ac42c6e2df568afd7b0d53c1b8f6943d.ppt
- Количество слайдов: 102



Общая характеристика моногенной патологии. Клиника и генетика отдельных форм моногенных болезней Проф. кафедры Павлишин Г. А.

Удельный вес наследственной патологии среди населения мира

Удельный вес наследственных нарушений обмена веществ среди наследственной патологии

Моногенные болезни – это разнородная по клиническим проявлениям группа заболеваний, обусловленных мутациями на генном уровне

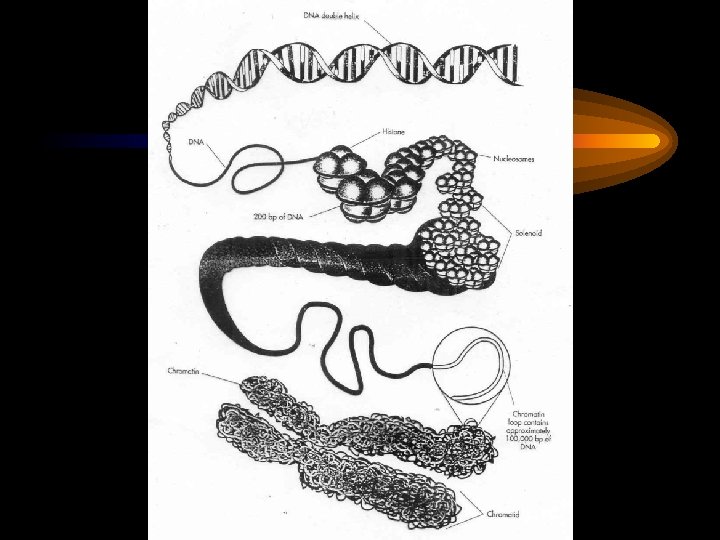
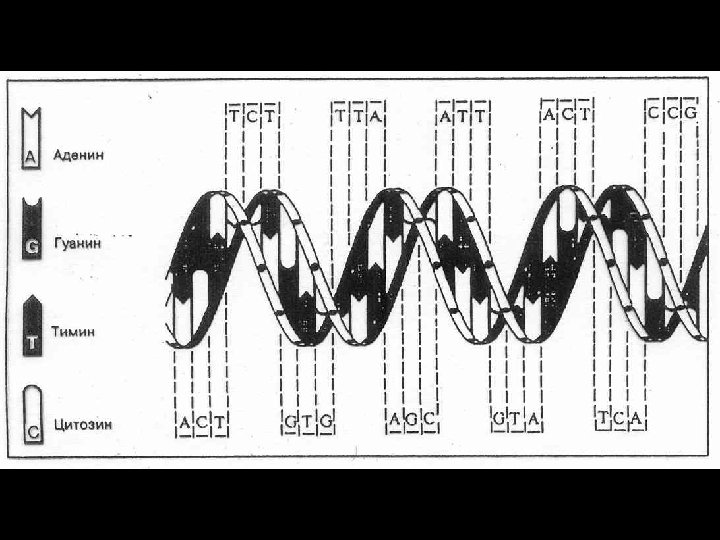
 Нарушения")
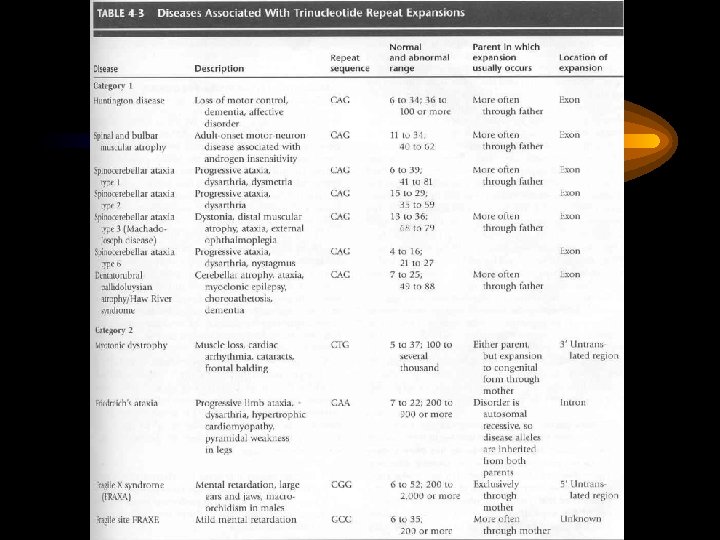
Виды генных мутаций • • Миссенс-мутации Нонсенс-мутации Сдвиг рамки считывания Делеции Вставки (инсерции) Нарушения сплайсинга Увеличение числа (экспансия) тринуклеотидных повторов

Частота генетически детерминированных болезней Формы наследственной патологии 1. Моногенные аутосомно-доминантные аутосомно-рецессивные Х-сцепленные 2. Хромосомные болезни 3. Мультифакториально обусловленные* 4. Врожденные пороки развития* ВСЕГО Примечание: * вклад генетических факторов варьирует Частота на 1000 4, 5 -15, 0 2, 0 -9, 5 2, 0 -3, 5 0, 5 -2, 0 5, 0 -7, 0 -10, 0 19, 0 -22, 00 35 -54

