953b6c6a3a7ccd4499dfc315eaf99307.ppt
- Количество слайдов: 49



Notes on Zing Conference Chao Wang, Bin Ling

Overview • • Key words: Chemical shift, docking, interaction Freq: – – – • Conclusion: 3 Method: 30 App: 9 Discovery: 15 New Insight: 7 Server: 5 Chao’s suggestion: – No pure prediction, contact-assisted prediction – Know about all direct-use tools – Refinement

John Moult, 10 CASP experiments: Successes, Bottlenecks, and Challenges in Protein Structure Prediction • • • GDT_TS remains the same In CASP 10, the number of overline increases Methods that not worked: threading (single-domain), approximate potential function, folding pathways (including discrete way, statistic zip and funnel) Stay the same: SS-prediction, Loop modeling Methods that worked: Multi-templates method. Consensus method, structure clustering. Knowledge based method, for example, fragment assembly and refinement. Next: contact prediction (distance-assisted prediction), conformational sampling, hierarchical potential, complexes (refer to CAPRI communitywide experiment on the comparative evaluation of protein-protein docking for structure prediction)

Nick Griphin, ECOD, Evolutionary Classification of Protein Domains • • • Expand evolutionary core Focus on Inter-domain docking With sequence information from the SCOP ‘‘multi-domain proteins’’ class Methods: – Identifying number and general position of domains. – Refinement of domain boundaries. – Sequence continuity and alternate domain definitions. TMscore: between superfamily and fold in SCOP

Andriy Kryshafovych, New Developments in the Assessment of Contact Predictions in CASP, and How to Quantify Improvements in Model Accuracy Due to Incorporation of Contact Information • The contact-assist protein structure prediction accuracy will be largely improved. • a prediction that satisfied the contact constraint does not mean that the prediction is a good prediction. • The predicted contact can help the structure prediction, too. • But the report didn’t answer how to deal with the low accuracy of contact prediction. • More details in the 4 Proteins papers.

Andrei Korostelev, Modeling the Structures of Large Macromolecular Complexes • • • Main method: simulate annealing Apply multistart simulated annealing crystallographic refinement to a 70 S ribosome-RF 1 translation termination complex that was recently solved at 3. 2 Å resolution. The analysis improves the interpretability of the electron density map of this 2. 5 -MDa ribonucleo-protein complex and provides insights into its structural dynamics.

Samuel Flores, Reliable Evaluation of Mutations in Protein-Protein Interfaces • Here we describe a community-wide assessment of methods to predict the effects of mutations on protein-protein interactions. • The most successful methods considered the effects of mutation on monomer stability in addition to binding affinity, carried out explicit side-chain sampling and backbone relaxation, evaluated packing, electrostatic, and solvation effects, and correctly identified around a third of the beneficial mutations.

Banu Ozkan, Novel Physics-Based Protein Structure Refinement Method Through Local Unfolding and Refolding • Pathway method is worked for single domain case. • Analog to Genetic Algorithm, swap parts of two decoys that share 90% contacts, then unfold and refold, using the zipping software by herself.

Faruck Morcos, Conformational Changes leave an Evolutionary Footprint across Protein Lineages • To determine a complete functional conformational landscape of proteins. • Methods: DCA • reveal a signature of functionally important states in several protein families, using direct coupling analysis, which detects residue pair coevolution of protein sequence composition. • DCA also predicts several intermediates or hidden states that are of functional importance.

Dominik Gront, Bioshell: Modular Platform for Biomolecular Modeling

Marcin Pawlowski, Protein Model Quality Assessment Prediction by using a Residue Specific Statistical Potential • Traditional QA: domain knowledge physical, knowledge-based potential, and consensus • The proposed method relies on an assumption that common substructure motifs among different protein folds can still share similar patterns of interaction with neighboring residues. • The new potential is a modification of DFIRE replace N(i, j, r) with N(i, j, AA, r, d).

