Kopia_Lk_3_KhROMOSOMNYE_BOLEZNI.ppt
- Количество слайдов: 71



НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ

ХРОМОСОМНЫЕ БОЛЕЗНИ • Это группа болезней, которые обусловлены изменением количества хромосом или их структуры. • Клинические проявления при хромосомных нарушениях наблюдаются с рождения и не имеют прогредиентного течения, поэтому правильнее называть эти состояния синдромами, а не заболеваниями. • Аномалии хромосом достаточно часто возникают как в половых, так и в соматических клетках.

Частота хромосомных болезней • Частота хромосомных синдромов составляет 5 – 7 на 1000 новорожденных (Иванов В. И. и др. , 2007). • Предполагается, что около 50% всех спонтанных абортов обусловлены наличием хромосомных перестроек у плода, в том числе: • около 30% зигот погибает в первые 10 дней после оплодотворения в связи с наличием хромосомных аномалий, которые нарушают координированную работу генов, экспрессирующихся в раннем эмбриогенезе (до имплантации); • В первом триместре беременности половина всех случаев спонтанных абортов связана с хромосомными перестройками у эмбрионов; • Во втором триместре хромосомные перестройки являются причиной самопроизвольных абортов в 25% случаев; • Гибель плода после 20 недель развития лишь в 10% случаев оказывается результатом хромосомных аномалий.

• Вероятность прерывания беременности и внутриутробной гибели плода зависит от номера хромосомы, вовлеченной в перестройку, и типа аномалии. Так моносомии по аутосомам, как правило, обладают летальным эффектом. Достаточно редко у живорожденных выявляются структурные перестройки хромосом 1, 2, 3. Наиболее жизнеспособны дети с аномалиями акроцентрических хромосом (13 -15 и 21 -22).

Строение хромосомы человека • • • Короткое плечо хромосом обозначают латинской буквой р, длинное — q. Каждое плечо хромосомы разделяют на районы, нумеруемые от центромеры к теломере. В некоторых коротких плечах выделяют один такой район, в других (длинных)—до четырех. Полосы внутри районов нумеруются по порядку от центромеры. Если локализация гена точно известна, для ее обозначения используют индекс полосы. Например, локализация гена, кодирующего эстеразу D, обозначается 13 р14 — четвертая полоса первого района короткого плеча хромосомы 13.

Описание нормального кариотипа человека
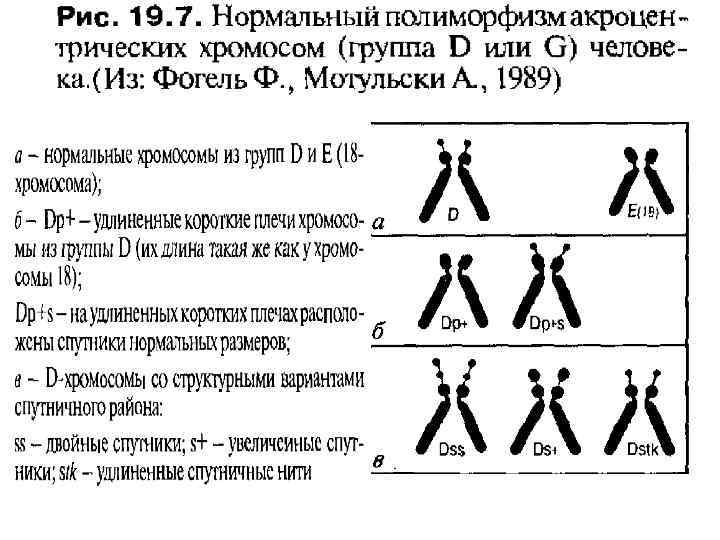

Схема записи численных и структурных аномалий хромосом • При описании аномального кариотипа используют дополнительные символы, указывающие тип и характер хромосомной перестройки. • Триплоидный кариотип: 69, ХХY или 69, ХХХ. • Трисомия по 21 хромосоме у мальчика: 47, ХY, + 21. • Моносомия по 13 хромосоме у девочки: 45, ХХ, – 13. • Моносомия по Х-хромосоме: 45, Х 0. • Трисомия по Х-хромосоме: 47, ХХХ. • Полисомия по Y-хромосоме: 47, ХYY. • Структурные аномалии: указывают общее количество хромосом, затем набор половых хромосом, запятая, описание характера структурной перестройки (del, inv, dup, t, r, ins), в скобках номер хромосомы – например, 46, ХY, del(2), а при межхромосомных обменах – номера хромосом, вовлеченных в перестройку. • Мозаицизм: 45, Х / 47, ХХХ (50% / 50%)

• Появление мозаицизма обусловлено возникновением мутаций в зиготе, сформированной нормальными гаметами в процессе клеточного деления во внутриутробном периоде. При обнаружении мозаицизма обозначения кариотипов различных клеточных клонов разделяются косой чертой (/). Например, запись кариотипа 45, Х/47, ХХХ (50% / 50%) означает, что при цитогенетическом исследовании препарата обнаружены два клона клеток – один с моносомией по Х-хромосоме, а другой с трисомией по Х-хромосоме, в соотношении 1: 1. (Иванов, с. 415

Классификация хромосомных аномалий у человека • В группе хромосомных аномалий принято выделять геномные и хромосомные мутации. • Геномные мутации характеризуются увеличением полного набора хромосом (полиплоидией) или изменением количества хромосом по одной из пар (анеуплоидия). • У человека описано два вида полиплоидий – триплоидия и тетраплоидия. Анеуплоидии выражаются в увеличении числа хромосом одной пары (трисомии и тетрасомии), и в их уменьшении (моносомии). • Полиплоидии, как правило, несовместимы с жизнью и встречаются у абортусов и мертворожденных. Летальным эффектом во внутриутробном периоде обладают и моносомии по всем аутосомам. • Наиболее частые геномные мутации у живорожденных – это трисомии по аутосомам и половым хромосомам и моносомия по Х-хромосоме. (Иванов, с. 411).

