Иммунодефициты.ppt
- Количество слайдов: 19



НОУ ВПО МИ «РЕАВИЗ» Кафедра медико-биологических дисциплин Тема: Иммунодефициты. Иммунодефицитные состояния. ВЫПОЛНИЛА СТУДЕНТКА 314 ГРУППЫ Кукушкина С. А. ПРЕПОДАВАТЕЛЬ: ПАНИН П. Ф

n Иммунодефицитные состояния и иммунодефициты — типовые формы патологии системы ИБН, характеризующиеся снижением эффективности или неспособностью иммунной системы организма к осуществлению реакций деструкции и элиминации чужеродного Аг. n В основе развития иммунодефицитных состояний и иммунодефицитов, как правило, находятся отсутствие или дефицит клеток иммунной системы и/или расстройства их функций. Это обусловливает высокую частоту развития при иммунодефицитах различных инфекционных, паразитарных, опухолевых и аллергических заболеваний. С другой стороны, при истощающих заболеваниях часто развиваются иммунодефицитные состояния.

Частота иммунодефицитов и иммунодефицитных состояний n Один из 500 младенцев рождается с дефектом иммунной системы. • Значительно большее количество лиц приобретают транзиторный или постоянный иммунодефицит в течение жизни.
 дефекты иммунной системы.")
Виды иммунодефицитных состояний n n n Первичные наследуемые и врождённые (генетические) дефекты иммунной системы. Вторичные — иммунная недостаточность развивается вследствие эндо- и экзогенных воздействий на нормальную иммунную систему (например, около 90% всех вирусных инфекций сопровождается транзиторной иммунодепрессией). Избирательные — вызваны селективным поражением различных популяций иммунокомпетентных клеток. Неспецифические — дефекты механизмов неспецифической резистентности организма (неспецифического иммунитета), фагоцитов и комплемента. Комбинированные — сочетанное поражение клеточных и гуморальных механизмов иммунитета (например, В- и Т-лимфоцитов).

В зависимости от преобладания дефекта иммуноцитов того или иного класса, иммунодефициты и иммунодефицитные состояния подразделяют на В-, Т-, А-зависимые (относящиеся к антигенпредставляющим клеткам) и смешанные.
 состояние недостаточности")
По преобладанию ведущих механизмов иммунопатологические процессы принято дифференцировать по видам: n 1) состояние недостаточности функции иммунной системы вследствие резкого снижения или полного отсутствия какого-либо типа молекул или клеток иммунной системы, т. е. иммунодефицит; 2) чрезмерная пролиферация какой-либо популяции, субпопуляции или клона лимфоцитов, приводящая к его экспансии и вытеснению всех других типов клеток, т. е. лимфопролиферативный процесс. Лимфопролиферативные процессы по своей сути относятся к опухолевым и, помимо специфической симптоматики (увеличение численности клона клеток, увеличение объёма ткани и т. д. ), сопровождаются признаками иммунодефицита. Механизм его развития связан с вытеснением из организма нормально функционирующих лимфоциты и продуктов их генерации опухолевым клоном; 3) состояние усиленной реактивности иммунной системы (гиперчувствительности, гиперергии), при которой иммунный ответ формируется на чужеродные молекулы-антигены (аллергены), не вызывающие в норме иммунного ответа вследствие элиминации физиологическими защитными реакциями. Это состояние называется аллергией; 4) состояние усиленной реактивности иммунной системы, при которой иммунный ответ развивается в отношении собственных клеток, тканей и молекул организма, т. е. характеризуется аутоагрессией. Такие процессы называются аутоиммунными.