Частота отдельных симптомов при гиперлизинемии ІІ типа

Частота отдельных симптомов при цитруллинемии

Опухоли и возраст детей

Объемные образования потенциально возможные у новорожденного

С косолапостью сочетаются: • • Артрогрипоз Синдром каудальной регрессии Детский церебральный паралич Краниокарпотарзальная дистрофия Дистрофическая карликовость Синдром Ларсена Миеломенингоцеле Прогрессирующая мышечная атрофия (малоберцовый тип) • Опухоль спинного мозга • Миотоническая дистрофия
 Хирургические болезни Всего больных Количество синдромов абс. % Ортопедическая")
Синдромология хирургических болезней (собственные данные) Хирургические болезни Всего больных Количество синдромов абс. % Ортопедическая патология (косолапость, деформация грудной клетки, сколиоз, лучевая косорукость, псевдоартрозы, врожденный вывих бедра) 76 44 58 Аноректативные дефекты (атрезия ануса и прямой кишки, стенозы и эктопия ануса) 19 12 62 Патология пищевода (атрезии и стенозы пищевода, желудочно-пищеводный рефлюкс) 26 9 34

Актуальность проблемы наследственных аномалий обмена веществ • Значительная распространенность патологии • Большое разнообразие наследственных ферментопатий • Большое количество наследственных ферментопатий заканчиваются летально или тяжелым умственным и физическим отставанием

Удельный вес наследственной патологии в детской смертности

Актуальность проблемы • Заболевания проявляются в разные периоды жизни ребенка • Диагностика патологии вызывает значительные трудности • Большие общие экономические затраты на содержание детей-инвалидов

Нет наиболее актуальной проблемы в медицинской и социальной сфере, чем предупреждение, выявление и лечение наследственной патологии у детей
 • Обусловленные влиянием")
Классификация заболеваний человека по Г. Харрису • Наследственные ( хромосомные, генные) • Обусловленные влиянием вредных факторов внешней среды • Обусловленные наследственной склонностью и действием провоцирующих факторов внешней среды
 Ген А Ген В Ген С Фермент")
Схема патогенеза наследственного нарушения обмена веществ (1) Ген А Ген В Ген С Фермент А Фермент В Фермент С Вещество А Вещество В Вещество С Вещество D
 Ген А Фермент А Вещество А Ген")
Схема патогенеза наследственного нарушения обмена веществ (2) Ген А Фермент А Вещество А Ген В Ген С Фермент В Вещество X Фермент С отсутствует Вещество С Вещество Y Вещество D Вещество. Z

Аномалии обмена аминокислот 1. Наследственные нарушения обмена АК, которые сопровождаются повышением их концентрации в крови и моче 2. Наследственные нарушения обмена АК, которые сопровождаются повышением их выделения с мочой без изменений уровня в крови 3. Наследственные нарушения системы транспорта АК 4. Вторичные гипераминоацидурии

Клинические признаки нарушения обмена АК у новорожденного ребенка • Появление уже через несколько часов после рождения сонливости, затруднений при кормлении, судорог и рвоты • Наличие выраженного рвотного синдрома после исключения хирургической патологии • Наличие в анамнезе близкородственных браков или случаев смерти детей в неонатальном периоде • Выявление при объективном обследовании неспецифических данных, которые свидетельствуют о поражении центральной нервной системы, гепатомегалию или необычный запах

Изменение запаха тела и экскрементов ребенка при болезнях обмена АК

Клинические признаки врожденного нарушения обмена веществ и АК в постнатальном периоде • Необъяснимое ничем отставание умственного и двигателього развития, судороги в анамнезе • Необычный запах, особенно во время острого заболевания • Интермитирующие эпизоды необъяснимой рвоты, ацидоза • Гепатомегалия • Почечные камни

Принципы диагностики наследственных нарушений обмена АК • • Генетические данные Сроки манифестации (ранняя и поздняя) Клинические характеристики Данные лабораторных исследований (увеличение содержания АК в крови и моче, увеличение мочевой экскреции продуктов нарушенного обмена АК, увеличение уровня амиака в крови)

Принципы лечения больных с наследственными нарушениями АК обмена • Ограничение поступления белка и соответственной аминокислоты с пищевыми продуктами • Дополнительное введение незаменимых аминокислот • Назначение препаратов, которые активируют альтернативные пути метаболизма • Назначение препаратов, которые усиливают связывание и выведение накопленных продуктов метаболизма • Назначение кофакторов энзимных реакций • Противосудорожная терапия и ноотропные средства • Интенсивная терапия в остром периоде с использованием гемофильтрации и перитонеального диализа

