НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ.ppt
- Количество слайдов: 29



НАСЛЕДСТВЕННЫЕ И ВРОЖДЕННЫЕ ЗАБОЛЕВАНИЯ

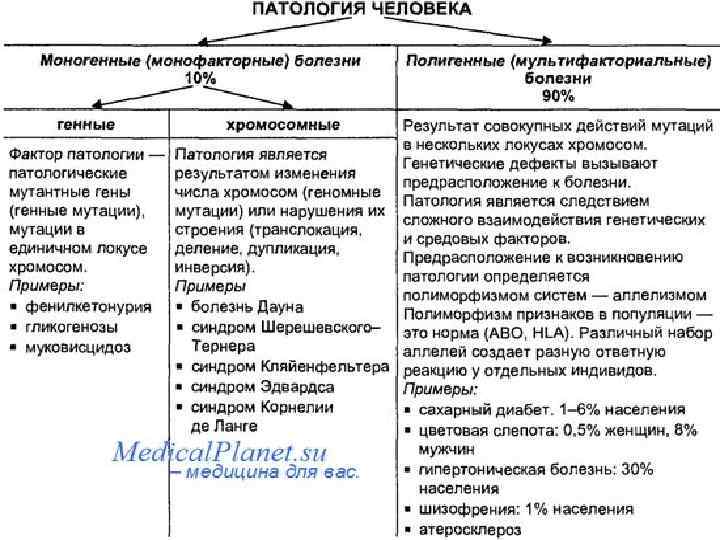
НАСЛЕДСТВЕННЫЕ ФАКТОРЫ И ФАКТОРЫ СРЕДЫ С генетической точки зрения все болезни в зависимости от роли наследственных и средовых факторов в их развитии можно подразделить на 3 группы. 1. Наследственные болезни. Фенотипическое проявление мутации как этиологического фактора практически не зависит от среды; последняя может только изменять выраженность симптомов и тяжесть течения болезни. Это генные и хромосомные наследственные болезни (гемофилия, фенилкетонурия, муковисцидоз, болезнь Дауна и др. ). 2. Болезни с наследственной предрасположенностью. Их в свою очередь можно подразделить еще на два вида. Болезни, наследственность при которых является этиологическим фактором, но для их проявления необходимо действие соответствующего фактора внешней среды (например, подагра, диабет). Болезни, этиологическими факторами при которых являются средовые влияния, однако частота возникновения и тяжесть течения болезней зависят от наследственной предрасположенности. К таким болезням относятся атеросклероз, гипертоническая болезнь, язвенная болезнь, псориаз и др. 3. Болезни, в происхождении которых наследственность не играет роли. Это, например, травмы, ожоги, инфекционные болезни. Генетические факторы в этом случае могут влиять только на течение патологических процессов (скорость регенерации, выздоровления, компенсации функций).

НАСЛЕДСТВЕННЫЕ И ВРОЖДЕННЫЕ БОЛЕЗНИ Наследственные болезни — заболевания человека, обусловленные хромосомными и генными мутациями. Нередко ошибочно термины «наследственная болезнь» и «врожденная болезнь» употребляются как синонимы, однако врожденными болезнями называют те заболевания, которые имеются уже при рождении ребенка и могут быть обусловлены как наследственными, так и экзогенными факторами. Таковы, например, пороки развития, связанные с воздействием на эмбрион и плод ионизирующего излучения, химических соединений, лекарственных средств, принимаемых матерью, а также внутриутробных инфекций. Однако далеко не все Н. б. относят к врожденным, поскольку многие из них проявляются уже после периода новорожденности (например, хорея Гентингтона и болезнь Альцгеймера клинически обнаруживаются после 40 лет).

БОЛЕЗНЬ ГЕНТИНГТОНА поражает специфические области мозга. Наиболее заметные ранние изменения затрагивают область базальных ганглиев. Накапливающиеся повреждения в этой области приводят к беспорядочным движениям. АЛЬЦГЕЙМЕРА БОЛЕЗНЬ, дегенеративное заболевание головного мозга, проявляющееся прогрессирующим снижением интеллекта.