Классификация геномных мутаций • Полиплоидия • Анеуплоидия: • моносомии по всем аутосомам летальны; • трисомии по 1, 5, 6, 11 и 19 приводят к гибели эмбриона на ранней стадии развития; • наиболее часто встречаются трисомии по 8, 13, 18 и 21 хромосомам; • дисбаланс половых хромосом не приводит к гибели эмбриона.

СИНДРОМЫ, СВЯЗАННЫЕ С АНОМАЛИЕЙ ЧИСЛА ХРОМОСОМ Причины аномалии числа хромосом: • нерасхождение хромосом; • утрата отдельных хромосом; • полиплоидия

• Аномалии числа хромосом могут быть вызваны разными причинами. • Наиболее важным механизмом является нерасхождение хромосом. Хромосомы, которые в норме должны делиться во время клеточного деления, остаются соединенными вместе и в анафазе отходят к одному полюсу. В результате формируются гаметы с неодинаковым количеством хромосом: на каждую гамету с одной добавочной хромосомой приходится гамета без одной хромосомы. Это происходит чаще во время мейоза, но может происходить и в митозе. Следует отметить, что у человека по неизвестным пока причинам акроцентические хромосомы имеют тенденцию чаще вовлекаться в нерасхождение. Если нерасхождение происходит во время митоза, наблюдается мозаицизм с наличием нормальных клеток, моно и трисомиков. • Второй механизм – утрата отдельных хромосом, вследствие отставания одной хромосомы во время анафазы. В результате формируется гамета с числом хромосом n – 1. • Полиплоидия. У человека обнаружена только триплоидия, 3 n =69.

Общие закономерности клинических проявлений хромосомных синдромов • Недостаток хромосомного материала приводит к более выраженным клиническим проявлениям, чем его избыток. Частичные моносомии (делеции) сопровождаются более тяжелыми клиническими проявлениями, чем частичные трисомии (дупликации), что обусловлено потерей ряда генов. Чаще обнаруживаются у абортусов и мертворожденных, т. к. часто затрагиваются гены, необходимые для роста и дифференцировки клеток. К гибели эмбриона на ранней стадии развития приводят полные моносомии по аутосомам, а также трисомии по 1, 5, 6, 11 и 19 хромосомам. Наиболее часто встречаются трисомии по 8, 13, 18 и 21. •

●Для большинства хромосомных синдромов, обусловленных аномалиями аутосом, характерны малый вес ребенка при доношенной беременности, пороки развития двух и более органов и систем, а также задержка темпов раннего психомоторного развития, олигофрения и снижение показателей физического развития ребенка. У детей с хромосомной патологией часто выявляют увеличение количества, так называемых, стигм дизэмбриогенеза или малых аномалий развития. В случае наличия пяти и более таких стигм говорят о повышении порога стигматизации у человека. К стигма можно отнести наличие санлавидной щели между первым и вторым пальцами на ногах, диастему (увеличение расстояния между передними резцами), расщепление кончика носа и другие.

• Для аномалии половых хромосом, в противоположность аутосомным синдромам, не характерно наличие выраженного интеллектуально дефицита, некоторые больные имеют нормальное умственное развитие. • У большинства больных с аномалиями половых хромосом возникает бесплодие и невынашивание беременности. • Необходимо отметить, что бесплодие и невынашивание беременности при аномалиях половых хромосом и аутосом имеет различные причины. • При аномалиях аутосом прерывание беременности часто обусловлено наличием хромосомных перестроек, несовместимых с нормальным эмбриональным развитием, или элиминацией несбалансированных по хромосомному материалу зигот, эмбрионов и плодов. • При аномалях половых хромосом в большинстве случаев наступление беременности невозможно по причине аномалии сперматозоидов или аплазии или резкой гипоплазии, как наружных, так и внутренних половых органов. В целом, аномалии половых хромосом приводят к возникновению менее выраженных клинических симптомов, чем аномалии аутосом. • Полные формы хромосомных аномалий характеризуются более тяжелыми клиническими проявлениями, чем мозаичные.

Показания к исследованию кариотипа Таким образом, учитывая все клинико-генетические данные больных с хромосомными синдромами, показания к исследованию кариотипа у детей и взрослых следующие: • • • Малый вес новорожденного при доношенной беременности. Врожденные пороки развития двух и более органов и систем в сочетании с олигофренией. Недифференцированная олигофрения. Бесплодие и привычное невынашивание беременности. Наличие сбалансированной хромосомной перестройки у родителей или сибсов пробандов.