Органические ацидемии • Состояния, обусловленные наследственной недостаточностью ферментов, которые осуществляют различные этапы превращения производных АК • Состояния, обусловленные нарушением биоэнергетических процесов (цикла Кребса) в метохондриях клеток • Заболевания, вызванные нарушением транспорта или митохондриального окисления жирных кислот • Пероксисомные заболевания
 и сопровождается")
Фенилкетонурия - группа заболеваний, которое возникает вследствие дефекта метаболизма аминокислоты фенилаланина (ФА) и сопровождается задержкой психомоторного развития и определенными специфическими проявлениями
 Фенил-гидроксилаза ФА Меланин Тирозин Адреналин")
Схема патогенеза фенилкетонурии (1) Фенил-гидроксилаза ФА Меланин Тирозин Адреналин
 Накопление фенилаланина фенил-пировиноградной кислоты фенил-уксусной кислоты фенил-молочной кислоты Фенил-гидроксилаза ФА")
Схема патогенеза фенилкетонурии (2) Накопление фенилаланина фенил-пировиноградной кислоты фенил-уксусной кислоты фенил-молочной кислоты Фенил-гидроксилаза ФА

Схема 1.

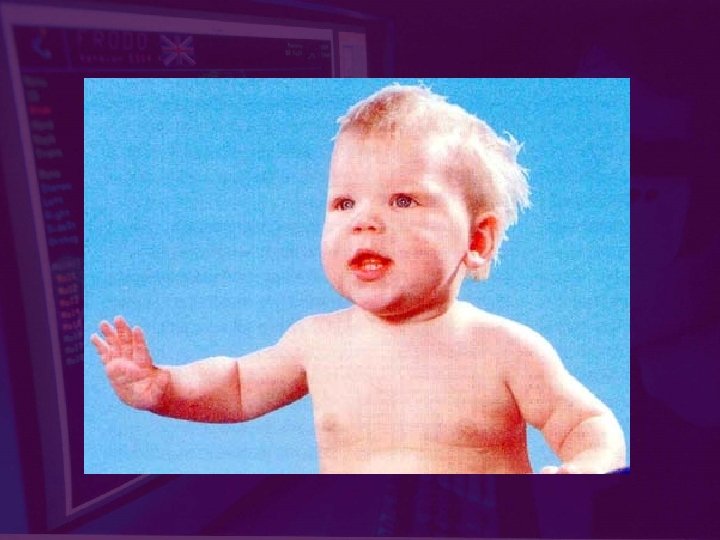
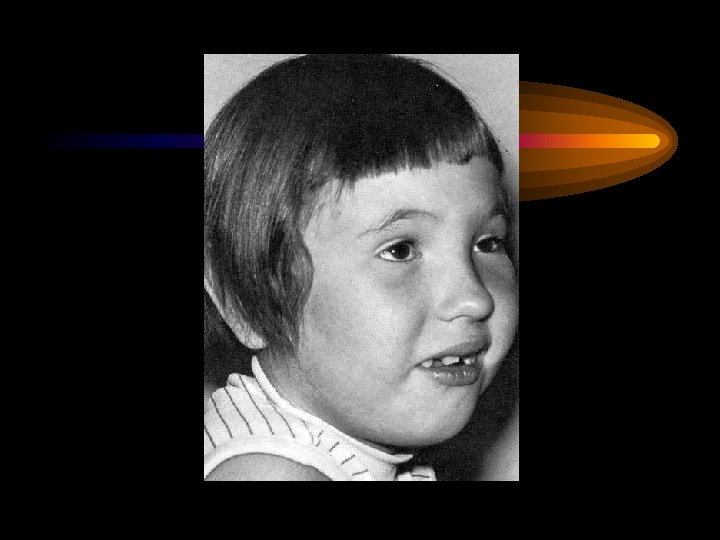
Клинические признаки ФКУ • Появление первых видимых признаков на 4 -6 месяце • Характерный внешний вид больного: бледная кожа, светлые волосы и глаза • Неврологические нарушения: повышение сухожильных рефлексов, гипертонус мышц, симптом Грефе, гидроцефалия или микроцефалия • Диспепсический синдром: частые рвоты, диарея • Кожный синдром: бледная кожа, экзематозные проявления, повышенная потливость • Специфический запах мочи и пота - мишиный, затхлый, плесени • Изменения в психической сфере : пассивность, замедление реакций, речевые нарушения, агрессивное поведение
")
Фенилкетонурия у ребенка 1 -го года. (значительная задержка психомоторного развития)

