гормоны.ppt
- Количество слайдов: 28



Недостаточная и усиленная выработка мужских половых гормонов

Недостаточная выработка или Гипогонадизм – синдром, сопровождающийся недостаточностью функций половых желез и нарушением синтеза половых гормонов. Гипогонадизм, как правило, сопровождается недоразвитием наружных или внутренних половых органов, вторичных половых признаков, расстройством жирового и белкового обмена (ожирением или кахексией, изменениями костной системы, сердечно-сосудистыми нарушениями). Различают мужской и женский гипогонадизм

Классификация гипогонадизма у мужчин Первичный и вторичный гипогонадизм может быть врожденным и приобретенным Первичный врождённый Одной из причин первичной недостаточности тестикул являются хромосомные нарушения, в результате которых тестикулярная ткань отсутствует или остается недоразвитой, не способной к секреции половых гормонов вообще, или в результате нарушения биосинтеза тестостерона в ней остается не способной к секреции половых гормонов в количестве, достаточном для развития андрогенозависимых органов и тканей и вторичных половых признаков. К гипогонадному состоянию яичек приводит патология их опущения. Для возникновения хромосомной и другой врожденной патологии пагубную роль играют инфекционные, токсические, лекарственные, химические, лучевые и другие вредные воздействия на организм матери.