Marek Cieplak, Energy Landscape and Dynamics of a Lattice Model of Proteins • • • funnel-like landscape, transition state theory, high temperature to unfold, two state protein P_N+P_D=1 square lattice tube model (Bonavar, Cieplak, Maritan 2004) An Ising-like model for beta-hairpin (2004) Consider as a two-dimension chain and the unfolding process is to change the direction

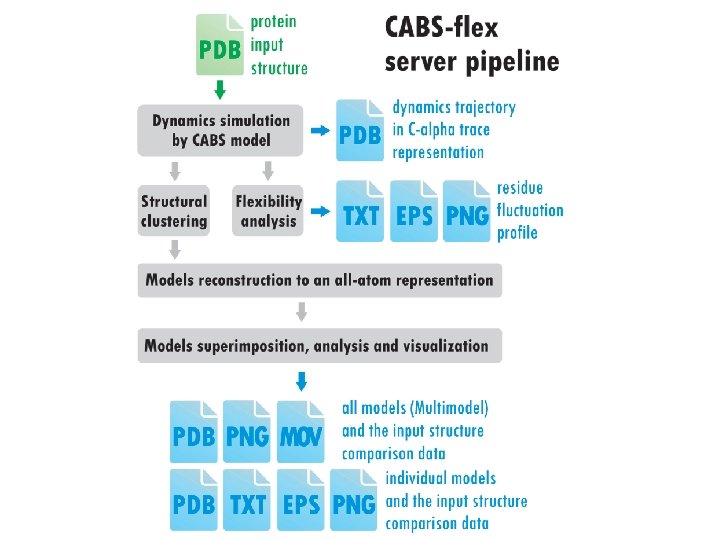
Sebastian Kmiecik, Fast Modeling of Protein Structure Flexibility • a computationally efficient alternative to all-atom molecular dynamics—a classical simulation approach.

Greg Chirikjian, Analysis of Preferred Orientations of Interacting Substructures in Proteins previous efforts to normalize angle distribution data did not include effect: helices can interact with each other in three distinct ways which we refer to as ‘‘line-on-line, ’’ ‘‘endpoint-to-line, ’’ and ‘‘endpoint-to-endpoint, ’’ and each of these interactions has its own geometric effects which must be included in the proper normalization of data For our H-form

Jie Liang, Sampling and Predicting Conformations of Single and Multiple Loops of Proteins • Previous: Sequential Chain Growth with importance sampling • Now: ab initio Distance-guided sequential chain growth • E_i_j_D: logodds • multiple loops: reject-sampling • local environment effect

Chris Sander, Protein structure prediction from sequence variation • Published on NBT • • • Evolutionary couplings contacts in proteins leave an evolutionary record Although evolutionary couplings show promise for the identification of functional sites, homomultimer contacts, alternative conformations and functional sites, many of the predicted contacts involved in these protein features may appear as false positives in the prediction of intradomain residue contacts.

Ron Elber, Predicting the Structure of Protein Switches • • • Our approach was to create 2 proteins that (i) are stably folded into 2 different folds, (ii) have 2 different functions, and (iii) are very similar in sequence. In this simplified sequence space we explore the mutational path from one fold to another. Two fold: 4 beta+alpha, 3 alpha Chao’s comment: this is used to compare the stability of A 1 -A 4 in helice. Just like the 88% sequence identity PNAS paper.

Brinda Vallat, Modeling Proteins using a Super. Secondary Structure Library and NMR Chemical Shift Information • we present a hybrid modeling algorithm that relies on an exhaustive Smotif library and on nuclear magnetic resonance chemical shift patterns without any input of primary sequence information. • In a test of 102 proteins, the algorithm delivered 90 homology-model -quality models, among them 24 high-quality ones, and a topologically correct solution for almost all cases.

Xiaoqin Zou, A Scoring Scheme for Predicting Protein Structures • • • In this study, we have developed a statistical mechanics-based iterative method to extract statistical atomic interaction potentials from known, nonredundant protein structures. Use coarse-grain atom pair distance to describe Iterative to extract effective potential

Drena Dobbs, Analyzing & Predicting RNA-Protein Interactions • • • Prediction improves using sequence info than structure info Hypothesis: conformational changes upon RNA binding confound structurebased Motif-based strategy: generate an RNA-protein motif lookup table RNA sequence: 4 -mer, Protein sequence: 3 -mer, 7 letter reduced alphabet SVM classifier

Shi-Jie Chen, Predicting Structure and Stability for RNA Complexes with Intermolecular Loop-loop Tertiary Contacts • Tetraloop-receptor is a frequently occurring tertiary motif • Obtaining motif by ss prediction • Finding motif-motif contacts

Ioan Andricioaei, Protein and RNA Folding Exhibits Universal Signatures of Granular Jamming Upon folding, proteins develop a peak in the interatomic force distributions that falls on a universal curve with experimentally measured forces on jammed grains and droplets.