Клиническая классификация Н. б. построена по органному и системному принципам. Согласно этой классификации, выделяют Н. б, нервной, эндокринной, дыхательной и сердечно-сосудистой систем, печени, желудочно-кишечного тракта, почек, системы крови, кожи, уха, носа, глаз и др. Такая классификация в значительной степени условна, т. к. большинство Н. б. характеризуется вовлечением в патологический процесс нескольких органов. В зависимости от того, где локализован патологический (мутантный) ген – в аутосоме или в половой хромосоме – и каковы его взаимоотношения с нормальным аллелем, т. е. является ли мутация доминантной (нормальный ген подавляется патологическим) или рецессивной (патологический ген подавляется нормальным), различают следующие основные типы наследования: аутосомнодоминантный, аутосомно-рецессивный и сцепленный с полом (или ограниченный полом). Тип наследования устанавливается путём анализа родословной. При составлении учитываются распространение в семье изучаемого заболевания и родственного отношения между больными. Построение и анализ родословной составляют предмет клинико-генеалогического исследования.

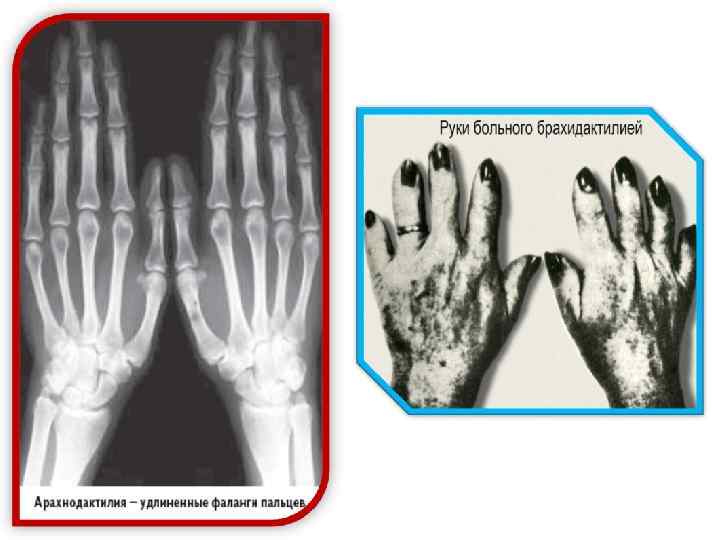
При заболеваниях, наследуемых по аутосомно-доминантному типу, мутантный ген проявляется уже в гетерозиготном состоянии; больные мальчики и девочки рождаются с одинаковой частотой; по крайней мере один из родителей больного также болен. По аутосомно-доминантному типу наследуются, например, арахнодактилия, ахондроплазия, брахидактилия, гипербилирубинемия, нейрофиброматоз, пельгеровская аномалия лейкоцитов, полидактилия, эктопия хрусталика и др.

Ахондроплази я Полидактили я эктопия хрусталика

При заболеваниях, наследуемых по аутосомно-рецессивному типу, мутантный ген проявляется лишь в гомозиготном состоянии; больные мальчики и девочки рождаются с одинаковой частотой; родители больных фенотипически здоровы, но являются гетерозиготными носителями мутантного гена. Вероятность рождения больных детей возрастает в случае кровного родства родителей.

Если один из родителей гомозиготен по патологическому рецессивному гену, а другой является его гетерозиготным носителем, то в половине случаев дети могут оказаться больными, и создаётся впечатление наследования заболевания по доминантному типу. Такое явление носит название псевдодоминирования. От истинного доминирования оно отличается тем, что больные с рецессивной мутацией в браке со здоровыми людьми всегда будут давать здоровое потомство, а здоровые в браке с гетерозиготными носителями с определенной частотой (25%) будут иметь больных детей. По аутосомно-рецессивному типу наследуются агаммаглобулинемия, алкаптонурия, альбинизм, амавротическая идиотия, гепатоцеребральная дистрофия, дистония мышечная деформирующая, муковисцидоз, серповидноклеточная анемия и др.

Серповидно-клеточная анемия это наследственная гемоглобинопатия, связанная с таким нарушением строения белка гемоглобина, при котором он приобретает особое кристаллическое строение - так называемый гемоглобин S. Альбинизм (от лат. albus — белый), отсутствие нормальной пигментации: у животных и людей — кожи, волос, радужной оболочки глаза.

Муковисцидоз - самое распространенное наследственное заболевание, при котором поражаются все органы, которые выделяют секреты (слизь). Это бронхолегочная система, поджелудочная железа, печень, потовые железы и др. Из-за дефекта (мутации) гена секреты во всех органах вязкие, густые, поэтому их выделение затруднено. В легких из-за вязкого, часто гнойного секрета, трудноотделяемого и скапливающегося в бронхах, довольно быстро, развиваются воспалительные процессы повторные бронхиты и/или пневмонии с постепенным формированием хронического бронхолегочного процесса.

