Нефротический синдром при наследственных нефропатиях.pptx
- Количество слайдов: 38



Нефротический при наследственных нефропатиях

Актуальность К проблеме генетическидетерминированного НС в настоящее время привлечено большое внимание. В какой-то степени это связано с учащением случаев гормонорезистентности у больных, наблюдаемых в нефрологическом стационаре. Причиной этих заболеваний могут являться мутации тех или иных генов в результате чего нарушается структура белка, расположенного на подоцитах, и развивается гормонорезистентный НС.
 2.")
НЕФРОТИЧЕСКИЙ СИНДРОМ — ЭТО КЛИНИКО-ЛАБОРАТОРНЫЙ СИМПТОМОКОМПЛЕКС, ДЛЯ КОТОРОГО ХАРАКТЕРНЫ: 1. ГИПОАЛЬБУМИНЕМИЯ(МЕНЕЕ 30 Г/Л) 2. ВЫСОКАЯ ПРОТЕИНУРИЯ (БОЛЕЕ 3, 5 Г/CУТ) 3. ГИПЕРЛИПИДЕМИЯ 4. ОТЕКИ 5. ЛИПИДУРИЯ 6. ПОВЫШЕНИЕ СВЕРТЫВАЕМОСТИ КРОВИ Симптом Частота встречаемости в % Периферическте отеки 89 Висцеральные отеки 83 Полостные отеки 47 Гиперкоагуляция 61 Дислипидемия 91 Распространенность факультативных симптомов при нефротическом синдроме
или вторичным(проявление общей системной болезни). первичные причины")
Причины НС НС может быть первичным(проявление заболевания почек)или вторичным(проявление общей системной болезни). первичные причины нефротического синдрома: • липоидный нефроз (нефропатия с минимальными изменениями). • мембранозный гломерулонефрит (идиопатический нефротический синдром взрослых). • наследственная нефропатия; • первичный амилоидоз почек. вторичные причины, способствующие развитию нефротического синдрома: • инфекционные болезни (постстрептококковый гломерулонефрит, эндокардит, вторичный сифилис, лепра, гепатит В, мононуклеоз, малярия, шистосомоз; • лекарственные средства: органические и неорганические соединения ртути, пеницилламин, героин, каптоприл, рентгеноконтрастные средства; • опухоли: лимфогранулематоз, лимфома, лейкоз, карцинома, меланома. Опухоль Вильмса; • полисистемные заболевания: системная красная волчанка, болезнь Шенлейна— Геноха, васкулит, синдром Гудпасчера, , саркоидоз, ревматоидный артрит; • семейно-наследственные заболевания: сахарный диабет, синдром Альпорта, серповидно-клеточная болезнь, болезнь Фабри, липодистрофия, врожденный нефротический синдром; • смешанные: преэклампсия, микседема, вазоренальная гипертензия и т. д.

ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА НС 1 этап 2 этап Нефропатия беременных Диабетическая нефропатия Хронические инфекционные заболевания и т д Медикаментозное поражение почек Паранеопластическая нефропатия Тромбоз почечных вен, нижней полой вены 3 этап Поражение при диффузных заболеваниях СТ Амилоидоз почек Злокачественный гломерулонефрит 4 этап Наследственные нефропатии Амилоидоз Злокачественный гломерулонефрит

ГЕНЕТИЧЕСКИ-ОБУСЛОВЛЕННЫЕ ПРИЧИНЫ НЕФРОТИЧЕСКОГО СИНДРОМА Нефропатии наследственные - заболевания почек, связанные с мутацией одного или нескольких генов. Наследственные нефропатии можно условно разделить на 3 группы: 1. Моногенно наследуемые(мутация одного гена) Влияние среды сказывается только в степени выраженности симптомов заболевания. • наследственный нефрит • синдром Альпорта • различные варианты почечных тубулопатий (синдром Фанкони, фосфат-диабет, первичная оксалурия) • наследственные аномалии органов мочевой системы • поликистозная болезнь. 2. Наследственные заболевания почек мультифакториальные(полигенные) Развиваются они при неправильном питании, ограничении жидкости, повторных инфекционных заболеваниях, токсических, аллергических воздействиях. • дисметаболические нефропатии, связанные с патологией щавелевой кислоты, пуринов, триптофанового обмена, и др. 3. Заболевания, где этиологическими факторами являются внешние воздействия (микробов, вирусов, токсинов), но реализация их воздействия происходит у индивидуумов с генетической предрасположенностью. • Гломерулонефрит в семье, где имеются иммунодефицитные состояния, может быть расценен как патология, развивающаяся при наличии предрасположенности.