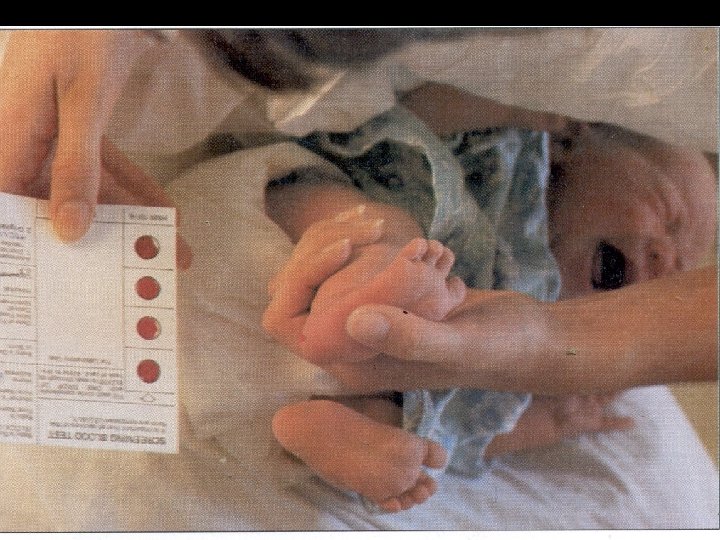
Первичная диагностика ФКУ- массовый скрининг новорожденных детей • Обследование всех новорожденных на 45 день жизни в роддоме до выписки при наличии начатого белкового питания ребенка • Недоношенных и больных в неонатальном периоде детей на 7 сутки жизни
 диагностика ФКУ • Метод тонкослойной и жидкостной хроматографии • Молекулярно-генетические методы диагностики")
Подтвержающая (вторичная) диагностика ФКУ • Метод тонкослойной и жидкостной хроматографии • Молекулярно-генетические методы диагностики генного дефекта • Определение наличия ФА и продуктов искаженного обмена в крови и моче ( проба Феллинга, индикаторные бумажные тесты) • Полуколичественные микробиологические тесты ( тест Гатри, метод ауксотрофных штаммов E. Coli K-12)

“ Следует всегда помнить, что мы не можем руководить событиями, а должны приспосабливаться к ним” Епиктет


Детские смеси, которые применяют при ФКУ


 Хроническое полисиндромное заболевание, которое характеризуется неспецифическими")
Целиакия ( глютеновая энтеропатия, идиопатическая стеаторея, нетропическая спру) Хроническое полисиндромное заболевание, которое характеризуется неспецифическими повреждениями слизистой оболочки тонкой кишки глютеном и проявляется нарушеннями абсорбции на поврежденных участках кишечника

Клинические критерии целиакии • • • Развитие во втором полугодии жизни Наличие объемных, с неприятным запахом, светлых испражнений больше 2 раз в сутки Увеличение окружности живота, боли в животе Рвота ежедневная или периодическая Отставание в массе тела и роста Осалгии, повышенная ломкость костей Раздражительность, агрессивность, неспокойный сон Наличие жирных кислот, мыл в копрограмме Дисбактериоз кишечника
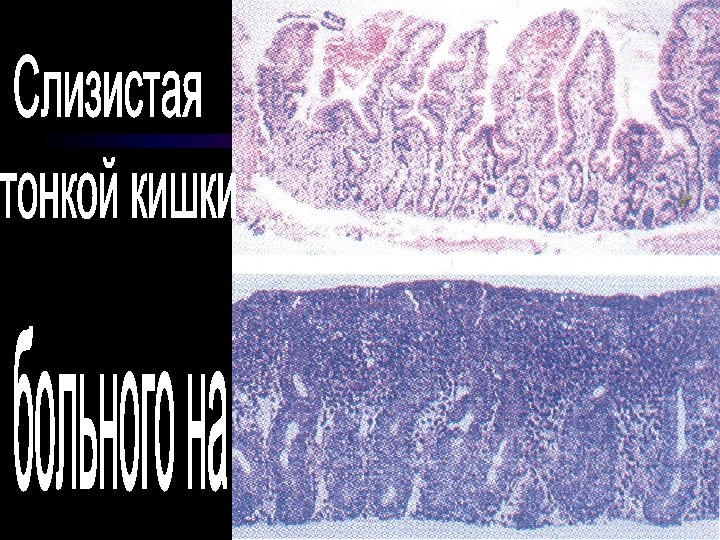

Лабораторные признаки целиакии • Анемия • Снижение уровня сивороточного железа и фолатов • Снижение содержания витамина В 12 в сиворотке крови • Гипоальбуминемия и гипогамаглобулинемия • Стеаторея • Плоская кривая при проведении пробы на толерантность к глюкозе • Рентгенконтрастное исследование тонкого кишечника - диффузное расширение кишечника, неровность складок слизистой оболочки • Рентгенография скелета - признаки остеопороза

Детские смеси, которые применяют при целиакии

Наследственные заболевания обмена углеводов и жиров. Особенности клиники, диагностики и лечения в детском возрасте.