ЭТИОЛОГИЯ n n n n Первичные иммунодефициты проявляются развитием инфекционных поражений организма вскоре после рождения, но могут не иметь клинических проявлений и до более позднего возраста. Генные и хромосомные дефекты (они приводят к многочисленным иммунодефицитам разных классов) Вторичные иммунодефициты, или иммунодефицитные состояния Причины иммунодефицитных состояний многообразны: Лекарственные средства с иммуносупрессивным действием (включая фенитоин [дифенин], пеницилламины, глюкокортикоиды). Недостаточность питания, полостного и мембранного пищеварения, а также кишечного всасывания. Наркотики и токсические вещества. Лучевые воздействия, химиопрепараты. Рост злокачественных опухолей. Вирусы (например, ВИЧ). Состояния, приводящие к потере белка (например, нефротический синдром). Гипоксия. Гипотиреоз. Уремия. Отсутствие селезенки (аспления).
 инфекционный синдром -")
Среди основных клинических синдромов, сопутствующих развитию иммунопатологических процессов выделяют: n 1) инфекционный синдром - развивается как следствие снижения способности иммунной системы к надзору за антигенным постоянством организма. Сопровождается учащением инфекционной заболеваемости, возможностью развития оппортунистических инфекций (вызываются представителями аутомикрофлоры и даже сапрофитой микрофлорой), более тяжёлым и затяжным течением, высоким риском хронизации инфекций, склонностью к генерализации инфекции и высокой летальностью при инфекциях, вызванных облигатно патогенными микробами; 2) опухолевый синдром – имеет те же причины и механизмы развития, что и инфекционный синдром; 3) синдром перенапряжения (или переактивации) иммунной системы – возникает вследствие неадекватной продукции цитокинов, клинические проявления различны и связаны с особенностями цитокинового профиля. Часто дополняется развитием массивного апоптоза клеток иммунной системы с последующим развитием (или усугублением имеющегося) иммунодефицита. Может сопровождаться клиническими признаками повышенной чувствительности, сходными с аллергией, но при преобладании в механизме развития эффектов цитокинов. В ряде случаев синдром сопровождается субфебрильной или фебрильной температурой, лимфаденопатией, увеличением СОЭ, изменением лейкоцитарной формулы; 4) синдром цитолиза развивается в результате иммуноопосредованного повреждения собственных клеток (тканей) со снижением выполняемой органом (тканью) функцией; 5) астеновегетативный синдром – сопровождается появлением слабости или чувством усталости, колебаниями настроения и вегетативным реакциями (изменение частоты сердечных сокращений, приступы потливости, изменение моторики желудочно-кишечного тракта и др. В основе этого синдрома – нарушений нейро-иммуно-эндокринных взаимодействия.

Синдром ретикулярной дисгенезии n Ретикулярная дисгенезия — редкое заболевание, характеризующееся дефицитом лимфоцитов и полиморфно -ядерных лейкоцитов. У больных в костном мозге отсутствуют гранулоциты и их предшественники. В периферической крови снижено содержание лимфоцитов, в органах кроветворения отсутствуют стромальные ретикулярные клетки. Патогенез ретикулярной дисгенезии связывают с нарушением дифференцировки стволовой клетки, возможно, клетки-предшественницы миелопоэза и лимфопоэза, но ни одно нарушение клеточного развития, взятое в отдельности, не может объяснить всех клинических проявлений указанной патологии. Дальнейший анализ случаев ретикулоцитной дисгенезии должен прояснить природу заболевания.

