Наследственные нефропатии.ppt
- Количество слайдов: 43



НАСЛЕДСТВЕННЫЕ НЕФРОПАТИИ

Наследственными нефропатиями называют заболевания, этиологическим фактором развития которых является мутация гена. ( «Нефрология» , 2000 г) 4 категории нефропатий: ç Моногенно -наследуемые (связаны с мутацией одного гена) - С-м Альпорта , финский тип, аутосомно -доминантная поликистозная болезнь; ç Полигенно -наследуемые нефропатии - заболевания почек, при которых этиологическим фактором является мутация нескольких генов, а также необходимы внешние неблагоприятные воздействия (подагрическая нефропатия); ç Мультифакториальная патология органов мочевой системы - зависит от воздействия вирусов, бактерий, охлаждения, но развивается лишь при генетическом предрасположении (наличие структурного дефекта почек, недостаточность фактора Н в системе комплемента); ç Экзогенные нефропатии - заболевания, которые связаны с влиянием внешних факторов - ожогов, отравлений, травм, укусов. Генетические факторы проявляют себя в процессе репарации.

МОНОГЕННО-НАСЛЕДУЕМЫЕ НЕФРОПАТИИ В ЗАВИСИМОСТИ ОТ ХАРАКТЕРА НАСЛЕДОВАНИЯ ü Аутосомно -доминантные нефропатии передаются «по вертикали» - от родителей детям (наследственный нефрит - с-м Альпорта, почечная глюкозурия); ü Аутосомно -рецессивные нефропатии определяются в «горизонтальных рядах» с «проскоком» через поколе- ние ( цистинурия , синдром Барттера , врожденный нефротический синдром финского типа и др. ); ü Доминантный сцепленный с Х-хромосомой - тип передачи от больного отца к дочери (классический вариант с-ма Альпорта, несахарный диабет).

СИНДРОМ АЛЬПОРТА - наследствен-ная неиммунная гломерулопатия , связанная с патологией коллагена базальных мембран, проявляющаяся гематурией и протеинурией , прогрессирующим сниже - нием функции почек, нередко сочетаю- щаяся с патологией слуха и зрения.

Органы-мишени при наследственном нефрите Базальные мембраны, страдающие при наследственном нефрите: çбазальные мембраны клубочковых капилляров; çбазальные мембраны кортиева органа; çбазальные мембраны капсулы хрусталика.

Состав коллагена в различных базальных мембранах Lamina densa Подоциты Эндотелий Субэндотелий и мезангий Lamina densa 1 (IV) 3 (IV) 2 (IV) 4 (IV) коллаген V типа 5 (IV) коллаген VI типа Дистальный каналец, Эпидермис капсула Боумена 1 (IV), 2 (IV) 5 (IV), 6 (IV) 3 (IV), 4 (IV) 5 (IV), 6 (IV)

Первое описание семьи, в которой отмечались случаи гематурии в нескольких поколениях принадлежало L. Cuthrie (1902), затем A. Hurst, продолжая наблюдение за этой семьей, проследил развитие уремии у некоторых ее членов (1923), а в 1927 г А. Альпорт описал эту же семью и отметил, что у некоторых родственников имелась тугоухость, и уремия развивалась раньше у мужчин, чем у женщин. В 1985 г L. Menlove et al. выявили ген, ответ- ственный за с-м Альпорта , который был обнаружен на длинном плече Х-хромосомы в зоне 21 - 22 q.

ЭПИДЕМИОЛОГИЯ СИНДРОМА АЛЬПОРТА Распространенность в России 17 : 100 000 [ Вельтищев Ю. В. , Игнатова М. С. 1996] Встречается у лиц в разных регионах мира, независимо от расы и национальной принад- лежности. Встречается чаще, чем выявляется.

ГЕНЕТИКА СИНДРОМА АЛЬПОРТА Генетическая основа наследственного нефрита - мутация гена COL 4 A 5 , кодирующего альфа-5 -цепь коллагена IV типа. Продукт гена - цепи коллагена IV типа. COL 4 A 3 и COL 4 A 4 находятся на 2 -й хромосоме. Их мутация характерна для аутосомно-рецессивного типа передачи наследственного нефрита. Аутосомно-рецессивный тип передачи выявляется у 6 % больных выявляется, а аутосомно -доминантный у 16 % больных [Игнатова М. С. , 2000]. Доминантный сцепленный с полом тип наследования характерен для классического с- ма Альпорта (наследственный нефрит с тугоухостью и ранним развитием почечной недостаточности).