Первичный приобретённый Причиной первичного приобретенного гипогонадизма нередко бывают острые и хронические инфекционные заболевания, перенесенные в допубертатном периоде. В частности, эпидемический паротит, осложненный орхитом в будущем в 60% случаев обусловливает нарушение спермограммы. Травматическое повреждение яичек, ранняя кастрация, варикоцеле, послеоперационная атрофия и гипоплазия яичек (после герниотомии, орхипексии, операций на органах мошонки) ведут к развитию симптомов евнухоидизма и бесплодия. Патогенез первичного гипогонадизма тесно связан со снижением уровня андрогенов в крови, компенсаторной реакцией надпочечниковых андростероидов, активацией секреции гонадотропных гормонов. Симптомы могут возникать при простатопатиях и других висцеропатиях (цирроз печени, гепатит и др. ). Яички повреждаются вторично. Однако патогенетические механизмы при этом развиваются по типу первичного (гипергонадотропного) дискорреляционного гипогонадизма.
 недостаточности")
Евнухоидизм заболевание, возникающее на почве врождённой или приобретённой (травмы, гонорейное или сифилитическое поражение) недостаточности половых желёз вследствие их непосредственного поражения или понижения продукции гипофизом гонадотропных гормонов. Е. чаще встречается у мужчин. Различают две формы Е. : евнухоидный гигантизм и евнухоидное ожирение; обычно наблюдается смешанный тип. Основные признаки Е. : нарушения роста — гигантизм и диспропорции скелета (чрезмерно длинные конечности, особенно нижние); тонкие кости (нередки вывихи и деформации в суставах — искривления, косолапость, плоскостопие), узкие плечи, широкие размеры таза (у женщин — узкий таз); дряблая мускулатура, значительное отложение жира в области груди, живота и бёдер; недоразвитие половых органов, слабо выраженные вторичные половые признаки, отсутствие растительности на лице, лобке и в подмышечных впадинах, высокий тембр голоса; половое чувство резко снижено. Евнухоиды большей частью медлительны, несамостоятельны, мало активны; интеллект обычно нормален. Лечение — длительная гормонотерапия
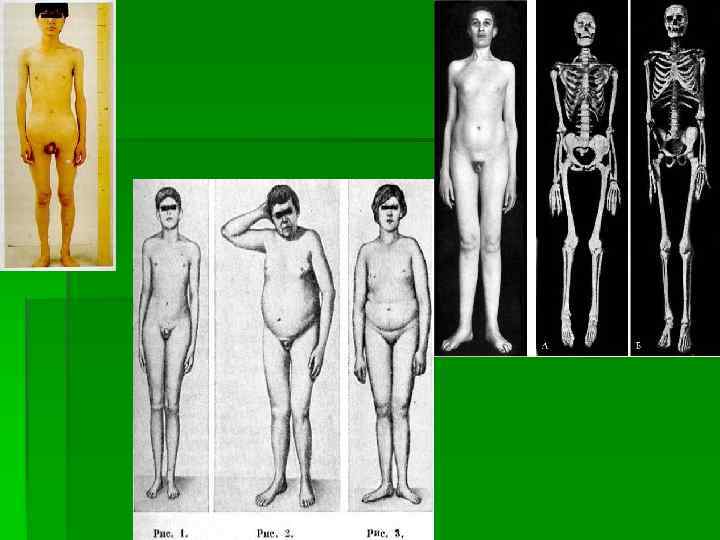
 — это результат первичной недостаточности гонадотропной функции аденогипофиза, которая может сочетаться")
Вторичный гипогонадизм (гипогонадотропный) — это результат первичной недостаточности гонадотропной функции аденогипофиза, которая может сочетаться с дефицитом выработки других тройных гормонов. Гонадотропная недостаточность, ведущая к гипоандрогении, связана либо с воспалительными, опухолевыми, сосудистыми нарушениями в гипофизе или в гипоталамусе, либо с нарушениями эмбрионального развития гипофиза. Клиническая симптоматика зависит от степени выраженности гипопитуитаризма и андрогенной недостаточности, а также возраста возникновения патологии (включая эмбриональный период). Различают эмбриональную, допубертатную и постпубертатную формы гипогонадизма. Термин мужской гипогонадизм охватывает все нарушения тестикулярной функции и ослабления действия тестостерона. В практическом отношении его целесообразно разделять на две основные группы: первичный и вторичный, которые имеют свои подгруппы.
 гипогонадизм 1. Врожденный: а) синдром анорхизма б) нарушения опущения яичек (крипторхизм, эктопия")
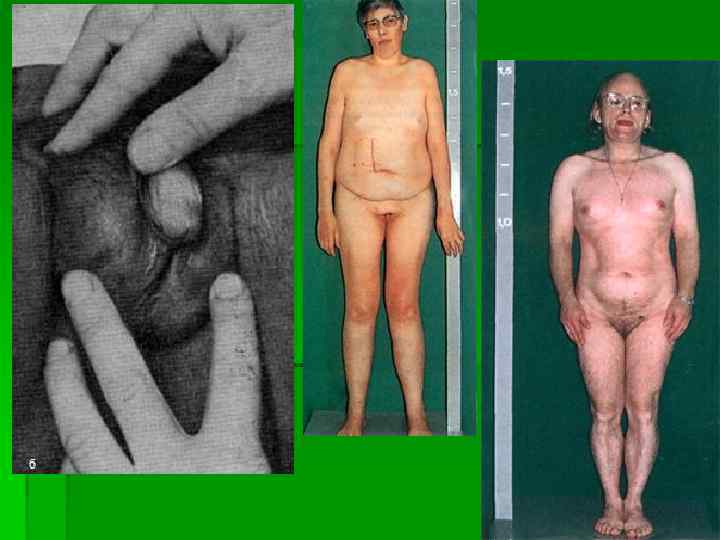
Первичный (гипергонадотропный) гипогонадизм 1. Врожденный: а) синдром анорхизма б) нарушения опущения яичек (крипторхизм, эктопия ) в) истинный хроматинположительный синдром Клайнфел-тера г) синдром Дель Кастильо д) синдром неполной маскулинизации е) синдром Шерешевского — Тернера у мужчин.
 заболевание, обусловленное аномалией половых хромосом, характерным симптомом которого является")
Синдром Клайнфелтера (дисгенезия семенных канальцев) заболевание, обусловленное аномалией половых хромосом, характерным симптомом которого является нарушение сперматогенеза Этиология. Причина заболевания неизвестна. Патогенез. Заболевание обусловлено хромосомной аномалией. У больных чаще всего есть одна лишняя X-хромосома, реже — несколько Х-хромосом (кариотип 47 XXY; 48 XXXY; 49 XXXXY). В ряде случаев выявляется полисомия по Y-хромосоме при моно-сомии по Ххромосоме, а также полисомия по Х- и Yхромосоме

