развиваются гепатоспленомегалия, у 30 % больных — катаракта, мышечная гипотония. В неонатальном периоде высока смертность детей в результате сепсиса, вызванного Е. coli. У нелеченых больных постепенно развиваются признаки печеночной недостаточности, что может привести к летальному исходу. Основными методами подтверждения диагноза галактоземии являются биохимические тесты, основанные на определении концентрации галактозы, галактозо 1 фосфата в крови Назначение безлактозных смесей приводит к быстрому купированию основных клинических симптомов и восстановлению функции печени Примерно у 20 % детей старшего возраста наблюдаются атаксия, интенционный тремор, гипотония. (IQ = 70— 80);
Галактоземия I типа l l l l 1 случай на 40 000— 70 000, описано более 180 различных мутаций, Следствием недостаточности фермента является накопление галактозы, галактозо 1 фосфата. Метаболиты токсически действуют на метаболизм многих тканей — мозг, печень, почки, кишечник. Первыми симптомами заболевания являются рвота, плохая прибавка массы тела, диарея. К 2 недельному возрасту развиваются гепатоспленомегалия, у 30 % больных — катаракта, мышечная гипотония. В неонатальном периоде высока смертность детей в результате сепсиса, вызванного Е. coli. У нелеченых больных постепенно развиваются признаки печеночной недостаточности, что может привести к летальному исходу. Основными методами подтверждения диагноза галактоземии являются биохимические тесты, основанные на определении концентрации галактозы, галактозо 1 фосфата в крови Назначение безлактозных смесей приводит к быстрому купированию основных клинических симптомов и восстановлению функции печени Примерно у 20 % детей старшего возраста наблюдаются атаксия, интенционный тремор, гипотония. (IQ = 70— 80);
 Спасибо за внимание !
Спасибо за внимание !