НОЗОЛОГИЧЕСКАЯ КЛАССИФИКАЦИЯ НЕФРОПАТИЙ НАСЛЕДСТВЕННЫХ І. При анатомических аномалиях строения почек и органов мочевыделения: Пороки развития почек (агенезия, аплазия, дополнительная почка, дистопия, нефроптоз, Sобразная, подковообразная почки и др. ). • Пороки развития мочеточников (изменения количества, калибра, др. ). • Аномалии мочевого пузыря, уретры. • Аномалии почечных сосудов. II. Нарушение дифференциации почечной ткани (гистологические аномалии): • С кистами (поликистоз почек и др. ). • Без кист (сегментарная или гипопластическая дисплазия, др. ). • III. Наследственный нефрит: • • Без глухоты. с глухотой (синдром Альпорта). IV. Тубулопатии: Первичные: • с полиурией (почечный несахарный диабет и др. ); • с деформацией костей (фосфат-диабет, болезнь де Тони-Дебре-Фанкони); • с поражения почек и костей (почечный тубулярной ацидоз); • с нефролитиазом (цистинурия, оксалурия, др. ). Вторичные - наследственная патология обмена веществ (Галактоземия, цистиноз, др. ). V. Дизметаболические нефропатии (оксалатно-кальциевая кристаллурия. VI. Амилоидоз почек (наследственный, при периодической болезни). VII. Эмбриональные опухоли почек (опухоль Вильмса).

ЧТО ХАРАКТЕРНО ДЛЯ НАСЛЕДСТВЕННЫХ НЕФРОПАТИЙ? Для наследственных нефропатий, несмотря на их нозологический полиморфизм, характерен ряд общих клинических признаков. 1. наличие однотипных заболеваний в семье, что отчетливо проявляется при сопоставлении родословных, 2. длительное латентное течение, нередко с "изолированным мочевым синдромом", что чаще расценивается как одна из форм латентного гломерулонефрита, если имеется гематурия и протеинурия, или латентного пиелонефрита при наличии лейкоцитурии. 3. Больным свойственны множественные внешние и соматические стигмы дизэмбриогенеза (малые аномалии) 4. Наконец, отмечается раннее снижение почечных функций, как правило, по тубулярному типу. Для некоторых вариантов патологии (дисплазия почек, тубулопатии, почечный аммлоидоз) характерно формирование почечной недостаточности уже в детском возрасте.