Группа наследственных заболеваний обмена углеводов включает различные нозологические формы заболеваний, обусловленных патологией метаболизма моносахаридов (глюкозы, фруктозы, галактозы), дисахаридов (лактозы, мальтозы, сахарозы) и полисахаридов (гликоген, крахмал)

Галактоземия Это заболевание, связанное с невозможностью использования организмом галактози и проявляется тяжелым поражением печени, нервной системы, глаз и других органов.
 Галактозо-1 -фосфат-уридилтрансфераза галактоза глюкоза")
Схема патогенеза галактоземии (1) Галактозо-1 -фосфат-уридилтрансфераза галактоза глюкоза
 Галактозо -1 -фосфат-уридилтрансфераза Накопление галактоза галактозо-1 -фосфата в эритроцитах, мышцах,")
Схема патогенеза галактоземии (2) Галактозо -1 -фосфат-уридилтрансфераза Накопление галактоза галактозо-1 -фосфата в эритроцитах, мышцах, сердце, хрусталике и коре головного мозга Клинические проявления
 Галактозо -1 -фосфат-уридилтрансфераза Инактивация реабсорбции Накопление Вторичная АК в галактозо-1")
Схема патогенеза галактоземии (3) Галактозо -1 -фосфат-уридилтрансфераза Инактивация реабсорбции Накопление Вторичная АК в галактозо-1 -фосфата галактоза почечных аминоацидурия канальцах АК Компенсаторное увеличение тестостерон Второстепенный метаболизм активности галактозы удфгп

Диагностические критерии галактоземии • • • Большая масса при рождении Диспепсические проявления сразу после рождения Быстрое развитие гипотрофии Желтуха с повышением неконьюгированного билирубина Гепатомегалия с дальнейшим развитием цирроза печени, портальной гипертензии Гипогликемия в анамнезе Отставание в психомоторном розвитии Геморрагический диатез Формирование катаракты на третьей неделе жизни

Пентада галактоземии • Желтуха за счет прямого билирубина в периоде новорожденности • Частая рвота без расстройства испражнений • Резкая степень истощения - гипотрофия • Катаракта • Отставание в психомоторном развитии

Диагностика галактоземии • Изучение генеологического анамнеза • Оценка анамнестических данных • Выявление клинических диагностических критериев • Определение гликемии с помощью различных методов • Выявление галактозурии • Снижение активности галактозо-1 фосфатуридилтрансферазы • Изменение функциональных печеночных проб • Оценка неврологического статуса • Офтальмологическое исследование

Определение прироста гликемии на нагрузку галактозой разными способами в диагностике галактоземии • Глюкозооксидазный метод отображает содержание истинно глюкозы в крови • Ортотолуидиновый метод отображает содержание глюкозы вместе с галактозой
 до 3 лет • Назначение безлактозных смесей (Энфамил")
Лечение галактоземии • Исключение молока (галактозы) до 3 лет • Назначение безлактозных смесей (Энфамил Соя, Нутрамиген, Симилак-изомил, Нутрисоя, Просоял) • Прикорм вводят на месяц раньше, чем обычно с постепенной заменой смеси • Медикаментозная терапия: 1. Препараты, которые улучшают обмен галактозы • 2. Урацил-4 -карбоновая ( оротовая) кислота • 3. Производные тестостерона • 4. Препараты, стимулирующие ЦНС • Сосудистые препараты • Гепатопротекторы • Антиоксиданты • Витаминотерапия
 • Первичная - генетически детерминирована (аутосомнорецесивная) или обусловленная мутацией гена, который отвечает")
Алактазия (гиполактазия) • Первичная - генетически детерминирована (аутосомнорецесивная) или обусловленная мутацией гена, который отвечает за синтез фермента лактазы • Вторичная как следствие поражения слизистой оболочки тонкой кишки

Роль лактозы в организме ребенка • Удовлетворяет 40 % энергетических потребностей организма • Способствует усвоению кальция и железа • Способствует становлению здоровой флоры кишечника • Замедляет рост патогенной микрофлоры кишечника • Наличие галактозы (следствие расщепления лактозы) способствует усовершенствованию ЦНС

Диагностические критерии алактазии • Начало клиники с рождения с развитием обезвоживания и токсикоза • Стойкий диспепсический синдром (рвота после кормления грудным молоком и пронос), который не поддается лечению обычными способами • Характер испражнений - малообъемные, частые, с кислым запахом • Прогрессирующая гипотрофия • Отсутствие прироста гликемии на нагрузку лактозой • Положительный водородный тест