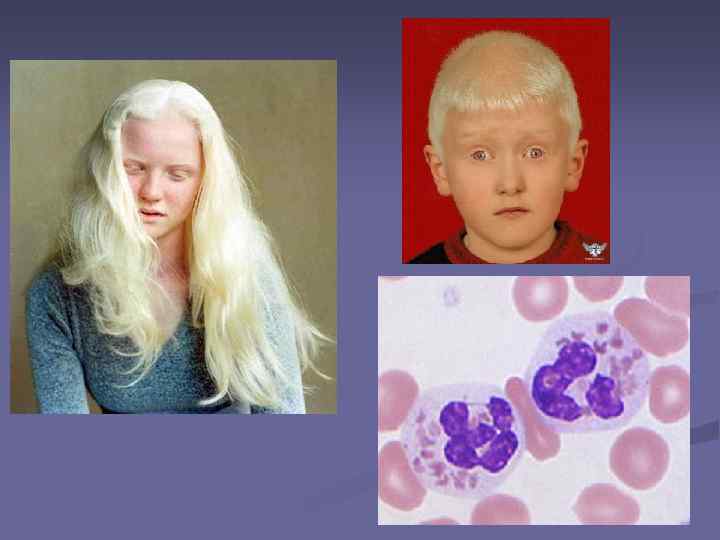
Синдром Чедиака–Хигаси n n редкое заболевание, характеризующееся тяжелыми повторными гнойными инфекциями, парциальным альбинизмом, прогрессирующей нейропатией, склонностью к кровотечениям, развитием лимфопролиферативного синдрома, а также наличием гигантских гранул во многих клетках, особенно в лейкоцитах периферической крови. Иммунодефицит при синдроме Чедиака-Хигаши обусловлен, в первую очередь, нарушением функции фагоцитоза в клетках гранулоцитарного и макрофагального ряда и проявляется склонностью к гнойным и грибковым инфекциям. Кровоточивость связана с дефектом высвобождения фомбоцитаркых гранул. Первое упоминание о синдроме Чедиака-Хигаши относится к 1943 году {Beguez Cesar). Дальнейшие описания мы находим у Steinbrinck 1948, Chediak 1952 и, наконец, у Higashi 1954.

Патогенез заболевания n связывают с аномалией структуры клеточных мембран, нарушением системы собирательных микротрубочек и дефектом взаимодействия последних с мембранами лизосом. Большая часть клинических проявлений может быть объяснена аномальным распределением лизосомальных ферментов. Частота и тяжесть пиогенных инфекций обусловлена снижением активности кислородного метаболизма и внутриклеточного переваривания микробов в фагоцитах вследствие задержки и непостоянного высвобождения гидролитических лизосомальных ферментов из гигантских гранул в фагосомы. Кроме того, у больных снижены активность естественных киллеров и антителозависимая цитотоксичность лимфоцитов. Заболевание относят к первичным иммунодефицитам.

Тяжёлый комбинированный иммунодефицит В классическом варианте отсутствует реакции как гуморального, так и клеточного иммунитета (нет Т-клеток и естественных киллеров); выявляются алимфоплазия или лимфопения (относится как к B-лимфоцитами, так и к T-лимфоцитам). Характерна низкая устойчивость к бактериальным, грибковым, протозойным, вирусным инфекциям. Введение живых вакцин таким лицам должно быть исключено. Смерть наступает к концу первого года жизни (если не проведена трансплантация костного мозга). Примерно у 70% больных В-лимфоциты присутствуют (в том числе при мутациях генов, недостаточности аденозиндезаминазы, синдроме голых лимфоцитов). Типы: • Недостаточность аденозиндезаминазы — причина 50% случаев тяжёлого комбинированного иммунодефицита. Проявления: B- и T-клеточный иммунодефицит, CD 4+-лимфопения, тромбоцитопеническая пурпура, гепатоспленомегалия, повторяющиеся бактериальные, вирусные, грибковые инфекции (преимущественно бронхолёгочные), часты различные дисплазии костного скелета. • Агаммаглобулинемия швейцарского типа • Недостаточность пуриннуклеозид фосфорилазы • Дефицит транскобаламина II транспортного белка витамина В 12. Проявления: тяжёлая мегалобластная анемия, агранулоцитоз, тромбоцитопения, геморрагический диатез, тяжёлая диарея, язвенный стоматит, повторные инфекции, агаммаглобулинемия. • Синдром голых лимфоцитов. Термин применяют по отношению к тяжёлому комбинированному иммунодефициту с отсутствием экспрессии ряда генов MHC класса II . Проявления: упорная диарея, синдром мальабсорбции, кандидоз, бактериальные инфекции, интерстициальная пневмония. Лабораторно: пангипогаммаглобулинемия, отсутствуют стимулируемая Аг пролиферация лимфоцитов и клеточноопосредованная цитотоксичность. • Сцепленная с хромосомой X форма. Слабое развитие лимфоидной ткани, множественного генеза инфекции; нормальное содержание Ig, В- и NK-клеток, уменьшено число CD 4+ и CD 8+ T-лимфоцитов, слабый их пролиферативный ответ на Аг и митогены.