Геномная организация генов шести типов коллагена IV типа на хромосомах 13, 2 и Х

КЛИНИЧЕСКАЯ КАРТИНА СИНДРОМА АЛЬПОРТА ü У женщин с наследственным нефритом часто отмечаются спонтанные аборты, мертворождения , нефропатия беременных; ü Первые клинические признаки могут быть выявлены в любом возрасте: ü гематурия (при случайном обследовании, м. б. изолированной); ü протеинурия (степень ее выраженности определяет прогноз заболевания. При большой протеинурии, а также развитии нефротического синдрома - прогноз неблагоприятный). ü абактериальная лейкоцитурия (как проявление тубулоинтер- стициальных изменений). ü Стигмы дизэмбриогенеза (характерна однотипность стигм у пораженных членов семьи: аномалия развития почек, аномалии хрусталика глаза и др. ); ü Симптомы интоксикации (бледность, вялость, мышечная гипотония, анорексия без явных причин);

КЛИНИЧЕСКАЯ КАРТИНА СИНДРОМА АЛЬПОРТА ü Головные боли, связанные с гипотензией (появление гипертензии при развитии ХПН); ü Снижение слуха, в результате неврита слухового нерва ü диагностика: аудиографическое исследование (снижение слуха на высоких частотах по звуковоспринимающему сигналу) ü У части больных к 8 -12 годам развивается тугоухость, которая м. б. первым симптомом, чаще тугоухость возникает у мужчин. ü Аномалия зрения ( 1/4 больных СА) ü лентиконус (передний и задний); астигматизм; сферофакия; катаракта; миопия и др. ü Интеллект соответствует возрастным нормам; ü Раннее развитие ХПН; ü Лабораторные данные: Ранние признаки ü азотемия; дизэлектролитемия; снижения функции почек

Синдром Альпорта. Неспецифические гломерулярные изменения, атрофичные канальцы, лимфоцитарная инфильтрация интерстиция, пенистые клетки

1 2 5 6 Неспецифические изменения при синдроме Альпорта 1. Нормальный клубочек 2. Мезангиальная пролиферация, утолщение капиллярной стенки 3. Пенистые клетки в интерстиции 4. Сегментарный фибриноидный некроз 5. Сегментарный склероз 6. Эпителиальная пролиферация 3 4
, м. б. утолщена, иметь слоистую структуру")
Базальная мембрана клубочка: истончена (особенно Lamina densa), м. б. утолщена, иметь слоистую структуру и неровные контуры. Ultrastructural appearance of GBM in hereditary nephritis (Alport syndrome). The basement membrane Синдром Альпорта is thickened, layered, and irregular in contour. (специфичные (Magnification +<CS: Hidden>{times}</CS> 9000. ) изменения)
")
Синдром Альпорта (специфичные изменения)

ДИАГНОСТИКА СИНДРОМА АЛЬПОРТА Предположительный диагноз устанавливается на основании данных родословной Критерии диагностики: [Kashtan C. et al. , 1993] ü Гематурия или летальный исход от ХПН в семье; ü Гематурия и/или протеинурия в семье; ü У больного специфические изменения гломерулярных базальных мембран при электронной микроскопии биоптата; ü Снижение слуха по данным аудиографии; ü Врожденная патология зрения. Для установления диагноза необходимо наличие 3 из 5 критериев Для подтверждения диагноза: электронная микроскопия биоптата и ДНК-исследование (идентификация Х-хромосомы у родственников).

ДИФФЕРЕНЦИАЛЬНАЯ ДИАГНОСТИКА СИНДРОМА АЛЬПОРТА ü С гематурической формой гломерулонефрита: ü При ГН нет семейных случаев одинаковых почечных заболеваний, тугоухости, ХПН; ü При морфологическом исследовании выявляется иммунный вариант ГН; ü При электронной микроскопии (ЭМ) отсутствуют специфические признаки с-ма Альпорта. ü С метаболической нефропатией (оксалатно-кальциевой кристаллурией: ü В анамнезе указания на наличие МКБ; ü При морфологическом исследовании выявляется тубулоинтер- стициальный нефрит и обнаруживаются кристаллы оксалатов в канальцах; ü При ЭМ нет специфических изменений. ü С болезнью тонких мембран: ü при ЭМ - равномерное истончение мембран гломерул; ü характерен благоприятный прогноз.