Полиплоидия • По данным Г. И. Лазюка в мировой литературе описано 32 наблюдения новорожденных детей с триплоидией (69, ХХУ или 69, ХХХ). По данным В. И. Иванова описаны также тетраплоиды. • Причина – диандрия (двойное оплодотворение) или отсутствие первого мейотического деления ооцита. • У матерей таких детей во 2 -й половине беременности наблюдают токсикоз и многоводие. • У детей выявляются большие дефекты в строении скелета и внутренних органов, половых органов. Продолжительность жизни таких детей в среднем равна 9 дням. • Некоторые мозаики мужчины выживают. Характеризуются глубокой умственной отсталостью, в сочетании с аномалиями плаценты, гениталий, ассиметрией.

Анеуплоидии. Синдром Дауна. Характерный фенотип
. Наиболее частое хромосомное заболевание человека. Его частота среди")
Синдром Дауна • Синдром Дауна (СД). Наиболее частое хромосомное заболевание человека. Его частота среди новорожденных 1 – 2/1000 (Фогель, Мотульски), 1, 5 – 5/1000 (Бердышев), 1/700– 800 новорожденных (Иванов). Эта болезнь впервые описана английским врачом Дауном в 1866 г. как своеобразная форма олигофрении, резкое снижение интеллекта, сочетающееся с характерным внешним видом. В 1959 г. французский ученый И. Лежен обнаружил в кариотипе больных лишнюю 21 -ю хромосому. • Таким образом, в 94% случаев болезнь Дауна обусловлена трисомией по 21 -й хромосоме. • В дальнейшем было показано, что в 4% случаев болезнь Дауна может быть обусловлена не только трисомией 21, но и транслокацией 21 -й хромосомы на другие (чаще 13, 14, 15, 21, 22).

• Анализ показал, что за развитие характерного фенотипа отвечает дистальный район длинного плеча хромосомы 21, а именно сегмент 21 q 22 (q – длинное плечо хромосомы, p – короткое). Трисомия только по 21 q 22 сегменту приводит к мягким проявлениям этой болезни. Например, у девочки, генотип которой содержал дополнительный сегмент 21 q 22, наблюдалась умеренная умственная отсталость, у нее отсутствовала большая часть признаков болезни Дауна. • В 2% случаев наблюдается мозаицизм, когда половина клеток имеет кариотип 46 хромосом, , другая половина – 47 хромосом. При синдроме Дауна, вызванном мозаицизмом, симптомы заболевания выражены менее резко. • Клиническая картина болезни Дауна, обусловленная трисомией 21 или транслокацией не различается. • Наиболее часто (примерно в 80% случаев) нерасхождение хромосом происходит во время первого мейотического деления, причем, в 2/3 случаев в женских половых клетках. (Иванов). • Предполагают, что ключевая роль в возникновении умственной отсталости при СД принадлежит увеличению дозы гена фермента супероксиддисмутазы, локализованной в области q 22. 3.

Синдром Дауна Трисомия по 21 хромосоме

Клинические синдромы СД • Клинические симптомы СД очень специфичны и позволяют диагносцировать болезнь уже в родильном доме. Наиболее важные характеристики синдрома Дауна следующие: • Это четко очерченное состояние. Диагноз редко вызывает сомнение. В первые годы жизни диагоностика бывает затруднена. В старшем возрасте диагносцируется легче. • Такие дети имеют некоторые общие морфологические черты. Они больше похожи один на другого, чем на своих родителей. • У больных наблюдаются характерные изменения со стороны лица и головы. У них круглая голова с уплощенным затылком, скошенным узким лбом, плоским лицом, косым, монголоидным разрезом глаз, толстыми губами, широким уплощенным языком с глубокой продольной бороздой. Ушные раковины уменьшены в вертикальном направлении с приросшей мочкой.

Дети с синдромом Дауна. А. Европеоид. Б. Темнокожий. Представитель азиатской расы В.

• У маленьких детей резко выражена гипотония, из-за чего в лежачем положении живот принимает форму «лягушачьего» . • Характерное изменение конечностей: укорочение и расширение кистей, стоп (акромикрия). На ладони одна поперечная борозда (четырехпальцевая). • В радужной оболочке отмечаются белые очаги (пятна Брушфильда), которые с возрастом становятся менее заметными. Рано развивается катаракта. • Отмечается неправильный рост зубов, высокое нёбо, низкий рост, румянец на щеках, низкая граница роста волос на шее. • Отмечаются изменения со стороны внутренних органов, особенно сердца (отмечаются у 53% больных, Иванов), пищеварительного канала (у 15%). Часто отмечают комбинированные пороки развития различных органов и систем, врожденные уродства.