Моногенные нефропатии, гены которых идентифицированы Гломерулярные Тубулярные Кистозные Периодическая болезнь Почечный несахарный диабет АДПБП 1 ого типа Синдром альпорта Цистиноз АДПБП 2 ого типа НС финского типа Первичная гипероксалурия 1 ого типа Нефрофтиз 1 ого типа Синдром Фрайзера
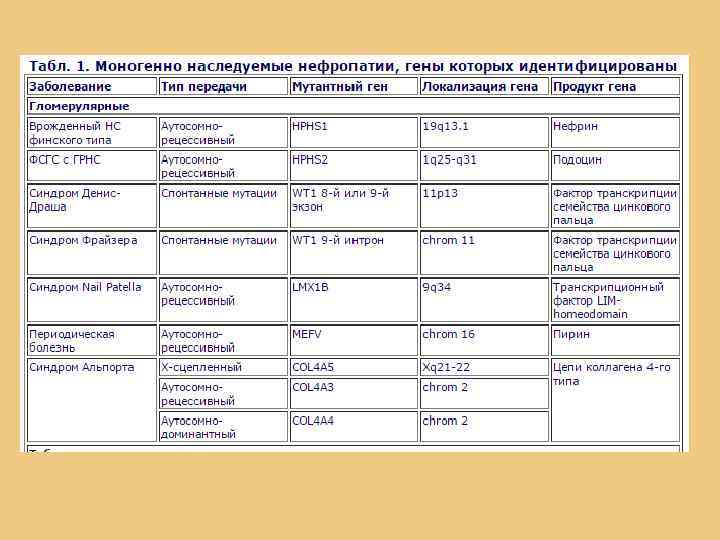
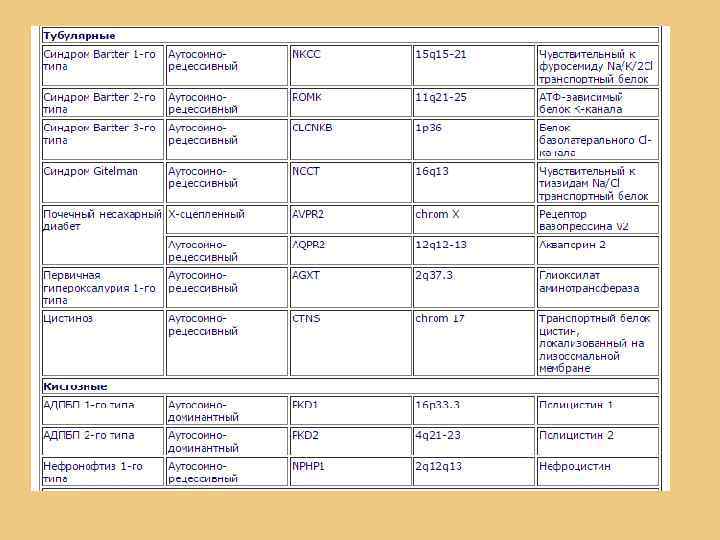

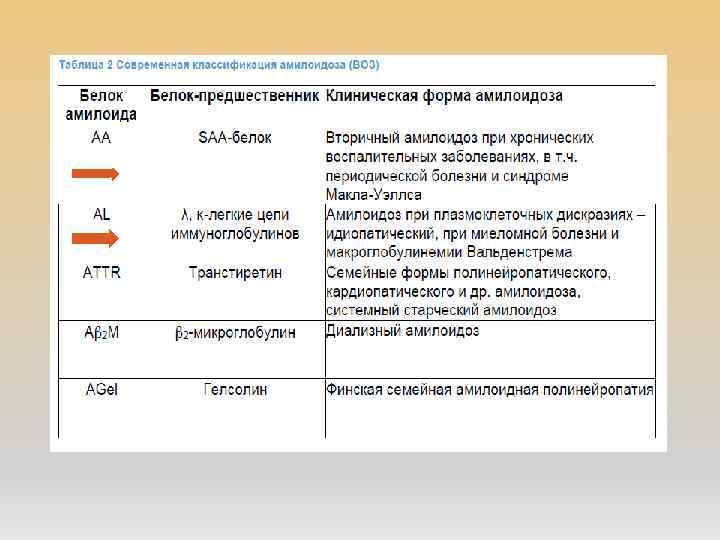
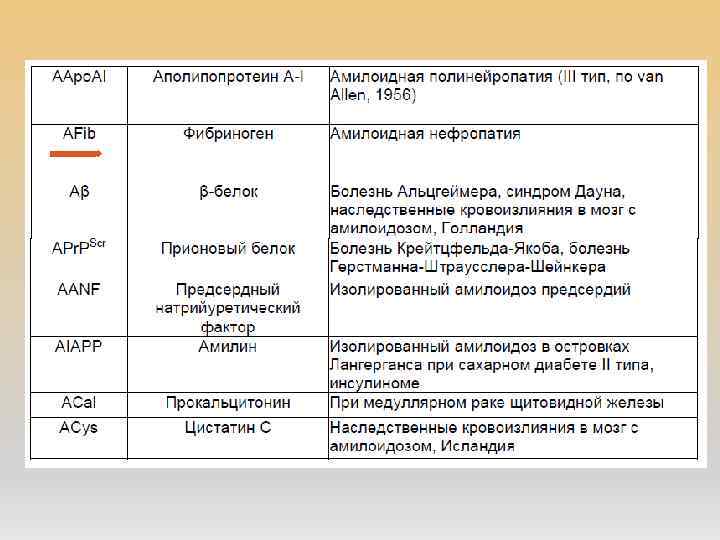
АМИЛОИДОЗ Амилоидоз – группа заболеваний, отличительным признаком которых является отложение в тканях и органах фибриллярного гликопротеида амилоида. Наиболее типичным проявлением амилоидоза почек является протеинурия более 0, 5 г/сут, чаще нефротического уровня. Критериями прогрессирования амилоидоза почек являются увеличение протеинурии(на 50% от исходного уровня, как правило на 1 г/сут и более), уровня креатинина (на 25% и более от исходного). Современная морфологическая диагностика амилоидоза предусматривает не только обнаружение, но и обязательное типирование амилоида, поскольку тип амилоида определяет терапевтическую тактику