Алкаптонурия - это наследственное заболевание, в результате которого моча при выходе становится черной. Охроноз - образование черного пигмента в соединительных тканях человека, например, в тканях хряща и кожи, что также является одной из характерных алкаптонурия - нарушение обмена тирозина вследствие пониженной активности фермента черт этого заболевания. Такая сине-черная пигментация обычно гомогентизиназы и накоплением в тканях организма гомотентизиновой кислоты; Также появляется после 30 лет. гомогентизиновая кислота выделяется с мочой, поэтому моча становится темной на воздухе.

Из заболеваний, сцепленных с полом, для клиники особое значение имеют болезни, обусловленные рецессивными мутациями в Х-хромосоме (Х-хромосомный тип). Женщины с такого типа мутацией фенотипически здоровы, т. к. рецессивному патологическому гену противостоит у них нормальный аллель другой Х-хромосомы. У мужчин же мутантный ген представлен в единственном числе и определяет патологию фенотипа. При болезнях, передающихся по Ххромосомному типу, действие мутантного гена проявляется только у гетерогаметного пола (т. е. у мужчин); в отягощенных семьях заболевает половина сыновей, а половина дочерей – носители мутантного гена; родители клинически здоровы. Болезнь часто обнаруживается у сыновей сестёр больного или у его двоюродных братьев по материнской линии. Больной отец не передаёт дефектный ген сыновьям. По Х-хромосомному типу наследуются гемофилия А, гемофилия В, периодический паралич, пигментный ретинит, фосфат-диабет, цветовая слепота и др.

Наследование по Х-хромосомному типу
 среди")
Пигментный ретинит (точнее, пигментная дистрофия сетчатки, так как воспаления при этом заболевании нет) среди наследственных болезней сетчатки глаза, приводящих к слепоте, в настоящее время - широко распространенное заболевание, занимающее по количеству больных одно из ведущих мест. В основе заболевания лежит врожденный дефект генетического кода. Первоначально страдают только палочки, но затем повреждаются и колбочки. Гемофилия — наследственное заболевание, связанное с нарушением процессом свёртывания крови; при этом заболевании возникают кровоизлияния в суставы, мышцы и внутренние органы. Резко возрастает опасность гибели пациента от кровоизлияния в мозг и другие жизненно важные органы. Гемофилия А (рецессивная мутация в X-хромосоме) вызвана генетическим дефектом, отсутствием в крови необходимого белка— так называемого фактора VIII (антигемофильного глобулина).

Хромосомные болезни Это группа болезней, в основе развития которых лежат нарушения числа или структуры хромосом , возникающие в гаметах родителей или на ранних стадиях дробления зиготы (оплодотворенной яйцеклетки). Классификация Х. б. основана на типах мутаций вовлеченных в них хромосом. Мутации в половых клетках приводят к развитию полных форм Х. б. , при которых все клетки организма имеют одну и ту же хромосомную аномалию. В наст. время описано 2 варианта нарушений числа хромосомных наборов тетраплоидия и триплодия. Другая группа синдромов обусловлена нарушениями числа отдельных хромосом – трисомиями (когда имеется добавочная хромосома в диплоидном наборе) или моносомия (одна из хромосом отсутствует ). Моносомии аутосом несовместимы с жизнью. Трисомии - более часто встречающаяся паталогия у человека. Ряд хромосомных болезней связаны с нарушением числа половых хромосом.

Наиболее часто у человека встречаются трисомии по 21 -й, 13 -й и 18 -й паре хромосом. Синдром Дауна - синдром трисомии 21 - самая частая форма хромосомной патологии у человека (1: 750). Цитогенетически синдром Дауна представлен простой трисомией(94%случаев), транслокационной формой (4%) или мозаицизмом (2% случаев). У мальчиков и девочек патология встречается одинаково часто. Дети с синдромом Дауна чаще рождаются у пожилых родителей. Если возраст матери 35 -46 лет, то вероятность рождения больного ребенка возрастает до 4, 1%. Возможность возникновения повторного случая заболевания в семье с трисомией хромосомы 21 составляет 1 -2% (с возрастом матери риск увеличивается). Три четверти всех случаев транслокаций при болезни Дауна обусловлены мутацией de novo. 25% случаев транслокации носят семейный характер, при этом возвратный риск гораздо выше (до 15%) и во многом зависит от того, кто из родителей несет симметричную транслокацию и какая из хромосом вовлечена.