Keith Dunker Advantages of Intrinsic Disorder for Protein Function • List functions of 90 loops • Chao’s comment: to build SSS, Smotif for threading

Sandor Vajda What Docking tells us about Protein-protein Association? • Classification of Protein Complexes based on Biophysics of Association “Tell me how you contact your partners, and I'll tell you who you are. ” • Docking will change disorder structure. • List 5 classes of complexes

Joel Sussman, Structure Based Drug Design: Can Crystal Structures Lead to Erroneous Predictions? • Conformational changes upon ligand binding thus involve preexisting equilibrium dynamics. • Consequently, rational drug design could benefit significantly from conformations monitored by MD simulations of native targets.

William Ray, Visualizing Co-Evolution: What does Structure look like, and How can we tell it from Function? • • • The evolutionary signature of structural constraints not the most strongly coevolving residues, as have been searched for, for decades by ever-morepowerful algorithms, but rather, diffuse cluster of weekly-co-evolving residues. Strongly co-evolving residues correlate more strongly with functional requirements for fitness. Mutual information, many alternatives Consider as a peculiar type of graph What’s necessary of protein function, Co-Evolution -> function Bin’s comments: Maybe we should change our focus from single site contact to motif contact. From single site co-relationship to window co-relationship

Jinbo Xu: A New Protein Statistical Potential Empowered by High-Throughput Sequencing • • • Position specific a context-specific potential. The context-specific means a local window profile This article takes a rather different view on the observed probability and parameterizes it by the protein sequence profile context of the atoms and the radius of the gyration, in addition to atom types. Experiments confirm that our position-specific statistical potential outperforms currently the popular ones in several decoy discrimination tests. Imply that, in addition to reference state, the observed probability also makes energy potentials different and evolutionary information greatly boost performance of energy potentials.

Jianzhu Ma: MRFalign: Protein Homology Detection through Alignment of Markov Random Field • Sheng has reported before • Markov Random Field to build alignment • Alignment Potentials: p(a) p(b) logodds, alignment logodds • Bin’s comments: The efficiency is the problem.

Daisuke Kihara, Evaluating Protein Structure Models by Predicted Pairwise Subunits • generate a series of predicted models (decoys) of various accuracies by our multiple protein docking pipeline, Multi-LZer. D, for three multi-chain complexes with 3, 4, and 6 chains. • analyze the decoys in terms of the number of correctly predicted pair conformations in the decoys. • term the fraction of correctly predicted pairs (RMSD at the interface of less than 4. 0Å) as fpair and propose to use it for evaluation of the accuracy of multiple protein docking.

• Residue contacts defined by Cβ−Cβ distance of 7. 0 Å work best overall among tested to identify proteins of the same fold. • effective contact definitions differ from fold to fold, suggesting that using different residue contact definition specific for each template will lead to improvement of the performance of threading.

George Stan, Topology-Dependent Mechanisms of Protein Unfolding and Translocation by AAA+ Nanomachines • conserved loop motif: G-Ar-Phi-G • Coarse grained model: Hydrophobic, Hodrophilic, Natrual loop • Unfolding HBP by force • Chao: Experimental Results

Chen Keasar, Scoring Individual Protein Models by a Purely Structural Function • MESHI energy function • MESHI is a software package for protein modeling. It is written solely in Java in strict object oriented design (OOD). We hope that the use of OOD will encourage other groups interested in protein modeling to use of the code and take part in its development. Please note though, that the MESHI is in a rather preliminary stage of development. Not all the features you may expect are already available (e. g. Molecular Dynamics), and documentation is less than perfect. We will gladly receive any comment and within our limited resources do our best to help users/developers. A legalistic remark: MESHI is intended to be free for use and development. We are still not sure about the exact format (GNU, Open-Source or anything else). • Prof Dong Xu suggests us to use MESHI energy function instead of ROSETTA

Phil Bradley, Predicting Protein-Nucleic Acid Structures and Interactions • • Conformational flexibility is likely important for design and template-based modeling, where non-native conformations need to be sampled and accurately scored. A successful application of such computational modeling techniques in the construction of the TAL-DNA complex structure is discussed.

Falk Hoffmann Protein Structure Prediction using Basin-Hopping Global Optimization • In the current work, we exploit chemical shifts by combining the basin-hopping approach to global optimization with chemical shift restraints using a penalty function. • We further show that our chemical shift restraint BH approach also works for incomplete chemical shift assignments, where the information from only one chemical shift type is considered.