Для амилоидоза почек характерно… 1. 2. 3. 4. 5. Выраженная протеинурия при скудном мочевом осадке Изменения мочевого осадка проявляются в микрогематурии, асептической лейкоцитурии без пиелонефрита (в основном за счет лимфоцитов, а не нейтрофилов), цилиндрурии (гиалиновой, реже зернистой), липидурии, наличием двоякопреломляющих кристаллов и капель в осадке мочи. При развитии ХПН не происходит уменьшения размеров почек. За счет отложения масс амилоида не развивается сморщивание почек. НС часто сохраняется и при развитии ХПН (даже в терминальной стадии – при критическом уровне клубочковой фильтрации <5 мл/мин). Выраженность ХПН в большей степени коррелирует с тубулоинтерстициальными нарушениями (интерстициальный фиброз), чем с количеством отложений амилоида в клубочках Идиопатический первичный амилоидоз-диагноз исключения!!!!!
 амилоидоз • Семейный TTR-амилоидоз возникает вследствие точковой мутации в гене, кодирующем синтез")
Транстиретиновый (TTR) амилоидоз • Семейный TTR-амилоидоз возникает вследствие точковой мутации в гене, кодирующем синтез транстиретина. Известно около 50 таких мутаций и, соответственно, столько же видов семейного амилоидоза, к которым относятся японский, шведский, португальский амилоидоз и т. д. Транстиретиновый семейный амилоидоз наследуется по аутосомно-доминантному типу. • Поражение почек с развитием амилоидной нефропатии встречается редко. Доминирование той или иной симптоматики зависит от типа генной мутации и, соответственно, от вида семейного транстиретинового амилоидоза. Чем ближе мутация локализована к N-концу белковой молекулы транстиретина, тем более выражены явления полинейропатии (30 Met); чем ближе мутация располагается к С-концу, тем более проявляется клиническая картина кардиопатии (121 Met). • http: //www. evrika. ru/show/129
 АА-амилоидоз AL-амилоидоз прогноз более благоприятен")
ПРОГНОЗ И ПРОДОЛЖИТЕЛЬНОСТЬ ЖИЗНИ (По данным клиники Мейо, 2014) АА-амилоидоз AL-амилоидоз прогноз более благоприятен серьезный средняя продолжительность жизни, месяцы 30 -60 13, 2 5 -летняя выживаемость 12% 7% 10 -летняя выживаемость 3% 1% Причины смерти почечная недостаточность сердечная недостаточность и нарушения ритма сердца (48%), уремия (15%), сепсис и инфекции (8%). Развитие ХПН В 86% случаев более чем у 60% умерших Источник: Нефрология: нац. рук. Авторы: Под ред. Н. А. Мухина 2009 г