Резкая гипотония

Синдром Дауна

Характерный фенотип больного Дауна

Основные клинические симптомы болезни Дауна • • • • Широкое плоское лицо Плоский затылок Косой монголоидный разрез глаз Маленькое и арковидное небо Большой складчатый язык Зубные аномалии Короткие широкие кисти На ладони одна поперечная борозда Врожденный порок сердца Комбинированные пороки развития Гипотоничные мышцы Задержка роста Умственная отсталость

Синдром Дауна

Взрослые больные с синдромом трисомии 21
 до идиотии. (Большинство")
У больных наблюдается глубокая умственная отсталость – от имбециальности (? ) до идиотии. (Большинство олигофренов имеют значение коэффициента интеллекта в границах 51– 70 баллов – дебилы, более тяжелые нарушения у имбецилов – 21– 51, и при значениях 0 – 20 баллов больные квалифицируются как идиоты). При синдроме Дауна коэффициент интеллекта колеблется в границах 20– 60 баллов, средние значения – 40 -50 баллов. Они послушны, повышенно внушаемы. Многих больных можно научить читать и писать, у них неплохо развита способность к подражанию. Хорошая память на людей, музыку, ситуации. Отсутствуют малейшие попытки к абстрагированию. Если письмо и чтение могут усвоить, то счет им недоступен. Многие больные не могут научиться даже простому арифметическому счету. К полноценной социальной адаптации способны единицы; как правило, больные не способны жить самостоятельно.

• При исследовании мозга погибших отмечают его недоразвитость, плохую выраженность борозд и извилин. Желудочки мозга часто недоразвиты, полости их уменьшены. Нервные клетки недостаточно дифференцированы. Нарушены процессы миелинизации головного и спинного мозга. • Описаны случаи (17 случаев, Фогель) беременности и родов у женщин, страдающих болезнью Дауна, обусловленной трисомией. Примерно у половины их детей развилась болезнь Дауна. Были дети с нормальным кариотипом. Однако и при нормальном кариотипе (46, ХУ) у ребенка наблюдался ряд отклонений от нормы, так как ребенок развивался в организме матери с лишней 21 хромосомой и, значит, с измененным белковым составом ее жидкостей. Но могут быть и здоровые дети. Такие случаи описаны, так, среди 19 детей, рожденных матерями – дауниками, 7 были больны

Продолжительность жизни с СД • При удовлетворительном уходе такие больные могут жить долго. В целом, продолжительность жизни больных сокращена. Примерно 31% больных умирает в конце первого ода жизни, 46% - в конце третьего года. Очевидно, у больных Дауна возможен дефект иммунной системы. С появлением антибиотиков и сердечной хирургии эти больные живут дольше, Считают, однако, что крайние значения продолжительности жизни для них не будут большими, они редко доживают до 40 – летнего возраста (Иванов). Предполагается, что больные с синдромом Дауна стареют быстрее, чем нормальные люди. Смерть часто наступает от сердечной недостаточности, инфекций, а в ряде случаев онкологических заболеваний, прежде всего, лейкемии.

Вероятность повторного рождения ребенка с СД • • В большинстве случаев в семье регистрируется только один больной. Кариотип родителей ребенка с синдромом Дауна, как правило, нормальный. Медицинские аборты и предшествующие беременности, как правило, не влияют на частоту рождения больных. При болезни Дауна, вызванной транслокацией, у одного из родителей обычно отмечается транслокация 21 хромосомы на одну из хромосом группы D(13, 14, 15) или на 22 хромосому. При этом частота рождения больных детей в данной семье может достигать 33%. Расчет: теоретически при транслокации, например, 21 на 13, можно ожидать 4 типа гамет: 13, 21; 13210; 132121 и 13, 0. При оплодотворении с нормальной гаметой 13, 21 можно ожидать 4 варианта зигот: 13, 21 + 13, 21 – норма; 13, 21 + 1321, 0 – зигота с транслокацией, как у одного из родителей (фенотип нормальный); 13, 21 + 1321, 21 – зигота с лишней 21 хромосомой , что проявляется как болезнь Дауна; 13, 21 + 13, 0 – зигота нежизнеспособна. Таким образом, вероятность рождения больного ребенка в этой семье составит 33%. При болезни Дауна, обусловленной мозаицизмом, последний может быть обнаружен у одного из родителей, но мозаицизм может возникнуть и на ранних стадиях дробления зигот. В этом случае у носителя можно обнаружить некоторые элементы, характерные для болезни Дауна – акромикрию, микроцефалию, четырехпальцевую ладонную борозду и др. В большинстве случаев в семье регистрируется только один больной, в очень небольшом числе семей наблюдаются повторные случаи рождения детей с болезнью Дауна.

