ГЕНЕТИКА))).pptx
- Количество слайдов: 29



Наследственные болезни, их профилактика и лечение ОМ 051 01: Омаров Алишер проверила: Таракова К. А

Содержание: § Понятие «генетика» § Понятие наследственные заболевания § Хромосомные наследственные заболевания § Генные болезни § Подгруппы наследственных заболеваний § Моногенные наследственные заболевания § Полигенные наследственные заболевания § Хромосомные аберрации § Синдром Дауна § Синдром Рейна § Постановка диагноз § Профилактика врожденных и наследственных заболеваний § Виды наследственных заболеваний § Заключение § Список используемой литературы

В 1905 году английский натуралист Уильям Бэтсон ввёл в употребление название новой научной дисциплины- генетика. В 1909 году датским ботаником Вильгельмом Йоханнсеном введён в употребление термин «ген» . Гены являются отмеченными участками ДНК или РНК – молекулы в которой закодирована вся генетическая информация. Генетика - ( от греч. - происходящий от кого-то) – наука Генетика о законах и механизмах наследственности и изменчивости.

Наследственное заболевание — заболевания, возникновение и развитие которых связано с дефектами в программном аппарате клеток, передаваемыми по наследству через гаметы. В основе наследственных заболеваний лежат нарушения (мутации) наследственной информации: хромосомные, генные и митохондриальные. От наследственных заболеваний следует отличать врождённые заболевания, которые обусловлены внутриутробными повреждениями, вызванными, например, инфекцией (сифилис или токсоплазмоз) или воздействием иных повреждающих факторов на плод во время беременности. Многие генетически обусловленные заболевания проявляются спустя некоторое время. Так, при хорее Гентингтона дефектный ген обычно проявляет себя только на третьемчетвёртом десятилетии жизни, проявление признаков спинальной мускульной атрофии (СМА) наблюдается в возрасте от 6 месяцев до 4— 50 лет (в зависимости от формы заболевания).

Хромосомные наследственные болезни Все хромосомные болезни с нарушением состояния хромосом можно разделить на две большие группы: изменение числа хромосом при сохранении структуры последних (геномные мутации); изменением структуры хромосомы (хромосомные мутации). У человека описаны все известные виды мутаций обоих типов. Численные нарушения могут состоять в изменении плоидности хромосомного набора и в отклонении числа хромосом от диплоидного по каждой их паре в сторону уменьшения (моносомия) или увеличения (полисемия). Геномные мутации составляют основную массу хромосомных болезней. Полные моносомии наблюдаются по Х-хромосоме, приводя к развитию синдрома Шэрешевского-Тернера.

Генные болезни делятся на две большие группы: болезни с выясненным первичным биохимическим дефектом болезни с невыясненным первичным биохимическим дефектом. К первой группе принадлежат наследственные болезни обмена веществ, биосинтеза белка, ферментов.

Среди наследственных болезней, развивающихся в результате мутаций, выделяют три подгруппы: § моногенные наследственные заболевания; § полигенные наследственные болезни ; § хромосомные аберрации.

Моногенные наследственные заболевания Моногенные болезни наследуются в соответствии с законами классической генетики Менделя. Генеалогическое исследование позволяет выявить один из трёх типов наследования: аутосомно-доминантный, аутосомнорецессивный и сцепленное с полом наследование. Это наиболее широкая группа наследственных заболеваний. В настоящее время описано более 4000 вариантов моногенных наследственных болезней. (например, фенилкетонурия, гемофилия частота серповидноклеточной анемии — 1/6000). Широкий круг моногенных болезней образуют наследственные нарушения обмена веществ, возникновение которых связано с мутацией генов. Дефект митохондриальных генов, передаваемых преимущественно яйцеклеткой.

Полигенные наследственные болезни Полигенные болезни характеризовались как болезни с наследственной предрасположенностью. Однако сейчас о них идёт речь как о мультифакториальных заболеваниях с аддитивно-полигенным наследованием с пороговым эффектом. К этим заболеваниям относятся такие болезни как рак, сахарный диабет, шизофрения, эпилепсия, ишемическая болезнь сердца, гипертензия и многие другие.