Лечение Целью терапии любого типа амилоидоза служит уменьшение количества белковпредшественников. Морфологическим критерием эффективности лечения считают уменьшение отложений амилоида в тканях. Кроме основных антиамилоидных терапевтических режимов, лечение должно включать симптоматические методы, направленные на уменьшение выраженности застойной недостаточности кровообращения, аритмии, отёчного синдрома, и др. Лечение АА-амилоидоза: Причина Тактика ревматоидный артрит пожизненное лечение различными схемами базисной терапии(ФНО-α, IL 1, IL-6, анти-СD 20 -агентам, ГКС. ) периодическая болезнь колхицин 2 мг/сут под контролем клинического анализа крови и уровня креатинина хронические нагноения хирургическое иссечение очагов, даже при отсутствии признаков активного воспаления в них.

Причина Тактика Идиопатическая плазмоклеточная дискразия подавление пролиферации клона плазматических клеток для уменьшения продукции легких цепей иммуноглобулинов: мелфалан+преднизолон Миеломная болезнь химиотерапия высоких доз с поддержкой аутологичными стволовыми клетками Макроглобулинемия Вальденстрема трехкомпонентная схема терапии: бортезомиб 1, 3 мг/м 2 1, 5, 8 и 11 -й дни, мелфалан 0, 15 мг/кг с 1 по 4 дни, дексаметазон 20 мг/сут 1, 5, 8 и 11 -й дни внутрь. всего 8 курсов Симптоматическая терапия амилоидоза противопоказано назначение сердечных гликозидов, недигидропиридиновых антагонистов кальциевых каналов, бетаблокаторов и ингибиторов АПФ. Ограничение в назначении доз салуретиков Идиопатический первичный амилоидоз? ? Лечение AL-амилоидоза

 – наследственное моногенное")
Периодическая болезнь Семейная средиземноморская лихорадка/периодическая болезнь (Familial Mediterranean Fever – FMF) – наследственное моногенное заболевание с аутосомно-рецессивным механизмом передачи, распространенное среди представителей определенных этносов (населяющих страны средиземноморского бассейна )и проявляющееся периодически возникающими немотивированными приступами лихорадки, сопровождающимися сильными ( «хирургическими» ) болями в животе и/или грудной клетке, с продолжительностью приступов от 12 до 72 ч. Частым осложнением FMF является развитие АА-амилоидоза. Причиной является мутация гена MEFV (Mediterranean Fe. Ver), в 16 -й паре хромосом.

В процессе отложения амилоида в почках при периодической болезни можно проследить стадийность. Выделяют 4 стадии амилоидоза почек: 1) латентную (диспротеинемическую), 2) протеинурическую, 3) нефротическую 4) уремическую (азотемическую). В отечную (нефротическую) стадию пораженными оказываются более чем 75% гломерул. Прогрессирует склероз интерстиция и сосудов, в пирамидах и интрамедиарной зоне склероз и амилоидоз имеют выраженный диффузный характер. Клинически эта стадия амилоидоза представлена полным НС, но может наблюдаться неполный НС. Протеинурия неселективная. Клиническими особенностями ХПН при амилоидозе, отличающими ее от ХПН вследствие других заболеваний, является сохранение НС с массивной протеинурией, часто определяются большие размеры почек, характерно развитие гипотензии.

Как поставить диагноз FMF? • • • диагноз FMF является в первую очередь клиническим!!! Выявление гомозиготного носительства мутаций М 680 I, М 694 V, V 726 А делает диагноз периодической болезни 100%-ным. Для постановки диагноза FMF необходимо соответствие ≥ 1 большим критериям, или ≥ 2 малым критериям, или 1 большому + ≥ 5 поддерживающим критериям, или 1 малому + ≥ 4 поддерживающим критериям из числа первых 5.

