Несовершенный остеогенез.ppt
- Количество слайдов: 71



НЕСОВЕРШЕННЫЙ ОСТЕОГЕНЕЗ СОБСТВЕННО КОСТНЫЕ ДИСПЛАЗИИ

Остеогенез продолжается в течение всей жизни Нарушения скелетогенеза создание функционально неполноценной кости, не обеспечивающей опорную функцию избыточное разрастание примитивной и пластинчатой костной ткани
 o")
Нозологические формы собственно костных дисплазий o врожденная ломкость костей (болезнь Лобштейна - Фролика) o врожденный системный гиперостоз (болезнь Камурати – Энгельмана) o мелореостоз o остеопойкилия o мраморная болезнь o миелосклероз o болезнь Педжета
 Наследственное заболевание с извращением и торможением остеобластических")
Врожденная ломкость костей (болезнь Лобштейна - Фролика) Наследственное заболевание с извращением и торможением остеобластических процессов с нарушением костеобразования. Проявляется потерей механической прочности костей и спонтанными множественными переломами Lobstein Johann Freidrich Georg Christian Martin (1834) – немецкий патолог и терапевт Vrolik Willem (1849) – голландский анатом
 – высокая летальность. Поздняя форма (в 7 – 12")
Ранняя форма (до 5 лет) – высокая летальность. Поздняя форма (в 7 – 12 лет). Основные клинические симптомы: § переломы костей § голубые склеры § прогрессирующая тугоухость § «янтарные» зубы аутосомно-доминантное наследование

Ранняя форма несовершенного остеогенеза у девочки 5 лет

Поздняя форма Деформация скелета у мальчика 14 лет

Голубые склеры «Янтарные» зубы

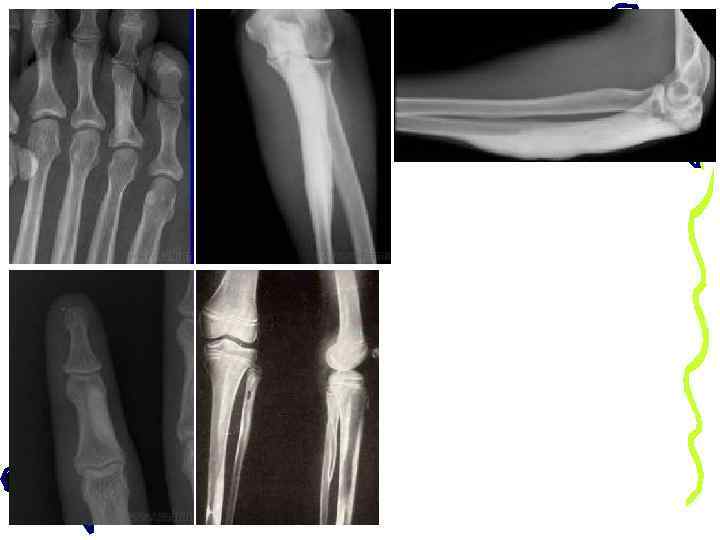
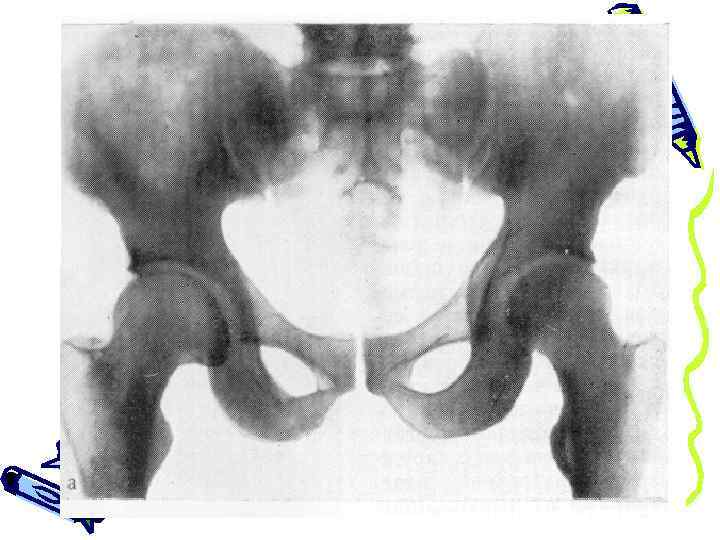
деформация костей таза и бедер, следы переломов бедренных костей деформация позвонков по типу рыбьих