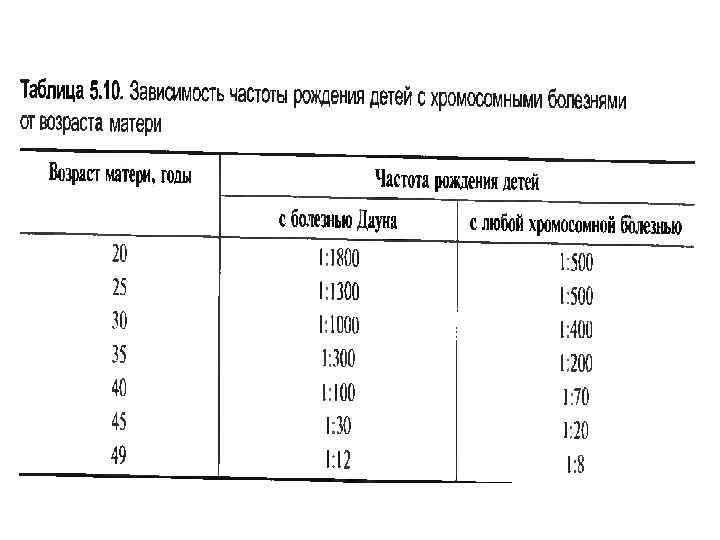
Причины рождения детей с СД • • Причины рождения детей с болезнью Дауна окончательно не выяснены Предполагается, что это могут быть перенесенные матерью перед зачатием инфекционные заболевания (гепатит, токсоплазмоз? ). Установлено, что частота синдрома увеличивается с возрастом матери. Эмпирический риск рождения больного ребенка в 19 лет – 1 : 1640; 40 – 41 год – 1 : 84; после 45 лет – 1 : 31 (Лазюк, 1979). В последние годы, благодаря флюорисцентному анализу показано, что нерасхождение хромосом может происходить и при сперматогенезе, так что и отец может быть «виновным» в возникновении болезни Дауна (в 20 -25% случаев). Особенно большой риск рождения ребенка с болезнью Дауна в семьях, где отцу больше 50 лет, а матери 40. Диагностика заболевания осуществляется на основании клинического осмотра и анализа кариотипа больного. В случае выявления регулярной трисомии по 21 -й хромосоме исследование кариотипа родителей больного не проводят, При обнаружении транслокационной формы заболевания этот этап медико-генетического консультирования семьи абсолютно необходим. Профилактика заболевания осуществляется на основании дороловой диагностики, основанной на цитогенетическом и ультразвуковом обследовании плода. Такое обследование необходимо проводить в двух случаях: – если беременная женщина старше 35 лет; –. Если у одного из родителей больного выявлена транслокация с вовлечением 21 хромосомы.


Синдром Патау Трисомия по 13 хромосоме
, 1/600")
• Трисомии по другим аутосомам описаны меньше. • Синдром Патау. 1/4000 (Бадалян), 1/600 (Иванов). • В 1961 г. К. Патау описал трисомию по 13 -й хромосоме. При синдроме Патау отмечаются значительные дефекты строения черепа, микроцефалия, низкий скошенный лоб, узкие глаза, запавшая переносица, низко расположенные ушные раковины, расщепление верхней губы и неба, дефекты сердечно-сосудистой системы, недоразвитие переднего отдела мозга, а также поли и синдактилия кистей и/или стоп, пороки развития почек, половых органов и кишечника. Дети погибают в первые 3 месяца после рождения (~ 70%), 95% погибают в возрасте до года, чаще погибают до рождения. Это подтверждается при исследовании кариотипа при спонтанных абортах. Причина смерти – сердечная недостаточность, нарушение дыхания, присоединяется пневмония.

Синдром Патау
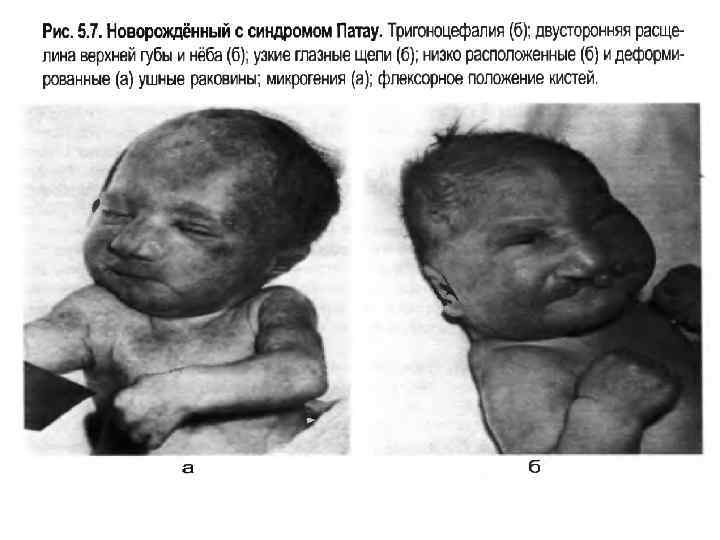

Синдром Патау Для СП характерны множественные врожденные пороки развития головного мозга и лица. Это патогенетическая единая группа ранних (и, следовательно, тяжелых) нарушений формирования головного мозга, глазных яблок, костей мозговой и лицевой частей черепа. Окружность черепа обычно уменьшена, встречается и тригоноцефалия. Лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположенные и деформированные. Типичный признак СП - это расщелины верхней губы и неба (обычно двухсторонние).

Синдром Эдвардса Трисомия по 18 хромосоме

• Синдром Эдвардса – трисомия по 18 хромосоме. Описана Дж. Эдвардсом в 1960 г. 1/3000 1/7000 (Иванов) Чаще бывает у мальчиков. Множественные врожденные пороки развития. Как и при синдроме Патау, у детей наблюдаются большие изменения со стороны черепа и скелета. Такие дети обычно рождаются переношенными, в асфиксии, с долихоцефалией, расщеплением неба и др. Отмечается деформация пальцев рук с характерным расположением пальцев (второй палец перекрывает третий, а пятый – четвертый), уплощение свода стопы (из-за чего стопа имеет форму качалки), пальцы ног и рук укорочены. Кожа очень подвижна, образует складки. Выражены дефекты внутренних органов (сердца, пищеварительного канала). • Мальчики погибают вскоре после рождения, девочки живут до года.