Лечение и прогноз FMF Основой лечения -колхицин. Максимальной суточной доза- 2 мг/сут. Основное осложнение терапии колхицином – диарея, тошнота и рвота. Могут нарушаться функции костного мозга, печени. В связи с этим в процессе лечения колхицином важно вести мониторинг показателей клинического анализа крови, а также АЛТ, АСТ. В качестве симптоматического средства во время атак используют НПВС. С развитием средств биологической терапии в лечении FMF стали использовать препараты, блокирующие функции ИЛ 1 (анакинра) и ФНОα (инфликсимаб). Следует подчеркнуть, что все вновь предлагаемые средства не заменяют, а лишь дополняют терапию колхицином и применяются в случае непереносимости или неэффективности данного препарата. Прогноз: При своевременной постановке диагноза и назначении колхицина прогноз ПБ благоприятен. При отсутствии терапии наибольшую опасность представляет развитие почечного амилоидоза, который является единственной причиной смерти больных с ПБ. При «естественном течении» периодической болезни приблизительно у 50% больных терминальная стадия почечной недостаточности развивается через 5 лет от момента появления протеинурии, у 75% — в течение 10 лет.

Миссенс-мутации

ПОЛИКИСТОЗ ПОЧЕК Критерий сравнения АД-ПКБ АР-ПКБ мутация Полицистин 1 и 2 в 16 и 4 хромосоме Фиброцистин 1 Локализация кист В корковом и мозговом веществе Мозговой слой Размер почек Значительно увеличен Нет ярких кистозных проявлений Темп развития ХБП Умеренный, близкий к медленному Быстрый Почечные проявления Боль в проекции почек, Макрогематурия(55%), протеинурия<1 г/сут, нефролитиаз(25%) Часто только на УЗИ: увеличена эхогенность медуллярного слоя с множеством кист небольших размеров Внепочечные проявления АГ в 65 %случаев Кисты других локализаций: панкреас, внутри черепные артерии, яичники Пролапс МК АГ в 35 %случаев Выраженная гипонатрийемия Гипоплазия легких Перипортальный фиброз и ПГ

ДИАГНОСТИКА • • Наличие в анамнезе семейных случаев болезниоснование для выполнения УЗИ. Сонографическими критериями болезни при наличии у родителей следует считать: >2 кист в одной или обеих почках в возрасте до 30 лет >=2 кист в каждой почке в возррасте 30 -59 лет >=4 кист в каждой почке в возрасте старше 60 лет Генетическая диагностика аутосомнодоминантного типа ПКБ основана на определении мутаций генов PKD 1(хромосома 16 р13. 3) PKD 2(хромосома 4 q 21), аутосомнорецессивного-PKHD 1(хромосома 6 р21). Генетическую диагностику следует считать целесообразной в случае отрицательных или сомнительных данных визуальных исследований. При диагностике у больных АБПБП следует провести скрининг на наличие конкрементов в мочевыделительной системе и гематурии.

Лечение Методы этиологического и патогенетического лечения поликистоза почек не разработаны: основой терапии является симптоматическая терапия, направленная на: • Замедление прогрессирования дисфункции почек • Контроль гипертензии • Снижение протеинурии • Профилактика и лечение сердечно-сосудистых заболеваний(и. АПФ+БРА) • Лечение болевого синдрома, обструкции, гематурии, сопуствующей инфекции мочевыводящих путей. • Декомпрессию кист почек, лапароскопическую хирургию, криоаблацию следует рассматривать как лечебную опцию при неэффективности анальгетиков про болевом синдроме. Так же показанием для хирургического лечения является малигнизация кист. • На поздних стадиях ХПН-трансплантация почки. Иногда нужно решать вопрос о предшествующей нефрэктомии. • В перспективе: применение антагонистов v 2 -рецепторов вазопрессина, соматостатин и ингибиторы m. TOR Приоритет медико-генетического консультирования!