Для больных характерны округлой формы голова с уплощенным затылком, узкий лоб, широкое, плоское лицо, запавшая спинка носа, косой (монголоидный) разрез глазных щелей, пятна Брушфильда (светлые пятна на радужке), толстые губы, утолщенный язык с глубокими бороздами, выступающий изо рта, маленькие, округлой формы, низко расположенные ушные раковины со свисающим завитком, недоразвитая верхняя челюсть, высокое нёбо, неправильный рост зубов, короткая шея. С самого раннего возраста отмечается отставание в умственном развитии. Среднее значение IQ составляет 50, но чаще встречается умеренная задержка умственного развития. Средняя продолжительность жизни при синдроме Дауна значительно ниже (36 лет), чем в популяции.
 - синдром трисомии 13 - встречается с частотой 1: 6000. Имеются")
Синдром Патау (СП) - синдром трисомии 13 - встречается с частотой 1: 6000. Имеются два цитогенетических варианта синдрома Патау: простая трисомия и робертсоновская транслокация. 75% случаев трисомии хромосомы 13 обусловлено появлением дополнительной хромосомы 13. Между частотой возникновения синдрома Патау и возрастом матери прослеживается зависимость, хотя и менее строгая, чем в случае болезни Дауна. 25% случаев СП - следствие транслокации с вовлечением хромосом 13 -й пары, в том числе в трех из четырех таких случаев мутация de novo. В четверти случаев транслокация с вовлечением хромосом 13 -й пары имеет наследственный характер с возвратным риском 14%. Дети с синдромом Патау рождаются с массой тела ниже нормы (2500 г). У 80% новорожденных встречаются пороки развития сердца: дефекты межжелудочковой и межпредсердной перегородок, транспозиции сосудов и др. Наблюдаются фиброкистозные изменения поджелудочной железы, добавочные селезенки, эмбриональная пупочная грыжа. Почки увеличены, имеют повышенную дольчатость и кисты в корковом слое, выявляются пороки развития половых органов. Для СП характерна задержка умственного развития.

У детей с синдромом Патау выявляются умеренная микроцефалия, нарушение развития различных отделов ЦНС, низкий скошенный лоб, суженные глазные щели, расстояние между которыми уменьшено, микрофтальмия и колобома, помутнение роговицы, запавшая переносица, широкое основание носа, деформированные ушные раковины, расщелина верхней губы и нёба, полидактилия, флексорное положение кистей, короткая шея. Большинство больных с синдромом Патау (98%) умирают в возрасте до года, оставшиеся в живых страдают глубокой идиотией.
- синдром трисомии 18. Дети с трисомией 18 чаще рождаются у пожилых")
Синдром Эдвардса (СЭ)- синдром трисомии 18. Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии хромосомы 21 и 13. Для женщин старше 45 лет риск родить больного ребенка составляет 0, 7%. У девочек встречается значительно чаще, чем у мальчиков, что связано, возможно, с большей жизнестойкостью женского организма. Дети с трисомией 18 рождаются с низким весом (в среднем 2177 г). Наиболее часто отмечаются аномалии мозгового и лицевого черепа. Нижняя челюсть и ротовое отверстие маленькие. Глазные щели узкие и короткие. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко, несколько вытянуты в горизонтальной плоскости. Мочка, а часто и козелок отсутствуют. Грудина короткая, из-за чего межреберные промежутки уменьшены и грудная клетка шире и короче нормальной. В 80% случаев наблюдается аномальное развитие стопы: пятка резко выступает, свод провисает (стопа-качалка). У всех больных наблюдаются гипоплазия мозжечка и мозолистого тела, изменения структур олив, выраженная умственная отсталость, снижение мышечного тонуса.

Продолжительность жизни детей с синдромом Эдвардса невелика: 60% детей умирают в возрасте до 3 мес, до года доживает лишь один ребенок из. десяти; оставшиеся в живых - глубокие олигофрены.