Пероральноя нагрузка лактозой • Введение перорально лактозы из расчета 1 г/кг массы тела ребенка • Измерение содержания глюкозы в крови через 30 и 60 минут после нагрузки
")
Лечение алактазии • Назначение безлактозных или низколактозных энпитов (Симилакизомил, Энфамил Соя, Нутрамиген, Нутрисоя, Просоял) • Введение кислых смесей • Симптоматическое лечение (ферменты, витамины, бактерийные препараты, озокеритотерапия)
 Группа заболеваний, которые передаются аутосомнорецессивным путем и обусловлены энзимным дефектом, который ведет")
Гликогенозы (GLYCOGENOSIS) Группа заболеваний, которые передаются аутосомнорецессивным путем и обусловлены энзимным дефектом, который ведет к нарушению синтеза и расщепления гликогена с его накоплением во внутренних органах
")
І тип гликогеноза (болезнь Гирке, гепаторенальный тип)
 Глюкозо-6 -фосфатаза гликоген Глюкозо-6 -фосфат глюкоза")
Схема патогенеза гликогеноза (болезнь Гирке) Глюкозо-6 -фосфатаза гликоген Глюкозо-6 -фосфат глюкоза
 Гипогликемия Глюкозо-6 -фосфатаза гликоген Накопление гликогена")
Схема патогенеза І типа гликогеноза ( болезнь Гирке) Гипогликемия Глюкозо-6 -фосфатаза гликоген Накопление гликогена в печени, почках Вторичное нарушение липидного обмена Увеличение липидемии Увеличение печени и почек Отложение жира в ПЖК
 •")
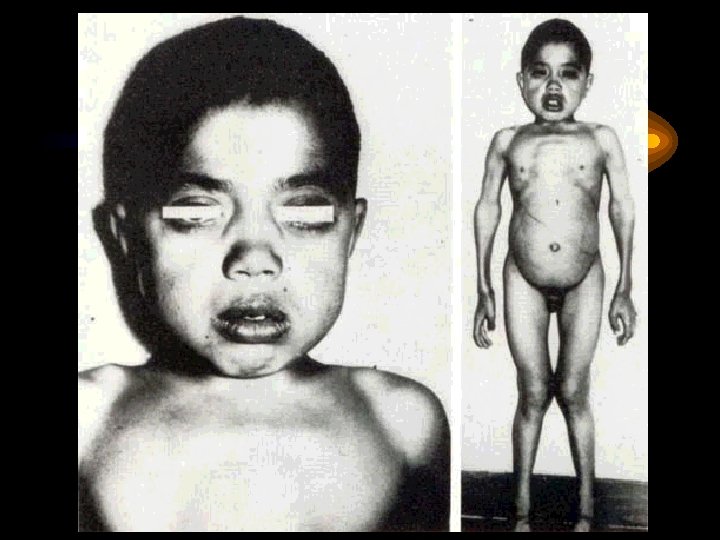
Диагностические критерии болезни Гирке • Клинические признаки гипогликемии (вялость, адинамия, рвота, судорожный синдром) • Увеличение размеров живота • Гепато-, спленомегалия, увеличение размеров почек • Характерный внешний вид ребенка: круглое «кукольное» лицо, маленький рост, короткие руки и шея, значительно увеличенный в размерах живот, умеренное отложение жира в области лица и туловища, мышечная сила снижена • Анорексия
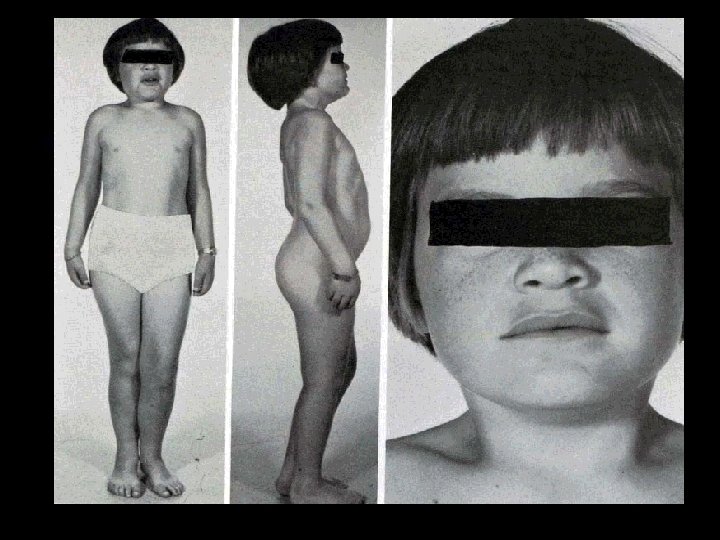

Диагностические критерии болезни Гирке • Появление гипогликемии при недлительном голодании, натощак, поэтому больные должны постоянно принимать еду • Гиперлипемия, гиперлактацидемия • Проба с адреналином или глюкагоном отрицательная по гликемии • Частая гиперкетонемия натощак с клиническими проявлениями кетозу, ацетонурия • Нарушение функциональных проб печени • Большое содержание гликогена в биоптатах печени и в клетках периферической крови, отсутствие, или снижения глюкозо-6 -фосфотазы
")
ІІ тип гликогеноза (болезнь Помпе, генерализованый гликогеноз, гликогенная кардиомегалия)