Синдром Эдвардса

Синдром Эдвардса
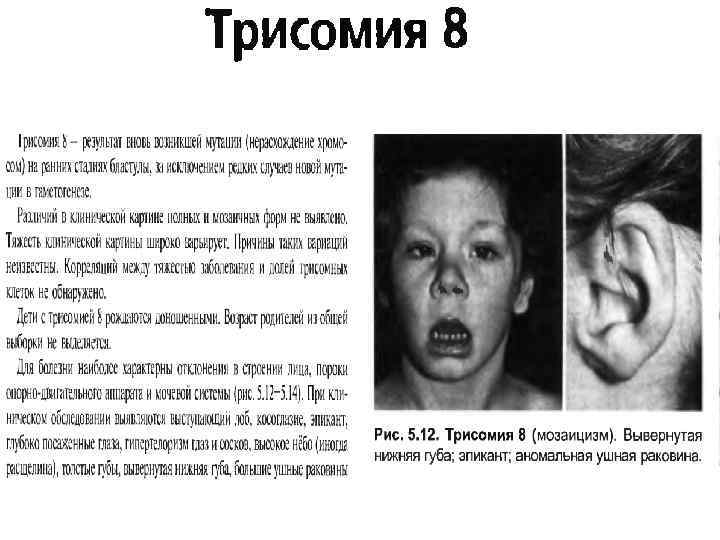

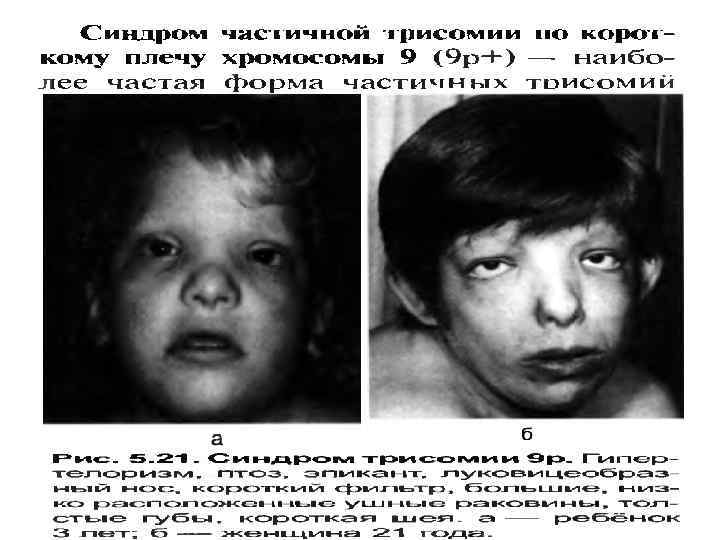
• Трисомия по 22 хромосоме. Описано около 30 случаев болезни. Эти дети – глубокие олигофрены. У них выражена микроцефалия, клювовидный нос, низко расположенные ушные раковины, расщепление неба, гипотония мышц. Такие дети рождаются у родителей с нераспознанным мозаицизмом (46/47). • В последние годы описаны трисомии по 3, 8, 9, 10, 14, 16, 20, 22 хромосомам (Фогель, Бердышев). Как и следовало ожидать, эти, весьма редкие, хромосомные аномалии вызывают тяжелые и комплексные пороки развития. Открытие 8, 9, 22 синдромов связано с освоением дифференциальной окраски хромосом. Описаны у спонтанных абортусов.

Синдромы, связанные со структурными аномалиями аутосом Пробелы и разрывы. • Разрыв в хромосоме – необходимое условие возникновения хромосомной перестройки любого типа. • Возникают во время интерфазы как до, так и после репликации ДНК.
 Инверсии (inv) Дупликация (dup) Кольцевая хромосома")
Классификация хромосомных перестроек • • • Делеции (del) Инверсии (inv) Дупликация (dup) Кольцевая хромосома (r) Инсерция (ins) Транслокация (t)

Синдром «кошачьего крика» • Синдром «кошачьего крика» . 1/40 000, 1/50 000. Описал Лежен с сотр. , 1963. Это делеционный синдром. Он обусловлен делецией короткого плеча хромосомы 5 (del 5 p-). Кроме обычных признаков аутосомных заболеваний (общее отставание в развитии, низкий вес, уродства развития, микроцефалия, короткая шея, четырехпалость, сердечно-сосудистые нарушения) у этих детей лунообразное лицо с гипертелоризмом (широко расставленные глаза). Вследствие аномалии развития гортани их плач напоминает мяуканье кошки. Большинство умирают, но описаны больные, которым больше 50 лет.
")
Синдром «кошачьего крика» 5(del 5 р –)
 • Задержка")
Синдром частичной трисомии проксимального отдела длинного плеча 14 хромосомы, 14(dup 14 q+) • Задержка психомоторного развития; • черепно-лицевые и скелетные аномалии, • удвоение проксимальной части 14 q хромосомы

ПОЛОВЫЕ ХРОМОСОМЫ. • Анеуплоидии по Х – хромосомам человека: ХХХ, ХХУ, Х 0. • Различие между Х-хромосомными и аутосомными анеуплоидиями: • Умственное развитие в среднем хотя и ниже нормы, но аномалии развития мозга выражены не столь отчетливо, как при аномалиях аутосом. Многие пробанды имеют нормальное умственное развитие. • Фенотипические нарушения затрагивают в большей степени развитие половых органов и гормонозависимый рост. • Наблюдаются и другие пороки развития, особенно при синдроме Тернера, но встречаются они реже и по масштабам менее тяжелые. • Объяснение, по-видимому, состоит в том, что в норме женщина имеет 2, а мужчина только 1 -у Х-хромосому, в связи с чем в эволюции сформировался механизм компенсации различий в дозе генов, сцепленных с Х-хромосомой. Этот механизм оказался фактором, благоприятствующим носителям Х-хромосомных анеуплоидий.