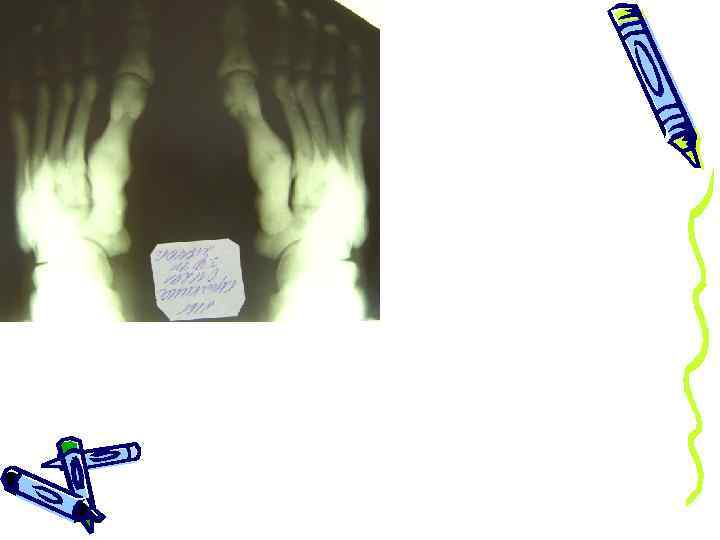
истончение и искривление берцовых и плюсневых костей, сросшийся перелом большеберцовой кости

Ложные суставы обеих бедренных костей, истончение и деформация костей голени

Дифференциальная диагностика q рахит q остеомаляция q ахондроплазия q врожденный сифилис

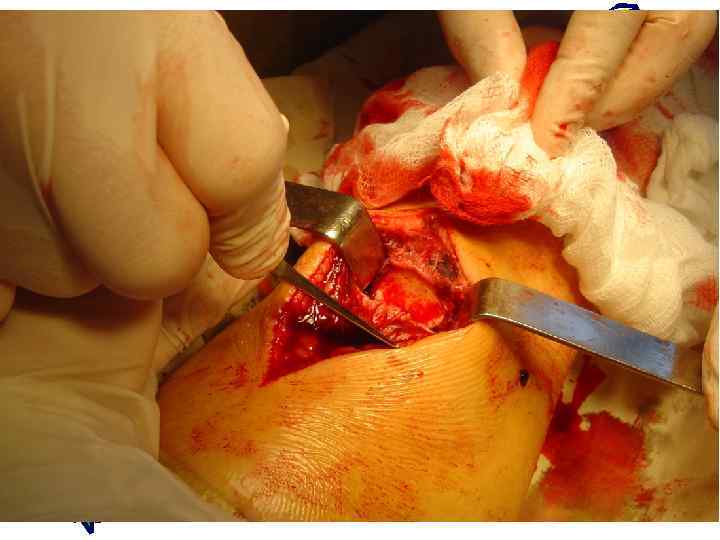
Переломы обычно хорошо срастаются с образованием большой костной мозоли, напоминающей картину злокачественной опухоли «Псевдосаркома» - избыточная костная мозоль при несовершенном остеогенезе

Лечение • рациональный режим • профилактика переломов • предупреждение • • • деформаций укрепление мышечной системы и скелета устранение развившихся деформаций ортезирование
 Наследственное (аутосомнодоминантное) нарушение периостального и эностального костеобразования,")
Врожденный системный гиперостоз (болезнь Камурати – Энгельманна) Наследственное (аутосомнодоминантное) нарушение периостального и эностального костеобразования, в результате чего развиваются периостозы и эностозы. Camurati Mario (1922) – итальянский ортопед Engelmann Guido (1929) – немецкий ортопед

Патологические сдвиги возникают в постнатальном периоде. Клинически проявляются после начала хождения, продолжаются и после окончания роста скелета. Усиление остеобластической функции надкостницы → утолщение коркового слоя за счет напластования на его поверхности новых костных слоев. Замедляется резорбция внутренних слоев кортикальной кости.