Синдром Незелофа n n n Заболевание наследуется по аутосомно-рецессивному типу. У детей с этим синдромом наблюдается недоразвитый эмбриоподобный тимус, который не способен поддерживать Т-клеточную дифференцировку. Заболевание проявляется в раннем детском возрасте. Характеризуется задержкой развития ребенка, его роста и массы тела. У таких детей наблюдаются вирусные, бактериальные и протозойные затяжные инфекции. Часто развивается сепсис с гнойными очагами в коже, легких и других органах, гемолитическая анемия с положительной реакцией Кумбса, аутоиммунная тромбоцитопения. Нередко бактериальный сепсис сочетается с грибковым. Иммунный статус таких больных характеризуется лимфоцитопенией, сниженным содержанием в периферической крови Т-лимфоцитов и низкой их функциональной активностью, резким угнетением кожных реакций. Уровень В-лимфоцитов и Ig – в норме или несколько снижен. Продукция специфических антител чаще подавлена. Фагоцитарная функция нейтрофилов при этой патологии не изменена или даже несколько повышена. Гибель таких детей наступает в первые недели жизни. Изучение структуры тимуса показало отсутствие кортикальных лимфоцитов и телец Гассаля. В лимфатических узлах наблюдается гипоплазия Т-зон.

Синдром Ди Джорджи n n n Описание: Синдром Ди Джорджи (син. : врожденное отсутствие вилочковой железы и паращитовидных желез) впервые описан A. M. Di. George — американским эндокринологом и педиатром. Причины Синдрома Ди Джорджи: Болезнь обусловлена врожденной аплазией (агенезией) тимуса и паращитовидных желез. Отсюда ее основными патогенетическими характеристиками являются нарушение фосфорнокальциевого обмена и иммунологическая Т-клеточная недостаточность. Приблизительно в 80% всех случаев синдром связан с хромосомной делецией, описываемой как del (qll. 2 q 1 l. 2). Предполагается аутосомный тип наследования с различной экспрессивностью. Симптомы Синдрома Ди Джорджи: Характерны внешние признаки: - гипертелоризм, антимонголоидный разрез глазных шелей, микрогнатия; - в периоде новорожденности могут наблюдаться гнпокальциемические судороги, ларингоспазм; - в более старшем возрасте наблюдаются признаки иммунодефицита: генерализованные инфекции, абсцессы, рецидивирующи пневмонии, грибковые инфекции; - характерна клиническая гетерогенность синдрома: кардиоваскулярные дефекты, краниофациалные аномалии, задержка физического и ментального развития. Клиническая диагностика трудна. Должна обращать на себя внимание гипокальциемия в сочетании с клиническими признаками иммунодефицита. Подтверждается диагноз исследованием концентрации паратиреоидного гормона (<10 pg/L) и компьютерной томографией - отсутствием вилочковой железы.

n Гипоплазия тимуса при синдроме Ди Джорджи

ОСЛОЖНЕНИЯ ИММУНОДЕФИЦИТОВ n n n Аутоагрессивные иммунные заболевания. • Сывороточная болезнь при лечении g-глобулином. • Злокачественные новообразования (например, при гипогаммаглобулинемии может развиться тимома). • Тяжёлые инфекции. • Реакция «трансплантат против хозяина» (обычно в результате проведения гемотрансфузии у пациентов с тяжёлым комбинированным иммунодефицитом).

Иммунодефициты.ppt