ЛЕЧЕНИЕ СИНДРОМА АЛЬПОРТА Специфического лечения не существует. Цель лечения: замедлить прогрессирование снижения функции почек. ü Исключение занятий спортом; ü Сбалансированная диета; ü Избегать контактов с инфекционными больными; ü Вакцинация только по эпид. показаниям; ü Санация очагов инфекции; ü Витаминотерапия (vit гр. В), анаболические стероиды (ретаболил); ü Гипербарическая оксигенация. Радикальный метод лечения - трансплантация почки
 Описан Фанкони и соавторами в 1951 г. Нефронофтиз")
МЕДУЛЛЯРНАЯ КИСТОЗНАЯ БОЛЕЗНЬ (НЕФРОНОФТИЗ ФАНКОНИ) Описан Фанкони и соавторами в 1951 г. Нефронофтиз Фанкони - генетически детерминированное заболевание, передаю- щееся аутосомно -рецессивным путем, про- являющееся наличием медуллярного кис-тоза, который всегда сочетается с развитием почечной недостаточности, отставанием в росте, выраженной анемией.

ГЕНЕТИКА НЕФРОНОФТИЗА ФАНКОНИ ü Наследуется аутосомно-рециссивным путем. Возможны спорадические случаи, генетичес- кая природа которых не установлена. ü Ген нефронофтиза Фанкони находится на 2 -й хромосоме (2 q 12 -q 13). ü Продукт гена - нефроцистин [Hildebrant F. et al. , 1998]

КЛИНИЧЕСКАЯ КАРТИНА НЕФРОНОФТИЗА ФАНКОНИ Встречается одинаково у лиц обоего пола (особенно в семьях, где имеются родственные браки) ç Полиурия; ç Полидипсия; ç Анемия (не соответствует уровню азотемии); ç Задержка роста и развития, проявляющаяся после 6 лет; ç Электролитные нарушения: Са , Na , K. ç Небольшая протеинурия; ç Остеодистрофия (при прогрессировании заболевания); ç Артериальная гипертензия возникает при развитии ХПН; ç ХПН развивается на 2 -м десятилетии жизни.

МОРФОЛОГИЯ НЕФРОНОФТИЗА ФАНКОНИ ü Кистозные изменения в собирательных трубочках и дистальных канальцах; ü Вокруг кист скопления белка Тамма-Хорсфолла; ü При ЭМ: утолщение базальных мембран канальцев и собирательных трубочек, м. б. истончение и расслоение базальной мембраны канальцев ; ü При иммунофлюоресцентном исследовании: нет фиксации ни иммуноглобулинов, ни комплемента.

ДИАГНОСТИКА НЕФРОНОФТИЗА ФАНКОНИ ç Нарушение роста и развития + ç полиурия, полидипсия, гипостенурия, изостенурия, анемия; ç Морфологическое исследование: медуллярно- кистозная трансформация почек; ç Данные УЗИ и компьютерной томографии - это поздний диагноз ( на стадии почечной недостаточности). ç Семейный анамнез.

ЛЕЧЕНИЕ НЕФРОНОФТИЗА ФАНКОНИ Специфического лечения нет. ç Диета (д. б. калорийной, содержать аминокислот- ные добавки); ç Достаточный водный и солевой режим; ç Коррекция остеодистрофии (применение препаратов кальция и витамина Д); ç Борьба с гипокалиемией; ç Коррекция анемии; ç Трансплантация почки (радикальное лечение).

ВРОЖДЕННЫЙ НЕФРОТИЧЕСКИЙ СИНДРОМ ФИНСКОГО ТИПА Врожденный нефротический синдром финского типа - это аутосомно-рецессивно наследуемая патология, проявляющаяся с первых дней жизни ребенка тяжелым нефротическим синдромом с большой протеинурией и резкой гипопротеинемией. При естественном течении летальный исход наступает в течение 1 года либо в результате септических ос- ложнений, либо развития почечной недостаточности. Впервые заболевание было описано в 1966 г - R. Norio.