синдром Дель Кастильо сочетание уменьшения размеров яичек и аспермии при нормальном или повышенном уровне гонадотропных гормонов в крови, наблюдающееся при недоразвитии яичек. синонимы: первичная герминативная аплазия, сертоликлеточный синдром, синдром одних клеток Сертоли); сочетание уменьшения размеров яичек и аспермии при нормальном или повышенном уровне гонадотропных гормонов в крови, наблюдающееся при недоразвитии яичек. Этиология и патогенез. В основе заболевания — аплазия зародышевой ткани яичка, что приводит к бесплодию.
; Является вариантом ложного мужского")
Синдром неполной маскулинизации (андроидный и евнухоидный варианты ложного мужского гермафродитизма); Является вариантом ложного мужского гермафродитизма Этиология и патогенез. Этиология неизвестна. В патогенезе развития синдрома неполной маскулинизации основную роль играет снижение тканевой чувствительности к андрогенам. В зависимости от степени маскулинизации наружных гениталий и выраженности вторичных половых мужских признаков выделяют андроидную и евнухоидную формы синдрома. Клиническая картина. У пациентов с кариотипом 46 XY и отрицательным половым хроматином яички расположены либо в расщепленной мошонке, либо по ходу паховых каналов. Внутренние половые органы мужские: придатки яичка, семявыносящие протоки, семенные пузырьки. Предстательная железа в большинстве случаев не определяется. Наружные половые органы представляют собой ту или иную степень неполной маскулинизации. Имеется искривленный недоразвитый половой член и урогенитальный синус, в который впадает слепое влагалище. Внутренние половые органы отсутствуют.