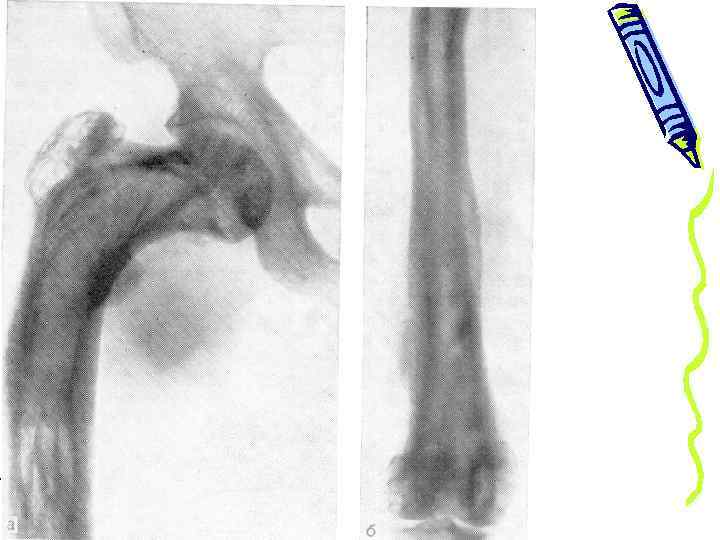
• утолщение конечностей, особенно бедер и голеней • «грызущие» боли в области диафизов костей • скованность движений системный гиперостоз у девушки 19 лет

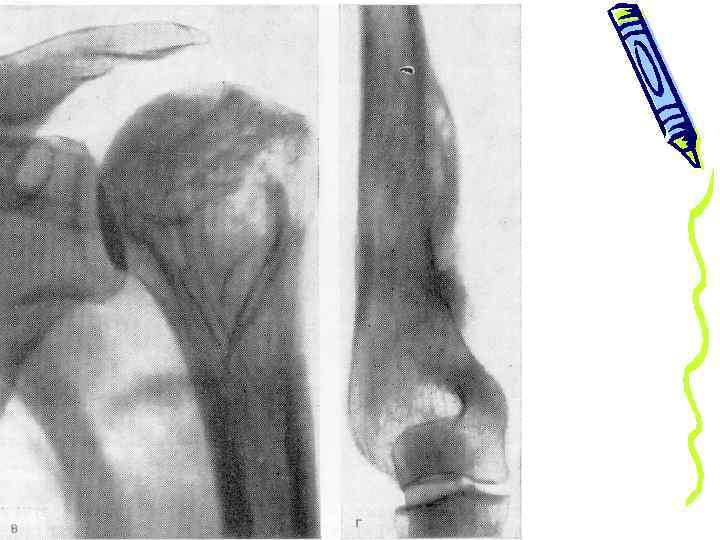
гиперостоз плечевых костей у ребенка 8 лет

a. гиперостоз бедренных и берцовых костей у больного 20 лет b. гиперостоз a b большеберц овых костей у больного 36 лет

o увеличение объема диафизов за счет утолщения коркового слоя с наружной и внутренней сторон. o гомогенная структура коркового слоя o костный канал сужен, но никогда не закрыт o трабекулы резко утолщены дифференциальный диагноз: гемангиома, болезнь Педжета

патогенетической терапии гиперостозов пока не существует!

Мелореостоз Врожденная неполноценность системы мезенхимального эмбриогенеза — извращение пери-, эндо- и интерстинального костеобразования с усилением остеобластического процесса и образованием избыточного количества компактной недифференцированной кости Leri Andre (1922), французский невропатолог

Нарушение остеогенеза возникает на раннем этапе эмбриогенеза, когда конечности выделяются из туловища и делятся на сегменты. Поражаются все сегменты конечностей и все отделы костей. Процесс продолжается в постнатальном периоде и выявляется иногда только у взрослых. Несмотря на системность, это мономелическая гиперостотическая дисплазия с односторонним метамерным поражением сегментов одной конечности.