ГЕНЕТИКА ВРОЖДЕННОГО НЕФРОТИЧЕСКОГО СИНДРОМА ФИНСКОГО ТИПА ã Врожденный нефротический синдром финского типа наследуется аутосомно-рецессивным путем. ã Мутантный ген локализован на 19 q 13, этот ген NPHSI - кодирует трансмембранный протеин - нефрин, присущий подоцитам. Нефрин находится на подоцитах. Его недостаточность вызывает протеинурию [Tryggvason K. , 1998]. МОРФОЛОГИЯ При гистологическом исследовании выявляются: Микрокистоз проксимальных канальцев в кортико-медул- лярной зоне, мультигломерулярность , пролиферация мезангиальных клеток, фиброзные изменения.

КЛИНИЧЕСКАЯ КАРТИНА ВРОЖДЕННОГО НЕФРОТИЧЕСКОГО СИНДРОМА ФИНСКОГО ТИПА ã Тяжелое течение беременности, преждевременные роды, масса плаценты составляет более 1/4 - 1/2 массы новорожденного; ã ребенок рождается с выраженными отеками или они появляются к концу 1 -го месяца жизни; ã протеинурия достигает 10 г/сутки; ã в крови: гипоальбуминемия, гиперлипидемия; ã множественные стигмы дизэмбриогенеза; ã дистрофия; ã гнойные осложнения (в результате снижения иммунитета); ã м. б. тромбоэмболии; ã АД в норме или снижено; ã в амниотической жидкости и в крови беременных содержится альфа-фетопротеин [Theile U. et al. , 1982 ].

ДИАГНОСТИКА И ЛЕЧЕНИЕ ВРОЖДЕННОГО НЕФРОТИЧЕСКОГО СИНДРОМА ФИНСКОГО ТИПА Диагностика: ç Выяснение этнических корней; ç Исключение вторичного нефротического синдрома, связанного с внутриутробной инфекцией; семейного нефротического синдрома; ç Крупная плацента; ç Морфологическое исследование: микрокистоз проксимальных канальцев; ç Наличие в крови беременных альфа-фетопротеина, при обнару- жении которого рекомендуется прерывание беременности. Лечение: ç Высокобелковая и высококалорийная диета; ç Поддержание водно-электролитного режима до 10 -12 месяцев жизни ребенка; ç ГКС и цитостатики не эффективны; ç Нефрэктомия и трансплантация почки.

СЕМЕЙНЫЙ НЕФРОТИЧЕСКИЙ СИНДРОМ Нефротический синдром, возникающий не менее, чем у 2 -х членов одной семьи. ç Заболевание развивается в любом возрасте (но чаще в детском); ç Генетика: передается аутосомно-рецессивным путем, мутантный ген на 1 -й хромосоме с маркером D 1 S 416 на длинном плече хромосомы [Niaudet P. et al. , 1998]; ç Клиника: проявляется нефротическим синдромом (отеки, гипопротеинемия, гиперлипидемия, протеинурия); ç Морфология: от минимальных, мезангально-пролифе- ративных до фокально-сегментарных и диффузного склероза в гломерулах;

СЕМЕЙНЫЙ НЕФРОТИЧЕСКИЙ СИНДРОМ çДиагностика: Обнаружение нефротического синдрома у 2 -х и более членов семей; çЛечение: w При возникновении нефротического синдрома показана терапия ГКС ( преднизолон в дозе 2 мг/кг /сутки в течение 4 недель); w При гормонорезистентности - проведение нефробиопсии , для уточнения варианта гломерулонефрита и определения дальнейшей тактики лечения (присоединение цито - статиков).
 Диффузная ангиокератома - моногенно-насле- дуемая лизосомная болезнь, связанная с")
ДИФФУЗНАЯ АНГИОКЕРАТОМА ФАБРИ (БОЛЕЗНЬ АНДЕРСОНА-ФАБРИ) Диффузная ангиокератома - моногенно-насле- дуемая лизосомная болезнь, связанная с не- достаточностью лизосомного фермента альфа- галактозидазы-А-церамид-тригексозидазы, проявляющаяся поражением сосудов кожи, глаз, почек и других органов. Дефект фермента состоит в нарушении метаболизма гликосфинголипидов, которые откладываются в сосудах различных органов. Встречаемость 1: 40 000 новорожденных