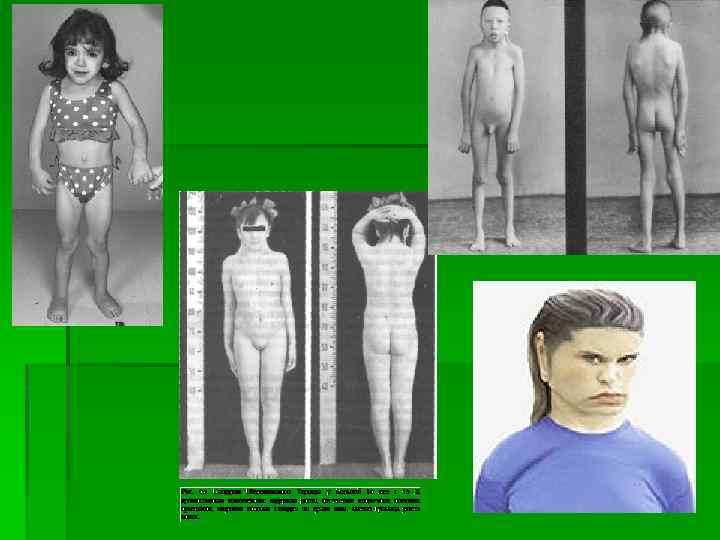
синдром Шерешевского — Тернера у мужчин Шерешевского-Тернера синдром - хромосомная болезнь, сопровождающаяся характерными аномалиями физического развития, низкорослостью и половым инфантилизмом. Характерными признаками синдрома Тернера при рождении являются избыток кожи на шее и другие пороки развития, особенно костно-суставной и сердечно -сосудистой систем, "лицо сфинкса", лимфостаз (застой лимфы, клинически проявляющийся крупными отеками). Для новорожденного характерны общее беспокойство, нарушение сосательного рефлекса, срыгивание фонтаном. В раннем возрасте у части больных отмечают задержку психического и речевого развития, что свидетельствует о патологии развития нервной системы. Наиболее характерным признаком является низкорослость. Рост больных не превышает 135 -145 см, масса тела часто избыточна.
 тотальный гипогонадизм (приобретенный евнухизм в результате ранней кастрации); б) недостаточность герминативного эпителия")
Приобретенный а) тотальный гипогонадизм (приобретенный евнухизм в результате ранней кастрации); б) недостаточность герминативного эпителия (ложный синдром Клайнфелтера); в) недостаточность тестикулярных эндокриноцитов. Как и первичный, вторичный (гипогонадотропный) гипогонадизм можно также разделить на врожденный и приобретенный.
 изолированный (идиопатический): тотальный изолированный дефицит лютропина, изолированный дефицит фолитропина; б) синдром Каллмена;")
Врожденный: а) изолированный (идиопатический): тотальный изолированный дефицит лютропина, изолированный дефицит фолитропина; б) синдром Каллмена; врожденное заболевание с аутосомнодоминантным или Х-сцепленным аутосомно-рецессивным типом наследования с различной степенью экспрессивности, чаще встречается у мальчиков. Основной характеристикой синдрома помимо гипогонадизма является аносмия вследствие агенезии ольфакторных долей. Ольфакторные доли являются местом внутриутробной закладки нейронов, секретирующих люлиберин, которые затем мигрируют в область гипоталамуса. Таким образом, агенезия ольфакторных зон приводит не только к аносмии (потеря обоняния), но и к гипоталамической форме гипогонадизма.
 гипофизарный нанизм; ГИПОФИЗАРНЫЙ НАНИЗМ - резкое отставание в росте и физическом развитии, связанное")
в) гипофизарный нанизм; ГИПОФИЗАРНЫЙ НАНИЗМ - резкое отставание в росте и физическом развитии, связанное с абсолютным или относительным дефицитом СТГ при патологии гипофиза, нарушениях не только биосинтеза гормона роста (гипофизарный нанизм), но и гипоталамической регуляции функции гипофиза (церебрально-гипофизарный, гипоталамогипофизарный нанизм), а также тканевой чувствительности к гормону роста в связи с недостаточностью инсулиноподобных ростовых факторов (ИРФ), особенно I типа (соматомединов) и тканевых рецепторов к СТГ.
 врожденный пангипопитуитаризм (краниофарингиома); состояние, сопровождающееся снижением концентрации циркулирующих гипофизарных гормонов с последующим развитием")
г) врожденный пангипопитуитаризм (краниофарингиома); состояние, сопровождающееся снижением концентрации циркулирующих гипофизарных гормонов с последующим развитием клинической картины гипотиреоза, гипокортицизма и гипогонадизма различной степени выраженности вплоть до развития комы, а также уменьшением продукции гормона роста (СТГ) и пролактина в случае первичного поражения гипофиза (разрушение эндокринных клеток).
 синдром Мэддока. Это редкое заболевание, возникающее в результате одновременной недостаточности гонадотропной и адренокортикотропной")
д) синдром Мэддока. Это редкое заболевание, возникающее в результате одновременной недостаточности гонадотропной и адренокортикотропной функций гипофиза. Клиническая картина. У больных развивается клиника евнухоидизма в сочетании с признаками хронической надпочечниковой недостаточности центрального генеза, поэтому отсутствует гиперпигментация кожных покровов и слизистых, что характерно для вторичного гипокортицизма. В литературе имеется описание единичных случаев этой патологии. Лечение таких больных проводится наряду с гонадотропинами по обычной схеме: препаратами, стимулирующими функцию коры надпочечников (кортикотропинами), или с помощью заместительной терапии глюкокортикоидами.
 адипозогенитальная дистрофия Адипозогенитальная дистрофия — заболевание, связанное с поражением гипоталамо-гипофизарной системы и")
Приобретенный: а) адипозогенитальная дистрофия Адипозогенитальная дистрофия — заболевание, связанное с поражением гипоталамо-гипофизарной системы и характеризующееся недоразвитием половых желез и ожирением. Заболевание чаще всего возникает у мальчиков и обычно выявляется в возрасте 6— 7 лет, но особенно часто в 10— 13 лет. Этиология. Заболевание может развиться вследствие внутриутробной инфекции (токсоплазмоз), родовой травмы, острых (скарлатина, тифы, вирусные инфекции) и хронических (туберкулез, сифилис) инфекций и травматических поражений мозга в раннем детском возрасте. Причиной адипозогенитальной дистрофии могут быть опухоли (краниофарингиома, хромофобная аденома), водянка III желудочка мозга, тромбозы, эмболии, кровоизлияния. Нередко причину заболевания установить не удается.