Клинические проявления незначительны – заболевание часто диагностируется поздно • • • ноющие боли в костях и суставах быстрая утомляемость «тяжесть» в пораженной конечности утолщение кости контрактуры в суставах иногда склеродермия

мелореостоз медиального полуцилиндра бедренной кости периостоз и эностоз в костях стопы

эностоз в губчатых структурах таза и правой бедренной кости мелореостоз в берцовых костях у девочки 12 лет

17 лет. Больна в течении 5 лет. Жалобы на боль в правом тазобедренном суставе, ограничение движений, хромоту. Лечилась 5 лет с диагнозом «правосторонний коксартроз» без эффекта. Объективно: легкая атрофия мышц правого бедра, ограничение ротации и отведения в правом тазобедренном суставе. Мелореостоз крыла правой подвздошной кости Лечение: манжеточное вытяжение, ЛФК, массаж, гипербарическая оксигенация, электромиостимуляция с положительным эффектом
 и больного мелореостозом (справа): на пораженной кисти отмечается")
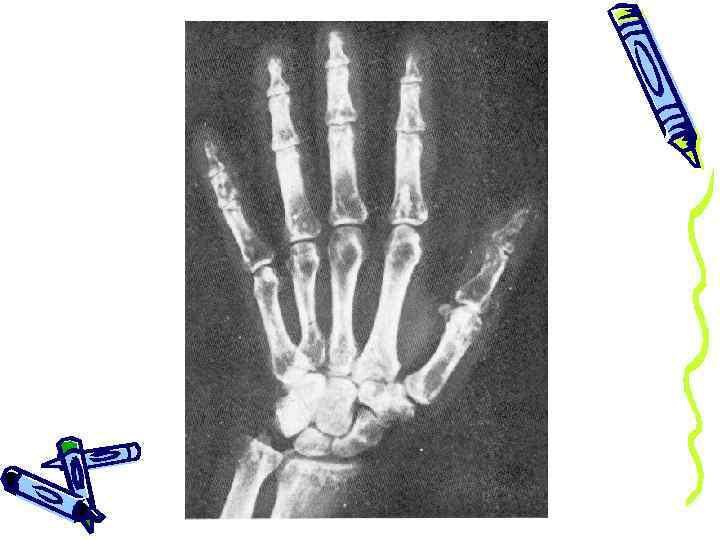
R-граммы кистей рук здорового человека (слева) и больного мелореостозом (справа): на пораженной кисти отмечается уплотнение и утолщение фаланг IV, V и частично III пальцев.

Остеопойкилия • Остеопойкилия (греч. osteon кость + poikilia пестрота, пятнистость; синоним: гиперпластическая остеопойкилия, диссеминированная остеопатия, врожденная пятнистая рассеянная склерозирующая остеопатия) — системное врожденное заболевание скелета, характеризующееся наличием во всех костях, кроме ключиц, округлых или овальных гомогенных склеротических плотных включений. Впервые описано в 1905 г нем. хирургом Штидой (A. Stieda) и подробно изучено нем. хирургом и рентгенологом Альберс. Шенбергом (Н. Е. Albers-Schonberg) в 1915 г. Современные исследователи относят О. к остеохондродисплазиям. Возможны семейные формы заболевания.

Нарушение остеогенеза лишь в эностальном компоненте – извращение дифференцировки губчатого вещества вследствие замедленного и неполного процесса резорбции. Периостальное костеобразование не нарушено. Форма и размеры костей не меняются. Жалоб больные не предъявляют Клинических проявлений нет

Это врожденное системное заболевание опорнодвигательного аппарата выражается в том, что во всех костях в губчатом веществе разбросаны более или менее густо располагающиеся округлые или овальные гомогенные склеротические плотные островки величиной от 2 до 8— 10 мм.