КЛИНИЧЕСКАЯ КАРТИНА БОЛЕЗНИ ФАБРИ ç Телеангиэктазии: на коже туловища, бедер, ягодиц в виде темно-красных точек и пятен, симметричных и не исчезающих при надавливании; ç Могут быть боли в суставах, животе; ç Поражение сердечно-сосудистой системы (вследствие отложения сфинголипидов в миокарде, аорте, коронарных сосудах); ç Поражение глаз: дистрофия роговицы; ç Неврологическая симптоматика: от эпизодов парестезий до цереброваскулярных изменений (ишемический или геморрагический инсульты); ç Нефропатия : протеинурия , эритроцитурия , гипостенурия и признаки почечного несахарного диабета ( полиурия , полидипсия, никтурия, дегидратация, гипернатриемия и др. ). Течение заболевание постепенное с исходом в ХПН.

БОЛЕЗНЬ ФАБРИ Генетика: Заболевание сцеплено с полом, встречает- ся только у мужчин. Мутантный ген расположен на длинном плече Х-хромосомы в зоне Xq 13 - 22 DXS 17 и DXYS 1 [Mac Dermot K. et al. , 1987]. Морфология: Отложение гликосфинголипидов в почках приводят к тубулоинтерстициальным изменениям, и постепенно прогрессируя, приводят к нефросклерозу. Диагностика: Клиническая картина и определение уровня церамидтригексозидазы в крови и моче, перинатальная диагностика: определение активности альфа-Д-галактозидазы в амниоцитах и лейкоцитах. Лечение: Симптоматическая терапия. При развитии ХПН - заместительная.

Light microscopic appearance of Fabry disease. The visceral epithelial cells are enlarged and finely vacuolated <IT+>(arrows)<IT->. (Periodic acid. Ц<CS: Hidden>{endash}</CS>Schiff; magnification +<CS: Hidden>{times}</CS> 500. )