Классификация Х-хромосомных анеуплоидий • В общем, число добавочных Х-хромосом увеличивает степень умственной отсталости. Число телец Х-хроматина (тельце Бара) всегда на одно меньше, чем число Х-хромосом. • 47, ХХY – синдром Клайнфельтера, 1/700 мужчин. • 48, ХХХY – вариант синдрома Клайнфельтера, 1/2500 мужчин. • 49, ХХХХY – глубокая умственная отсталость, сильное недоразвитие половых органов и другие пороки развития. Встречается очень редко. • 47, ХХХ – трисомия по Х-хромосоме, иногда легкая олигофрения, непостоянно нарушена функция гонад, 1/1000 женщин. • 48, ХХХХ – физически нормальные. Редкий. • 49, ХХХХХ – тяжелая умственная отсталость. Редкий. • 45, Х 0 – синдром Тернера. 1/2500 женщин при рождении. • 47, ХYY – синдром дисомии по Y-хромосоме, высокий рост, иногда аномалии поведения. 1/800 мужчин. • 48, ХХYY – высокий рост, в остальном сходен с синдромом Клайнфельтера.

Синдром Шерешевского – Тернера; 45, Х 0 • Кариотип 45, Х 0; 1/1000 женщин • Шерешевский Н. А. , 1925 • Тернер Х. Х. , 1938 • Форд Г. К. в 1959 г. установил, что у этих больных имеется только одна Х-хромосома

Синдром Шерешевского – Тернера • • • Синдром Шерешевского – Тернера. Кариотип 45, Х 0. Описан Н. А. Шерешевским в 1925 г. , а затем в 1938 г. – Тернером. В 1959 г. К. Форд установил, что у этих больных имеется только одна Х-хромосома. Низкий рост, аномалии половых органов, недоразвиты матка и яичники, половой инфантилизм. Месячных не бывает или они однократны. Грудные железы отсутствуют, на их месте иногда определяются складки жира. Ушные раковины деформированы, располагаются низко, шея короткая, по ее бокам отмечается широкая кожная складка. Отмечаются изменения со стороны внутренних органов. Рост прекращается в 15 – 18 лет. Интеллект нарушен мало или вообще не страдает. Резко снижено выделение эстрогенов, повышена экскреция гонадотропина. При рождении не всегда установить правильный диагноз. Часто рождаются недоношенными с малым ростом. Обычно диагноз устанавливается позже, когда наблюдается отставание девочки в росте и половой инфантилизм. Важнейшим для диагностики является исследование полового хроматина. Если его не обнаруживают, то это свидетельствует о моносомии по Х-хромосоме. При мозаицизме 46, ХХ/45, Х 0 могут быть матерями. Возраст родителей роли не играет. Но чаще рождаются у родителей низкого роста с нормальным кариотипом (Бердышев). «Виновными» могут быть как мать, так и отец. Причиной могут быть инфекционные болезни во время беременности, например, краснуха.

Х 0 – синдром. Синдром Шерешевского – Тернера

Моносомия по Х-хромосоме
. Кариотип")
Синдром полисомии по Ххромосоме • Синдром полисомии по Х-хромосоме у женщин (или трипло-Х-синдром). Кариотип 47, ХХХ. Фенотипических изменений может и не быть, т. к. две Х-хромосомы спирализованы и представляют собой половой хроматин (Х -хромосома в виде гетерохроматина). Может наблюдаться умственная отсталость. Могут иметь здоровое потомство. • Описаны случаи с 4 -мя и 5 -ю Х-хромосомами. Чем больше Х-хромосом в кариотипе, тем больше выражен дефект умственного развития, а также изменения фенотипа и половой инфантилизм. При кариотипе 48, ХХХХ дети маложизнеспособны и рано умирают.

Синдром Клайнфельтера • ХXY – высокий рост, иногда аномалии поведения. 1/700 -800 мужчин • ХХXY – вариант синдромома Клайнфельтера. 1/2500 мужчин XXXXY – глубокая умственная отсталость. Очень редкий ХXY/XY и ХXY / ХX – сходен с синдромом Клайнфельтера, иногда с более сглаженными симптомами. 5 – 15% от больных с синдромом Клайнфельтера. • • д

Синдром Клайнфельтера • Описан Клайнфельтером в 1924 г. как синдром первичного мужского гипогонадизма. Кариотип 47, ХХУ. В клетках букального эпителия обнаружен половой хроматин, как у нормальных женщин. До достижения половой зрелости синдром может быть не обнаружен. При половом созревании отмечают признаки евнухоидности. У больных недоразвиты половые железы при хорошем развитии полового члена, отсутствует растительность на лице , выражена гинекомастия, отложение жира на бедрах по женскому типу. Больные обычно высокого роста за счет удлиненных конечностей, рост волос на лобке по женскому типу, высокий голос. • При гистологических исследованиях отмечена склеротизация семенных канальцев и др. отклонения. Бесплодны (из-за отсутствия сперматогенеза). • Дебильность разной степени. Описаны случаи синдрома Клайнфельтера с тремя и четырьмя Х-хромосомами. Чем больше Ххромосом в кариотипе, тем больше выражены симптомы заболевания и степень дебильности. • Причина появления болезни Клайнфельтера неясна. ? . Такие дети обычно рождаются у пожилых матерей с нормальным кариотипом. Больные не оставляют потомства.