очаговая форма остеопойкилии

очаговая форма остеопойкилии

полосчатая форма остеопойкилии
.")
Мраморная болезнь Системное, строго симметричное, генерализованное поражение скелета — резкое уплотнение костной ткани (остеопетроз). Торможение резорбции и усиление бластической функции приводит к тому, что кость стоится преимущественно из компактного губчатого вещества Albers-Schonberg (1907), немецкий хирург, рентгенолог

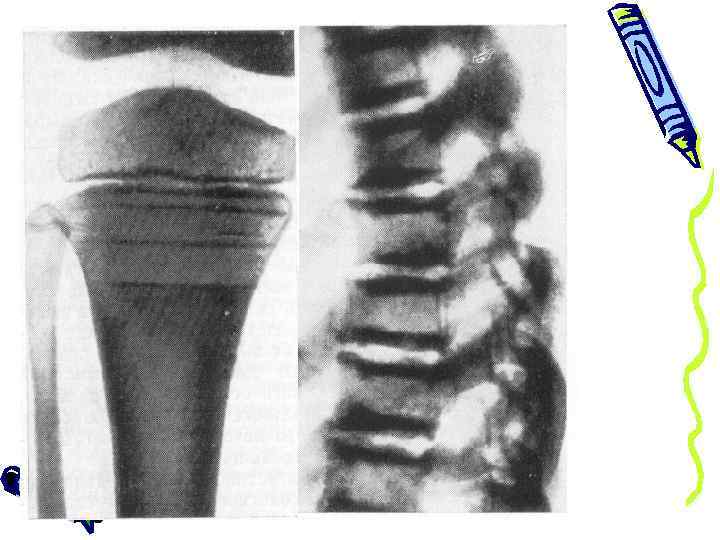
• Кость макроскопически на распиле напоминает мрамор • Кровоснабжение плотной кости резко понижено • Периоды развития остеосклероза меняются ремиссиями → слоистое строение кости

Классическая триада 1. генерализованный остеосклероз – наибольшие изменения в костях туловища и нижних конечностей 2. патологические переломы – обычно без смещения, с ровными краями, замедленной консолидацией 3. нарушение гемопоэза – эритропения, анизоцитоз, пойкилоцитоз, сдвиг лейкоцитарной формулы влево. От этого зависит тяжесть заболевания и прогноз

Перелом надколенника у 29 -летней пациентки (до этого было 6 переломов плюсневых, пястных и лучевых костей)

Заблуждение С. А. Рейнберга «Просверлить эту кость дрелью или распилить ее гораздо легче, нежели кость нормальную»
 Первично системное замещение активного кроветворного костного мозга фиброзной тканью, костные изменения")
Миелосклероз (остеосклеротическая анемия) Первично системное замещение активного кроветворного костного мозга фиброзной тканью, костные изменения развиваются как вторичные. В костях возникает склероз: очаговый или сплошной (картина «мраморности» )

Рентгенографическая картина подобна мраморной болезни, но o поражаются главным образом кости таза и позвоночника o кисти, стопы, череп не изменены o патологических переломов нет o симметричности костных изменений нет

Необходимо дифференцировать § нефропатии различного генеза § анемии § лимфогранулематоз § миеломатоз
 Временное количественное несоответствие между функцией остеокластической резорбции и")
Деформирующая остеопатия деформирующий остит (болезнь Педжета) Временное количественное несоответствие между функцией остеокластической резорбции и остеобластическим костеобразованием, завершающееся созданием неполноценного костного вещества. Paget James (1877), английский хирург

Этиология неизвестна Сегодня наиболее популярная теория - медленная вирусная инфекция костной ткани, вызываемая дефектными парамиксовирусами

§ патологический процесс протекает только в кости § резорбция спонгиозного вещества и каналов остеона пролиферация фиброретикулярной ткани и заполнение ей всех костных пространств препятствие созданию новых костных структур § в кости создаются три последовательно сменяющихся зоны: зона нормальной кости, подвергающейся резорбции; зона фиброретикулярной ткани; зона вновь образующихся костных балок
 1. Начальная (стадия")
Ранний клинический диагноз болезни Педжета невозможен Три стадии заболевания (клиникоренгенологические данные) 1. Начальная (стадия разряжения) 2. Промежуточная (стадия уплотнения 3. Стадия стабилизации или груботрабекулярной перестройки.

• Заболевание лиц преимущественно зрелого и пожилого возраста • у мужчин чаще • частота семейных форм от 1, 1 до 30%.