Peter Rogen Which Distance Measure is best for Training and Testing Protein Pair Potentials? • 4 metrics: rmsd, MTP mean Tods Potential, GDT_TS, Q* 1 -the fraction of native contacts

Joanna I. Sulkowska

Ilya Vakser Knowledge-Based Modelling of Protein-Protein Interactions • • Training sets of protein-protein matches were generated based on bound and unbound forms of proteins taken from the DOCKGROUND resource. Each residue was represented by a pseudo-atom in the geometric center of the side chain.

Michael Widom Folding Kinetics of Riboswitch Transcriptional Terminators and Sequesterers • • employ kinetic Monte Carlo simulation to model the time-dependent folding during transcription of riboswitch expression platforms. both that riboswitch transcriptional terminator sequences have been naturally selected for high folding efficiency, and that sequesterers can maintain their function even in the presence of significant misfolding.

Leon Martinez Rd. HMM Score as a Reliable Objective Function for the Prediction of the Three-Dimensional Structure of Proteins • • • The approach described in this work begins by generating a large number of amino acid sequences using ROSETTA [Dantas G et al. (2003) J Mol Biol 332: 449– 460], a program with notable robustness in the assignment of amino acids to a known threedimensional structure. The resulting sequence-sets showed no conservation of amino acids at active sites, or protein-protein interfaces. Hidden Markov models built from the resulting sequence sets were used to search sequence databases. Surprisingly, the models retrieved from the database sequences belonged to proteins with the same or a very similar function. Given an appropriate cutoff, the rate of false positives was zero.

Jianlin Cheng

Andrzej Joachimiak, Structure Determination of Transcriptional Factors and their Complexes with DNA • • • We have determined the crystal structures of Het. R complexed with palindromic DNA targets, 21, 23, and 29 bp at 2. 50 -, 3. 00 -, and 3. 25 -Å resolution, respectively. The highest-resolution structure shows fine details of specific protein–DNA interactions. The lower-resolution structures with longer DNA duplexes have similar interaction patterns and show the flap domains interact with DNA in a sequence nonspecific fashion.

Jarek Meller Ultrafast Clustering of Macromolecular Structures • • • consider a fast alternative, in which structural similarity is assessed using 1 D profiles, e. g. , consisting of relative solvent accessibilities and secondary structures of equivalent amino acid residues in the respective models. the new approach, dubbed 1 D-Jury, allows to implicitly compare and rank N models in O(N) time, as opposed to quadratic complexity of 3 D-Jury and related clusteringbased methods. In addition, 1 D-Jury avoids computationally expensive 3 D superposition of pairs of models.

Dong Xu, MUFOLD • Mini-threading, Use structural fragments of all sizes and broad confidence levels • build distance constraints, Distance matrix>MDScaling • Zscore to assessment: Opus energy, modelevaluator score, rapdf score, dfire, hopp score, geometric score, DOPE • MESHI Casp 9 Ranking 2 nd of FM

• • • developed a database with predicted protein domains for five plant proteomes (http: //pfp. bio. nyu. edu) and used both protein structural fold recognition and de novo Rosetta-based protein structure prediction to predict protein structure for Arabidopsis and rice proteins. Based on sequence similarity, we have identified ; 15, 000 orthologous/paralogous protein family clusters among these species and used codon-based models to predict positive selection in protein evolution within 175 of these sequence clusters. results show that codons that display positive selection appear to be less frequent in helical and strand regions are overrepresented in amino acid residues that are associated with a change in protein secondary structure.

• • • show that the performance of three mirrortree-related methodologies depends on the set of organisms used for building the trees, and it is not always directly related to the number of organisms in a simple way. Certain subsets of organisms seem to be more suitable for the predictions of certain types of interactions. This relationship between type of interaction and optimal set of organism for detecting them makes sense in the light of the phylogenetic distribution of the organisms and the nature of the interactions.

• • We present a knowledge-based function to score protein decoys based on their similarity to native structure. A set of features is constructed to describe the structure and sequence of the entire protein chain. The features we use are associated with residue–residue distances, residue –solvent distances, pairwise knowledge-based potentials and a four-body potential. This new approach enables us to obtain information both from decoys and from native structures.
953b6c6a3a7ccd4499dfc315eaf99307.ppt