Electron microscopic appearance of Fabry disease. Numerous zebra bodies are present in visceral epithelial cells. Note similar structures also in an endothelial cell of the glomerular capillary in the lower left corner. (Magnification +<CS: Hidden>{times}</CS> 5500. )
 Наследственная остеоониходисплазия - нас- ледственное заболевание, характеризующееся гипоплазией или отсутствием")
ОСТЕООНИХОДИСПЛАЗИЯ (синдром nail-patella) Наследственная остеоониходисплазия - нас- ледственное заболевание, характеризующееся гипоплазией или отсутствием надколенников, дистрофией ногтей и поражением почек. s Заболевание впервые было описано в 1897 г английским врачом E. Little. s В 1971 г. М. Ben- Bassat et al. описали ультраструктуру базальной мембраны клубочков при остеоонихо - дисплазии. Частота встречаемости около 22 случаев на 1 млн. [Kemper R. , 1994] Генетика: Наследуется по аутосомно-доминантному типу. Локус гена находится на длинном плече 9 -й хромосомы (9 q 34).
 Классическая тетрада: r Дисплазия ногтей (80 -90 % больных)")
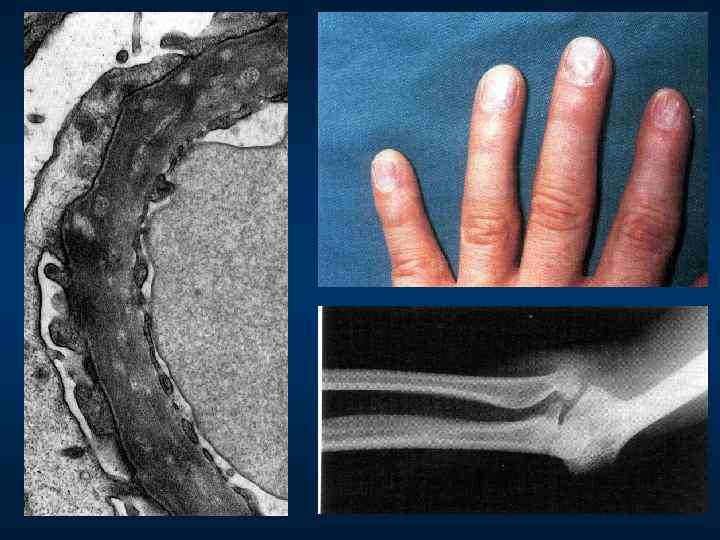
КЛИНИЧЕСКАЯ КАРТИНА ОСТЕООНИХОДИСПЛАЗИИ (синдрома nail-patella) Классическая тетрада: r Дисплазия ногтей (80 -90 % больных) - симметрична, чаще поражаются пальцы рук (большой и указательный) r. Отсутствие или дистрофия ногтевых пластинок; r. Разнообразие патологии ногтей: продольные бороздки, ложкообразные ногти, обесцвечивание; r Гипо- или аплазия надколенника (60 %) r Дислокация головки лучевой кости r iliac horns у 80% (подвздошный рог - двусторонние симметричные костные выросты на передней поверхности крыльев повздошных костей) Патогномоничный признак
 r Поражение глаз (гетерохромия радужки). r Артериальная гипертензия")
КЛИНИЧЕСКАЯ КАРТИНА ОСТЕООНИХОДИСПЛАЗИИ (синдрома nail-patella) r Поражение глаз (гетерохромия радужки). r Артериальная гипертензия (редко). r Нефропатия развивается у 30 % носителей гена: rизолированная протеинурия, м. б. нефротический с-м; rреже микрогематурия. r Около 1/3 входит в терминальную ХПН, а у 2/3 больных течение доброкачественное.
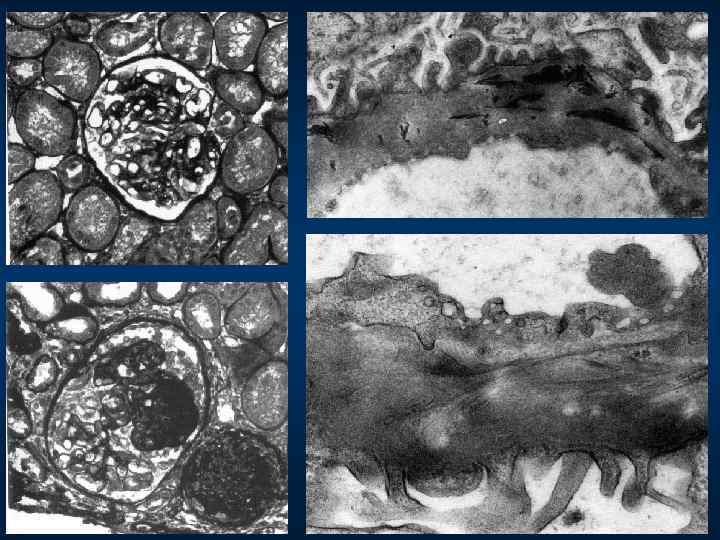
 Световая микроскопия: ü У больных с сохранной функцией почек")
МОРФОЛОГИЯ ОСТЕООНИХОДИСПЛАЗИИ (синдрома nail-patella) Световая микроскопия: ü У больных с сохранной функцией почек и min протеинурией изменений в клубочках не обнаруживается; ü У больных со сниженной функцией почек и нарастающей протеинурией изменения в клубочках соответствуют фокально- сегментарному или диффузному гломерулосклерозу. Электронная микроскопия: ü Неравномерное утолщение гломерулярной базальной мембраны с мелкими очагами разрежения (картина «изъеденности молью» ГБМ). Иммуногистохимическое исследование: ü У большинства больных свечения нет, но при наличии гломеруло- склероза имеются депозиты Ig. M и C 3, которые располагаются как вдоль ГБМ, так и в мезангии.
 üВыявление гипоплазии надколенников и локтевых суставов; üПодвздошный рог; üСемейный")
ДИАГНОСТИКА ОСТЕООНИХОДИСПЛАЗИИ (синдрома nail-patella) üВыявление гипоплазии надколенников и локтевых суставов; üПодвздошный рог; üСемейный анамнез; üБиопсия почки с ЭМ ЛЕЧЕНИЕ ОСТЕООНИХОДИСПЛАЗИИ (синдрома nail-patella) ü В связи с тем, что в основе заболевания лежит патология коллагена, то лечения иммунодепрес- сантами нецелесообразно; ü Показана симптоматическая терапия (коррекция АГ, при нефротическом синдроме - диуретики) ü Радикальное лечение - трансплантация почки.
Наследственные нефропатии.ppt