Хромосомные аберрации Хромосомные болезни обусловлены грубым нарушением наследственного аппарата — изменением числа и структуры хромосом. Причина в частности — алкогольная интоксикация родителей при зачатии ( «пьяные дети» ). Сюда относятся синдромы Дауна, Клайнфельтера, Шерешевского — Тернера, Эдвардса, «кошачьего крика» и другие.

Синдром Ангельмана — генетическая аномалия. Для него характерны задержка психического развития, нарушения сна, припадки, хаотические движения (особенно рук), частый смех или улыбки. При синдроме Ангельмана отсутствуют некоторые гены из 15 -й хромосомы (в большинстве случаев — частичная делеция либо мутация 15 хромосомы). При синдроме Ангельмана страдает материнская хромосома; в случае повреждения отцовской хромосомы возникает синдром Прадера-Вилли. Диагностика Синдром диагностируется путем генетического анализа (15 хромосома), рекомендуемого для новорожденных с пониженным мышечным тонусом (гипотонусом), отставанием в развитии общей моторики и в развитии речи. Родители и врачи должны обратить внимание на случаи мелкого тремора, хаотические, порывистые движения конечностей, походку с негнущимися ногами; в ряде случаев специфическое выражение лица, слишком частый смех. Возможные методы анализа: процесс флуоресцентной гибридизации in situ, метилирование ДНК в области 15 q 11 -q 13, анализ мутации импринтингового центра, анализ прямой мутации гена UBE 3 A. Существует небольшая группа людей, у которых результаты всех вышеописанных анализов в норме, однако наблюдаются все внешние проявления синдрома Ангельмана. Наука пока не дает ответ на вопрос, как это возможно. Лечение Синдром Ангельмана является врожденной генетической аномалией и, следовательно, не может быть излечен. Однако некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии). Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом. Нарушения сна корректируются назначением легких снотворных. Д-р Вагстафф (США) считает, что назначение 0. 3 мг мелатонина за 30 минут-1 час перед сном улучшает сон пациентов с синдромом Ангельмана. Нарушения стула регулируются назначением легких слабительных.

Синдрома Шэрешевского-Тернера Синдром развивается при полной Х-моносомии, когда все клетки или их большинство имеют хромосомный набор. Клиническими проявлениями этого синдрома являются: §отсутствие у женщин обычных вторичных половых признаков; §низкий рост, §сближенные соски, §нарушения скелета, §бесплодие, §разнообразные пороки внутренних органов.

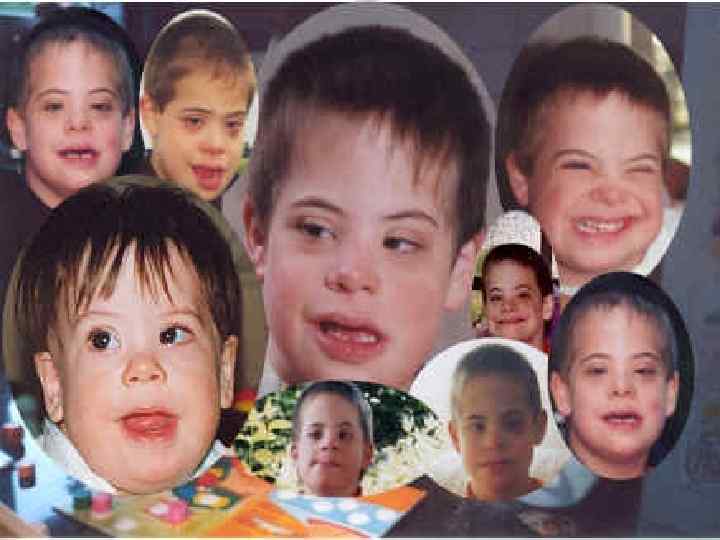
Синдром Дауна Болезнь Дауна, названа так по имени врача, впервые описавшего ее в 1866 году, вызванная не расхождением хромосом. Симптомы болезни Дауна: §пониженная сопротивляемость болезням, §врожденные сердечные аномалии, §короткое коренастое туловище и толстая шея, § характерные складки кожи над внутренними углами глаз. Синдром Дауна и другие сходные аномалии чаще встречаются у детей, рожденных немолодыми женщинами. Точная причина этого неизвестна, но она как-то связана с возрастом яйцеклеток матери. Структурные перестройки хромосом, какого бы вида они ни были, вызывают нарушения развития организма вследствие или недостатка части материала по данной хромосоме (частичная моносомия) или его избытка (частичная трисомия). Пример можно привести Х-полисомию при отсутствии У-хромосомы. Такие организмы имеют хромосомный набор 47, XXX и хотя внешне женщины выглядят нормально и они плодовиты, но у них отмечается умственная отсталость.