Жалобы на утомляемость, сонливость, резкую прибавку массы тела, снижение работоспособности и т. д. Кожа нередко сухая, бледная. Лицо округлое. У мальчиков отложение жира по женскому типу (шея, плечи, грудь, живот, область таза и бедер, ягодицы). Волосы на лице и теле отсутствуют. Отмечается гинекомастия, нередко наблюдается задержка роста. Внутренние органы обычно не изменены. У мальчиков малые размеры мошонки, полового члена, яичек. Нередко наблюдается крипторхизм. Вторичные половые признаки отсутствуют.
синдромы Лоренса—Муна—Барде—Бидля, Прадера— Вилли Для синдрома Прадера–Вилли характерна выраженная мышечная гипотония, наблюдаемая при рождении")
б)синдромы Лоренса—Муна—Барде—Бидля, Прадера— Вилли Для синдрома Прадера–Вилли характерна выраженная мышечная гипотония, наблюдаемая при рождении и сохраняющаяся в течение первого года жизни ребенка, задержка психомоторного развития, гипогонадотропный гипогонадизм, крипторхизм, сниженный интеллект, низкий рост, нарушение сна и терморегуляции. Фенотипические особенности включают: относительно небольшие размеры кистей и стоп (акромикрия), долихоцефалию, миндалевидный разрез глаз, низкорасположенные ушные раковины, широкую переносицу, маленький рот с тонкой верхней губой.

Пациенты с синдромом Лоуренса–Муна– Барде–Бидля часто при рождении имеют добавочные пальцы кистей и стоп (полидактилия), мышечную гипотонию. Ожирение развивается со второго года жизни ребенка, характерен гипогонадизм. Из других аномалий развития часто встречается поликистоз почек, пигментный ретинит. Интеллект снижен. Предполагается, что прогрессивный набор веса у этих детей обусловлен дисфункцией центра насыщения в гипоталамусе, что приводит к полифагиихней губой.
 гипогонадизм при гипоталамическом синдроме (в результате травматического, инфекционно-воспалительного или опухолевого поражения гипоталамо-гипофизарной области);")
в) гипогонадизм при гипоталамическом синдроме (в результате травматического, инфекционно-воспалительного или опухолевого поражения гипоталамо-гипофизарной области); г) гиперпролактинемический синдром. Гиперпролактинемический гипогонадизм - клинический синдром, обусловленный избытком пролактина, включающий той или иной степени гипогонадизм и патологическое отделяемое из молочных желез (необязательный признак).
 выделения гормонов половых желез и характеризующийся")
ГИПЕРГОНАДИЗМ Клинический синдром, вызванный избытком (относительно возрастной нормы) выделения гормонов половых желез и характеризующийся преждевременным (у девочек раньше 8, а у мальчиков раньше 10 лет) половым созреванием.
")
Различают следующие формы: 1. Изосексуальное преждевременное созревание, обусловленное гиперпродукцией половых гормонов, соответствующих полу: а) истинное изосексуальное созревание обусловлено у девочек патологией головного мозга; у мальчиков - патологией головного мозга или врожденной дисфункцией коры надпочечников. К преждевременному повышенному выделению гонадотропных гормонов может привести поражение гипоталамогипофизарной системы вследствие врожденных аномалий, воспалительных изменений, опухолей мозга, гидроцефалии; б) ложное преждевременное изосексуальное созревание обусловлено гормональноактивными опухолями половых желез.

2. Гетеросексуальное преждевременное созревание обусловлено гиперпродукцией гетеросексуальных гормонов. У девочек частой причиной является врожденная дисфункция коры надпочечников (реже - вирилизирующие опухоли яичников: адренобластома, липоидноклеточная опухоль и др. ), что приводит в период внутриутробной жизни к развитию клиники ложного женского гермафродитизма, а в постнатальном - к преждевременному половому созреванию (оволосение по мужскому типу, увеличение и вирилизация клитора, усиленное развитие мускулатуры, снижение тембра голоса, вторичные женские половые признаки развиты слабо). У мальчиков развивается крайне редко и обусловлено феминизирующей опухолью коры надпочечников (кортикостерома). Развиваются истинная гинекомастия, половое оволосение по женскому типу, гипоплазия полового аппарата, феминное распределение подкожной жировой клетчатки.

гормоны.ppt