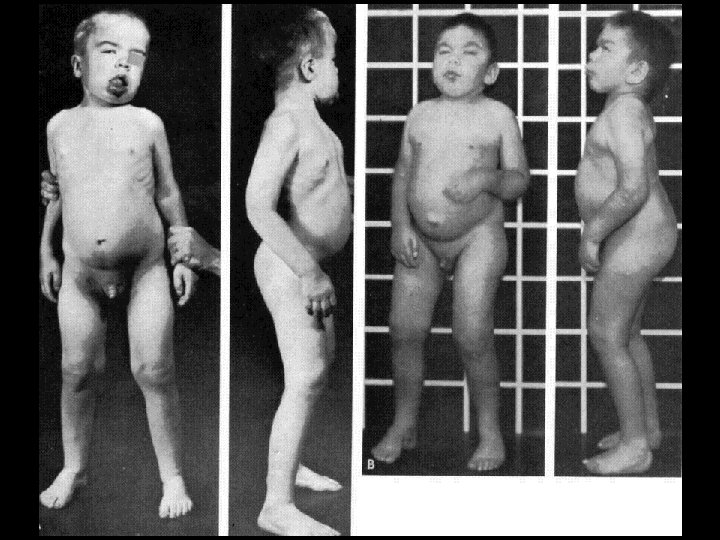
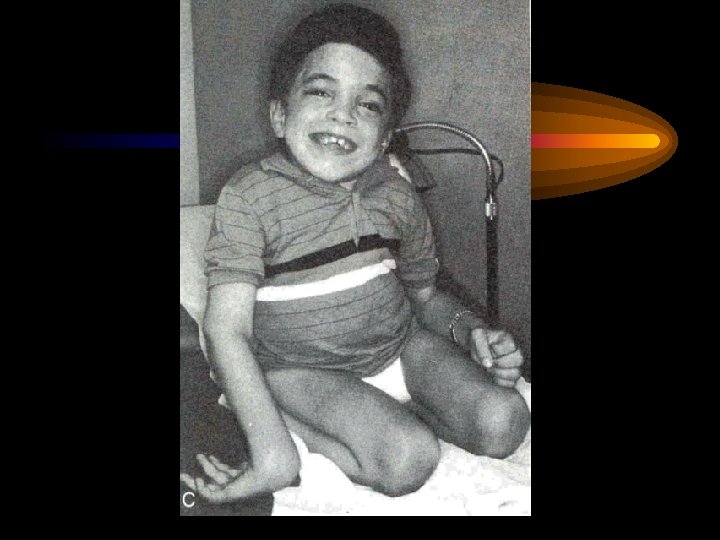
Дагностические критерии болезнь Помпе • Появление клиники в раннем грудном возрасте • Шарообразная кардиомегалия • Сердечная недостатность по правожелудочковым типом • Частые пневмонии на фоне ателектазов • Характерный внешний вид ребенка: круглое, пастозное лицо, увеличенный язык, мышечная гипотония, задержка физического развития
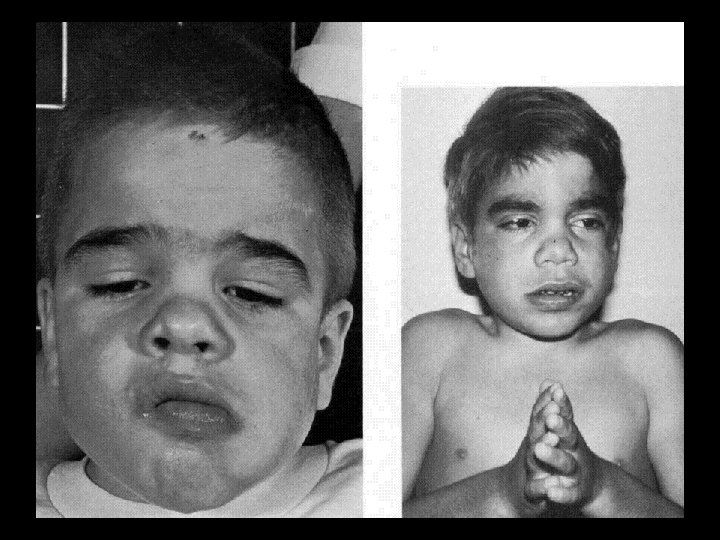
 обусловленный отсутствием фермента амино-1, 6")
ІІІ тип гликогеноза (гликогеноз печени и мышц, болезнь Форбса) обусловленный отсутствием фермента амино-1, 6 -глюкозидазы или олиго-1, 1 -глюкотрансферазы, что приводит к накоплению гликогена в печени, мышцах скелета и сердца

Наследственные болезни обмена липидов • Гиперлипопротеидемии • Липодистрофии • Липидозы
")
Гиперлипопротеидемии • Характеризуются нарушением жизненноважных химически разнообразных структур (глицериды, холестерол и его экстеры)
 • Проявляется вушкольном возрасте • атрофия жировой ткани")
Липодистрофия прогрессирующая сегментарная (болезнь Барракера. Симондса) • Проявляется вушкольном возрасте • атрофия жировой ткани на ограниченных участках лица, верхней половины туловища • дебильность • кистозные изменения костей • отосклероз
 Группа заболеваний, в основе которых лежит дефект определения фермента, который ведет к")
Липидозы (lipidosis) Группа заболеваний, в основе которых лежит дефект определения фермента, который ведет к накоплению повышенного количества липидных субстанций в клетках и тканях ЦНС и внутренних органов тип наследования аутосомно-рецессивный

Болезнь Гоше - характеризуется отложением в печени, селезенке, костях цереброзида, который содержит глюкозу (в N содержится галактоза) Формы: • Инфантильная • Ювенильная • Хроническая висцеральная

Диагностические критерии инфантильной формы • С первых месяцев жизни отставание в психомоторном и физическом развитии • Спленогепатомегалия • Дисфагия, стридор • Опистотонус, конвульсии • Поражение костного мозга (анемия, лейкопения, тромбоцитопения) • Геморрагический синдром