Методы лечения Формы синдрома Дауна Примерно в 91 % случаев возникает ненаследственный вариант болезни — простая полная трисомия 21 хромосомы, обусловленная нерасхождением хромосом во время мейоза. Примерно у 5 % больных наблюдается мозаицизм (не все клетки содержат лишнюю хромосому). В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21 й хромосомы. Как правило, такие транслокации возникают в результате слияния центромеры 21 -й хромосомы и другой акроцентрической хромосомы. Фенотип больных определяется трисомией 21 q 22. Повторный риск рождения ребенка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1 % при обычной трисомии у ребенка. Информация об этих редких формах значима для родителей, так как риск рождения других детей с синдромом Дауна различен при разных формах. Тем не менее, для понимания развития детей эти различия не так важны. Хотя профессионалы склонны считать, что дети с мозаичной формой синдрома Дауна отстают в своём развитии меньше детей с другими формами этого синдрома, достаточно убедительных сравнительных исследований на эту тему пока нет

Синдром Дауна
 (синонимы: болезнь кошачьего крика, синдром Лежена по имени описавшего")
Синдром кошачьего крика (Cri-Du-Chat Syndrome) (синонимы: болезнь кошачьего крика, синдром Лежена по имени описавшего в 1963 г. французского ученого). Генетика Кариотип 46 XX или ХУ, 5 р-. Диагноз подтверждается кариологическим исследованием с применением одного из методов идентификации хромосом. Хромосомно синдром кошачьего крика объясняется частичной моносомией; он развивается при делеции (с утратой от трети до половины, реже полная утрата) короткого плеча пятой хромосомы. Для развития клинической картины синдрома имеет значение не величина утраченного участка, а конкретный незначительный фрагмент хромосомы. Изредка отмечается мозаицизм по делеции или образование кольцевой хромосомы-5. Частота синдрома примерно 1: 45000. Соотношение полов М 1 : Ж 1, 3. Клиническая картина синдрома и продолжительность жизни людей с этим синдромом довольно сильно варьирует по сочетанию врождённых пороков развития органов. Лечение симптоматическое. Показаны средства, стимулирующие психомоторное развитие, лечебный массаж и гимнастика.


Нейропатология Болезнь характеризуется потерей нейронов и синаптических связей в коре головного мозга и определённых субкортикальных областях. Гибель клеток приводит к выраженной атрофии поражённых участков, в том числе к дегенерации височных и теменной долей, участков фронтальной коры и поясной извилины. Как амилоидные бляшки, так и нейрофибриллярные клубки хорошо заметны под микроскопом при посмертном анализе образцов мозга больных. Бляшки представляют собой плотные, в большинстве случаев нерастворимые отложения бета-амилоида и клеточного материала внутри и снаружи нейронов. Внутри нервных клеток они растут, образуя нерастворимые закрученные сплетения волокон, часто называемые клубками. У многих пожилых людей в мозге образуется некоторое количество бляшек и клубков, однако при болезни Альцгеймера их больше в определённых участках мозга, таких как височные доли.