клиника • первые проявления: боль и деформации. • для нижних конечностей характерна саблевидно-варусная деформация, утолщение метафизарных областей. • свод черепа становится бугристым, его диаметр увеличивается. • местная гипертермия • ограничение движений, артрозы

R-диагностика • рентгенологический диагноз в большинстве случаев поздний • может быть поражена одна кость (монооссальная форма), ее сегмент, несколько костей - полиоссальная форма). • в большинстве случаев процесс начинается в метаэпифизе

прогрессирование процесса на протяжении 20 лет

Типы перестройки: гомогеннопросветвлен ный ячеистотрабекулярн ый гомогенноуплотненный

«банановый» перелом бедренной кости, его сращение, косой перелом

• КТ - обязательна при подозрении на малигнизацию или метастатическое поражение • РНО (радионуклидное обследование) - не специфично, но позволяет оценивать активность патологического процесса

биохимические отклонения • Щелочная фосфотаза: N при монооссальной форме, ↑↑ в 3 - 5 раз при множественном поражении и высокой активности • Гидроксипролин в суточной моче: ↑↑ в 2 - 5 раз

Озлокачествление при болезни Педжета возникает не менее чем в 30% случаев. И. Г. Лагунова, 1989.

Консервативное лечение - использование препаратов угнетающих остеокластическую резорбцию костной ткани: • бифосфонаты: этидронат, клодронат, памидронат • тириокальцитонин (миакальцик, цибакальцин) Оперативное лечение при развитии осложнений : озлокачествление, компрессионный спинальный синдром, вторичные остеоартрозы, деформация костей и др.

Синдром Марфана Какая система органов первично поражается при синдроме Марфана? Глаза, опорно-двигательный аппарат, сосудистая система, но могут наблюдаться заболевания кожи и дыхательной системы. У всех пациентов выявляется дефект в гене фибриллина. Встречаемость — более 6 случаев на 100 000 человек. Пораженные органы — это те, в которых эластические волокна (фибриллин) выполняют важную функцию.

Синдром Марфана Синдромом Марфана, или Марфана. Ашара, обозначают сочетания врождённых дефектов соединительной ткани арахнодактилии, гигантизма, долихостеномегалии, гиперхондроплазии, мезодермальной дисплазии и другие. Впервые этот синдром описан в 1896 г. французским педиатром Антонином Бернардом Марфаном (A. B. Marfan, 1858 - 1942), , и в 1902 г. французским терапевтом Эмилем Шарлем Ашаром (E. Ch. Achard, 1860 1944).
, когда нарушается синтез коллагена и")
Синдром Марфана Наследственная болезнь соединительной ткани (аутосомно-доминантный тип наследования), когда нарушается синтез коллагена и эластина из -за повреждения гена 15 пары хромосом, который отвечает за продукцию фибриллина белка, являющегося важным компонентом соединительной ткани, формирующим её эластичность и сократимость.

Синдром Марфана Фибриллина много в стенке аорты, связочном аппарате различных органов. При синдроме чаще всего поражается восходящая часть аорты. Нередко эти изменения являются причиной внезапной смерти взрослых от разрыва аорты, когда они даже не подозревали о своей болезни. Наиболее яркий пример - случай со звездой американского волейбола, членом олимпийской сборной Flo Hyman, который умер от этого осложнения в 1986 г.

Синдром Марфана Характерным является внешний вид больных: длинные и тонкие конечности с такими же пальцами, «птичье лицо» (большой нос и маловыраженный подбородок), кифосколиоз, переразгибание в суставах, чрезмерная растяжимость кожи. Могут отмечаться различные нарушения зрения (подвывих хрусталика, миопия и сходящееся косоглазие). Нарушения сердечнососудистой системы включают поражение клапанного аппарата сердца и аневризму аорты.

Синдром Марфана Высокий выброс адреналина, характерный для заболевания, способствует не только развитию сердечно-сосудистых осложнений, но и появлению у некоторых лиц особой силы духа и умственной одаренности. Способы лечения неизвестны. А. Линкольн Г. Х. Андерсен Н. Паганини К. И. Чуковский

Несовершенный остеогенез.ppt