Диагностические критерии ювенильной формы • • Начало преимущественно на втором году жизни Отставание в умственном развитии Гепатоспленомегалия Анемия, лейкопения Гиперрефлексия Хореатетоз Конвульсии

Диагностические критерии хронической висцеральной формы • Проявляется у подростков или у взрослых • Без поражения ЦНС и задержки умственного развития • Гепатоспленомегалия • Анемия, лейкопения • Клетки Гоше, которые находят в красной пульпе селезенки, лимфатических узлах, костном мозге, синусоидах печени

Наследственные нарушения обмена соединительной ткани Группа различных по происхождению нозологических форм, которые объединяют первичное или вторичное вовлечение в патогенез заболевания соединительной ткани и обусловленные нарушением ферментных систем, которые контролируют синтез структурных белков

Синдром Марфана • Тип наследования аутосомнодоминантный • Встречаются с частотой 1: 50 000 • Включает два типа: астенический (детский) и неастенический

Скрининг-тест для диагностики синдрома Марфана • Длина среднего пальца кисти руки находится в пределах 10 см и больше • Размах рук превышает длину тела на 5 см и больше

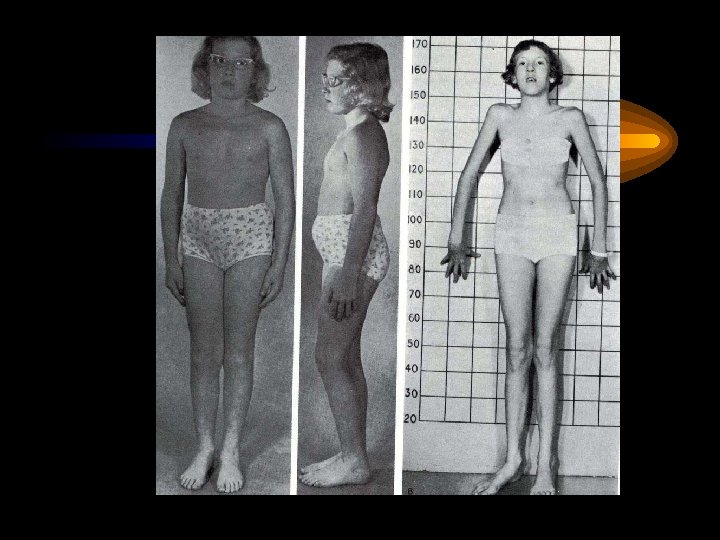
Два брата. Слева: нормальный мальчик 10 лет. Справа 8 -летний мальчик с синдромом Марфана (симптомы: подвывих хрусталика (он в очках), высокий рост, отсутствие подкожной жировой клетчатки, сколиоз, деформация грудной клетки)
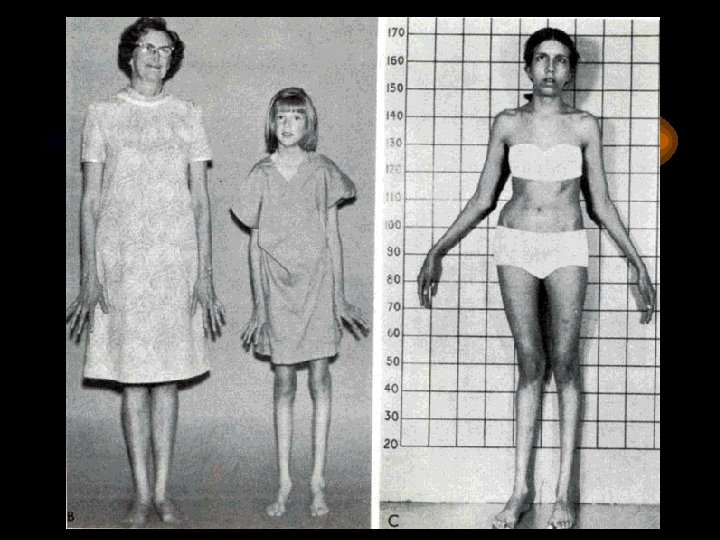

Диагностические критерии синдрома Марфана • Нарушение мышечно-скелетной системы: арахнодактилия, высокий рост, длинные конечности, искривления позвоночника, деформация передней грудной клетки, ненормальная подвижность суставов, мышечная гипотония • Кожный синдром: атрофичные стрии, паховые грыжи • Патология глаз: миопия, вывих или подвывих хрусталика обеих глаз • Сердечно-сосудистый синдром: расширение аорты, пролабирование митрального клапана, нарушение проведения возбуждения • Бронхолегечной синдром: эмфизема, пневмоторакс
 и при синдроме Марфана (справа)")
Ладони и ступни в норме (слева) и при синдроме Марфана (справа)

Диагностические критерии арахнодактилии при синдроме Марфана
")
Николо Паганини (1784 -1840)

Спасибо за внимание
ac42c6e2df568afd7b0d53c1b8f6943d.ppt