АНОМАЛИИ СОЧЕТАНИЯ ПОЛОВЫХ ХРОМОСОМ При нормальном течении мейоза у женского организма образуется один тип гамет, содержащих Х-хромосому. Однако при нерасхождении половых хромосом могут образовываться еще два типа гамет - XX и 0 (не содержащая половых хромосом). У мужского организма в норме образуется два типа гамет, содержащих Х- и Y-хромосомы. При нерасхождении половых хромосом возможны варианты гамет XY и 0. Рассмотрим возможные комбинации половых хромосом в зиготе у человека (их 12) и проанализируем каждый вариант. XX- нормальный женский организм. XXX- синдром трисомии X. Кариотип 47, ХХХ. В настоящее время имеются описания тетра-и пентосомий X. Трисомия по Х-хромосоме возникает в результате нерасхождения половых хромосом в мейозе или при первом делении зиготы. Синдрому полисемии X присущ значительный полиморфизм. Женский организм с мужеподобным телосложением. Могут быть недоразвиты первичные и вторичные половые признаки. В 75% случаев у больных наблюдается умеренная степень умственной отсталости. У некоторых из них нарушена функция яичников (вторичная аменорея, дисменорея, ранняя менопауза). Иногда такие женщины могут иметь детей. Повышен риск заболевания шизофренией. С увеличением числа дополнительных Х-хромосом нарастает степень отклонения от нормы.
. Частота встречаемости 1: 2000 -1: 3000. Кариотип 45, Х.")
ХО синдром Шерешевского-Тернера (моносомия X). Частота встречаемости 1: 2000 -1: 3000. Кариотип 45, Х. У 55% девочек с этим синдромом обнаруживается кариотип 45, X, у 25% - изменение структуры одной из Ххромосом. В 15% случаев выявляется мозаичность в виде двух или более клеточных линий, одна из которых имеет кариотип 45, X, а другая представлена кариотипами 46, XX или 46, XY. Третья клеточная линия наиболее часто представлена кариотипом 45, Х, 46^ХХ, 47, ХХХ. Риск наследования синдрома составляет 1 случай на 5000 новорожденных. Фенотип женский. У новорожденных и детей грудного возраста отмечаются признаки дисплазии: короткая шея с избытком кожи и крыловидными складками, лимфатический отек стоп, голеней, кистей рук и предплечий, низкорослость. В подростковом возрасте выявляются отставание в росте (рост взрослых 135 -145 см) и в развитии вторичных половых признаков.

XY- нормальный мужской организм. XXY и XXXY- синдром Клайнфелтера. Частота встречаемости 1: 500. Кариотип 47, XXY у 80% мальчиков с синдромом Клайнфелтера. Возвратный риск для синдрома Клайнфелтера не превышает общепопу-ляционные показатели и составляет 1 случай на 2000 живорожденных детей. Фенотип мужской. Клиника отличается широким разнообразием и неспецифичностью проявлений. У мальчиков с этим синдромом рост превышает средние показатели, характерные для данной семьи, у них длинные конечности, женский тип телосложения, гинекомастия. Слабо развит волосяной покров, снижен интеллект. Вследствие недоразвития семенников слабо выражены первичные и вторичные половые признаки, нарушено течение сперматогенеза.

СИНДРОМЫ ЧАСТИЧНЫХ АНЕУПЛОИДИЙ Помимо полных трисомий и моносомий известны синдромы, связанные с частичными трисомиями и моносомиями практически по любой хромосоме. Однако эти синдромы встречаются реже одного случая на 100 000 рождений. Синдром трисомий по короткому плечу 9 -й хромосомы (9 р+) - наиболее частая форма частичных трисомий. Для больных с трисомией 9 р+ характерны умственная отсталость, задержка роста, микроцефалия, антимонголоидный разрез глазных щелей, глубоко посаженные глаза, опущенные уголки рта, нос с характерным округлым кончиком, низко расположенные оттопыренные ушные раковины, недоразвитие ногтей и дистальных фаланг пальцев рук. Синдромы частичных моносомий распространены примерно с такой же частотой, как и синдромы частичных трисомий. Наиболее известные из них - синдромы Вольфа-Хиршхорна, кошачьего крика, Орбели.
 обусловлен делецией короткого плеча хромосомы 4. Популяционная частота заболевания -")
Синдром Вольфа-Хиршхорна (4 р-) обусловлен делецией короткого плеча хромосомы 4. Популяционная частота заболевания - около 1 случая на 100 000. Дети с синдромом Вольфа- Хиршхорна обычно рождаются у молодых родителей, доношенные, но со значительно сниженным весом. У них наблюдаются умеренно выраженная микроцефалия, клювовидный нос, выступающее надпереносье, деформированные, низко расположенные ушные раковины, вертикальные складки кожи впереди ушных раковин, гипотония мышц, значительное снижение реакции на внешние раздражения, судорожные припадки. Отмечаются также расщелины верхней губы и нёба, деформации стоп, аномалии глазных яблок, эпикант и маленький рот с опущенными уголками. Большинство детей с синдромом 4 р- умирает на 1 -м году жизни.
НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ.ppt