Дисомия по Y-хромосоме; 47, ХYY • • 1/800 мужчин Кариотип 47, ХУУ. Описан как у фенотипически здоровых мужчин, так и у мужчин с гипогонадизмом, крипторхизмом, умственной отсталостью и развитой мускулатурой. • Несбалансированность по половым хромосомам приводит к особенностям развития нервной системы, снижению интеллекта, что может явиться причиной агрессивности и криминальных поступков. • Зарубежные ученые обнаружили кариотип 47, ХУУ у агрессивных мужчин, преступников-рецидивистов, с высоким ростом, правильным телосложением, большими ушными раковинами и надбровными дугами, хорошо развитой мускулатурой, нормальным развитием половых желез, превалированием лицевого скелета над мозговым.

Интерсексы • • • Различают три типа интерсексов: Истинный гермафродитизм: присутствуют половые клетки обоих полов. Мужской псевдогермафродитизм, 46, ХУ, но признаки женского пола. а) неполноценная У-хромосома (У-хромосома содержит гены, которые контролируют развитие по мужскому типу). Б) тестикулярная феминизация (Ж. д’Арк) – у лиц, генетически мужского пола (ХУ) соматические клетки не реагируют на мужской половой гормон тестостерон из-за дефекта белкового рецептора (имеются данные, что склонность к мужскому гермафродитизму наследуется аутосомно-рецессивно). Женский псевдогермафродитизм. Кариотип 46, ХХ. Предпалогают, что в Ххромосоме существуют гены репрессоры, не позволяющие ХХ-организму развиваться по мужскому типу, В присутствии У-хромосомы действие этих генов нейтрализуется. В результате мутации гена-репрессора генотип ХХ может формироваться по мужскому типу. (Каминская Э. А. , 1992). (женские половые гормоны – эстрогены и прогестерол (образуе(ю)тся в яичниках), мужской половой гормон – тестостерон (образуется в семенниках). Продукт превращения тестостерона – дигидротестостерон обусловливает развитие по мужскому типу
 наблюдают избыточное выделение андрогенов при гиперплазии надпочечников (женский псевдогермафродитизм?")
Псевдогермафродитизм • Врачи (по Бердышеву) наблюдают избыточное выделение андрогенов при гиперплазии надпочечников (женский псевдогермафродитизм? ). 1) Первопричиной является недостаточное образование кортизола в надпочечниках при нормальном образовании половых гормонов. Кортизол в норме оказывает ингибирующее действие на гипофиз через гипоталамус. При отсутствии этого ингибирующего действия в гипофизе вырабатывается избыточное количество АКТГ, который вызывает гиперплазию надпочечников, избыточное выделение андрогенов, от которых и зависит мускулинизация женщины. Женский псевдогермафродитизм наблюдается также 2) при опухолях яичников во время беременности и чрезмерной продукции вирилизирующих гормонов. 3) Женский гермафродитизм может появиться у девочек, родившихся от матерей, получавших во время беременности гестогены с целью сохранения беременности

Другие случаи нарушения половой дифференциации • Некоторые изменения половых признаков появляются и при нормальном развитии. • Например, гинекомастия, приводит к развитию органа женского типа (например, груди) у мужчин, типичных (нормальных) в других ошениях. Возможно, что генотип тканей молочных желез таких мужчин вызывает измененную реакцию на нормальную смесь мужских и женских гормонов и реакцию, ведущую к образованию развитых молочных желез. • На такое объяснение наводит существование в выводках цыплят петушков, имеющих куриные хвосты. Оказалось, что гормоны семенников у них вполне нормальны, но что зачатки перьев реагируют на них аномально. •
 гипоспадия или смещение отверстия")
• Другая аномалия в развитии мужских особей (1/несколько сотен) гипоспадия или смещение отверстия мочеиспускательного канала с конца penis в направлении к его основанию. Это представляется изменением в сторону женского организма. Иногда нет оснований рассматривать их как явление генетического порядка, хотя в других случаях наблюдается как рецессивная, так и доминантная (реже) передача по наследству (Штерн, с. 313).

• Еще один пример генетически обусловленной половой аномалии – преждевременное половое созревание. В некоторых родословных у мужчин, а в других у женщин за годы до возраста половой зрелости развиваются признаки полового созревания – такие как рост волос под мышками или на лобке, развитие молочных желез. Имеется родословная, в которой 27 мужчин в 4 -х поколениях обнаруживали преждевременную половую зрелость – у некоторых из них даже в двухлетнем возрасте. Наследование доминантное, осуществляется и через мужчин и через женщин. У женщин признак не проявляется – неполная пенетрантность.

Спасибо за внимание
Kopia_Lk_3_KhROMOSOMNYE_BOLEZNI.ppt