МЕТОДЫ МЕДИКО-ГЕНЕТИЧЕСКОГО КОНСУЛЬТИРОВАНИЯ Медико-генетическое консультирование – это специализированный вид медицинской помощи. Его смысл – профессиональная оценка риска рождения в конкретной семье ребенка с наследственной болезнью. Поводом для медико-генетического консультирования могут быть: • Рождение ребенка с врожденными пороками развития, умственной и физической отсталостью, слепотой и глухотой, судорогами и др. • Спонтанные аборты, выкидыши, мертворождения. • Близкородственные браки. • Неблагополучное течение беременности. • Работа супругов на вредном предприятии. • Несовместимость супружеских пар по резус-фактору крови. • Возраст женщины старше 35 лет, а мужчины - 40 лет. • Осложненное течение беременности (угроза прерывания с ранних сроков, которая не поддается терапии, многоводие, маловодие). • Патология плода, выявленная при УЗИ.

Медико-генетическая консультация включает 3 этапа: 1 этап - диагноз. уточняется благодаря использованию современных генетических, биохимических, иммуногенетических и других методов. Составление родословных 2 этап — прогноз. Днкметоды(кариотипирование), цитогенетика 3 этап. Заключение и совет. Генетический риск до 5% считается низким и не является противопоказанием к повторному рождению ребенка в семье. Риск от 6 до 20% принято считать средним, и в этом случае для дальнейшего планирования семьи рекомендуется всестороннее обследование. Генетический риск свыше 20% принято относить к высокому риску. Дальнейшее деторождение в данной семье не рекомендуется. В согласовании с приказом Минздрава РФ № 316 от 30. 12. 93 г. в РФ работает 85 медикогенетических консультаций.

May 2015

http: //med-gen. ru/

http: //www. vigg. ru/genetika/ Европейское общество генетики человека - http: //www. eshg. org Американское общество генетики человека - http: //www. ashg. org Международная федерация обществ генетики человека http: //www. ifhgs. org

Медико-генетические консультации России В Уральском центре радиационной медицины осуществляет медицинское наблюдение людей, подвергшихся радиационному воздействию, их потомков, а также проводят лечение больных лучевой болезнью и гематологических больных.

ЛЕЧЕНИЕ НЕФРОПАТИЙ Первое, что должно быть сделано, это подтверждение предположения о генетической детерминированности заболевания. Путь к коррекции может быть двух типов. 1 — кардинальное воздействие на уровне ДНК клетки: генная инженерия 2— паллиативный, на других уровнях генетической информации. Паллиативные методы терапии врожденных и наследственных заболеваний включают хирургическую коррекцию. Примером является восстановление уродинамики при врожденных анатомических аномалиях строения органов мочевой системы. Целесообразно применение препаратов, стимулирующих, индуцирующих синтез ферментов. Следует помнить, что ГК для этой цели непригодны, особенно при наследственном нефрите. Чтобы стимулировать синтез ферментов тубулярного эпителия канальцев, назначают витамин В 6 до 150 мг/сут, АТФ, кокарбоксилазу. Источник: Клиническая нефрология под ред. Е. М. Тареева

Заместительная терапия При врожденных и наследственных нефропатиях пересадка почки оказывается высокоэффективной. В ЧОКБ есть центр по пересадке почек. Начал работать в 2009 году. За 5 лет успешно проведено 59 трансплантаций. Такой темп пересадок связан с зачаточным состоянием трансплантологии в России. (1 трансплантолог на 1, 5 млн. населения вместо должных 12 врачей на 1 млн. человек). Руководитель центра-Барышников А. А. В центре осуществляются различные формы пересадок, ведется сотрудничество с нефрологами, реаниматологами, терапевтами, и другими специальностями.

В заключении • Геном человека по-прежнему остаётся сложным и до конца не описанным объектом исследования. • 2015 стало доступным получить собственный геном в России (http: //www. genotek. ru/uslugi/analiz_genoma/sekvenirovanie_vse go_genoma). • Подобное развитие технологий позволяет говорить о наступлении эры персонализированной медицины. • Наиболее быстрого внедрения новых технологий можно ожидать в самой изучаемой и финансируемой области – лечении ЗНО • уже сейчас геномное секвенирование позволяет получить персонализированную медицинскую помощь • В. н. с. лаб. мутагенеза, к. м. н. , А. В. Лавров

Благодарю за внимание!
Нефротический синдром при наследственных нефропатиях.pptx