Синдром Клайнфельтера Синдром Клайнфельтера — генетическое заболевание. Клиника описана в 1942 году в работе Фуллера Олбрайта и Гарри Клайнфельтера. Генетической особенностью этого синдрома является разнообразие цитогенетических вариантов и их сочетаний (мозаицизм). Обнаружено несколько типов полисомии по хромосомам X и Y у лиц мужского пола: 47, XXY; 47, XYY; 48, XXXY; 48, XYYY; 48 XXYY; 49 XXXXY; 49 XXXYY. Наиболее распространен синдром Клайнфельтера (XXY). Общая частота его колеблется в пределах 1 на 500 -700 новорожденных мальчиков [1]. Для мужчин с синдромом Клайнфельтера характерны высокий рост, длинные конечности и относительно короткое туловище, евнухоидизм, бесплодие, гинекомастия, повышенное выделение женских половых гормонов, склонность к ожирению. Лишняя Х хромосома обусловливает различные нарушения психики. Больные очень внушаемы, вялы, апатичны, безынициативны, у них часто отмечается умственная отсталость (обычно дебильность). Нередко возникают параноидные, галлюцинаторно-параноидные, депрессивные психозы и навязчивые состояния, иногда наблюдаются антисоциальное поведение и алкоголизм. Клиническая картина начинает проявляться у мальчиков в период полового созревания. Диагностировать синдром Клайнфельтера, особенно у взрослых лиц, нетрудно, особенно при кариотипировании лишней Х хромосомы. Лечение проводится мужскими половыми гормонами для коррекции вторичных половых признаков

Дальтонизм Дальтони зм , цветовая слепота — наследственная, реже приобретённая особенность зрения, выражающаяся в неспособности различать один или несколько цветов. Названа в честь Джона Дальтона, который впервые описал один из видов цветовой слепоты на основании собственных ощущений, в 1794 году.

Диагностика Характер цветового восприятия определяется на специальных полихроматических таблицах Рабкина. В наборе имеется 27 цветных листов — таблиц, изображение на которых (обычно цифры) состоит из множества цветных кружков и точек, имеющих одинаковую яркость, но несколько различных по цвету. Человеку с частичной или полной цветовой слепотой (дальтонику), не различающему некоторые цвета на рисунке, таблица кажется однородной. Человек с нормальным цветовосприятием (нормальный трихромат) способен различить цифры или геометрические фигуры, составленные из кружков одного цвета. Дихроматы: различают слепых на красный цвет (протанопия), у которых воспринимаемый спектр укорочен с красного конца, и слепых на зелёный цвет (дейтеранопия). При протанопии красный цвет воспринимается более тёмным, смешивается с тёмно-зелёным, тёмно-коричневым, а зелёный — со светло-серым, светло-жёлтым, светло-коричневым. При дейтеранопии зелёный цвет смешивается со светло-оранжевым, светло-розовым, а красный — со светло-зелёным, светлокоричневым Лечение дальтонизма В настоящее время дальтонизм неизлечим. Однако разработана технология лечения дальтонизма за счет внедрения в клетки сетчатки недостающих генов с помощью методов генной инженерии с использованием в качестве вектора вирусных частиц. В 2009 г. в Nature появилась публикация об успешном испытании этой технологии на обезьянах, многие из которых от природы плохо различают цвета.

Многие люди с нарушением цветовосприятия не увидят на этом изображении число 83 • Многие люди с нарушением цветовосприятия не увидят на этом изображении число 56
 — расстройство")
Синдром туретта. Синдром Туре тта (болезнь Туретта, синдром Жиль де ла Туретта) — расстройство центральной нервной системы, в виде сочетания тикообразных подёргиваний мышц лица, шеи и плечевого пояса, непроизвольных движений губ и языка с частым покашливанием и сплевыванием, копролалией. Болезнь может иметь наследственный характер. Синдром обусловлен изменением структуры полосатого тела мозга, но может носить и функциональный характер. Впервые описан Жоржем Жилем де ла Туреттом в 1885 году. Наблюдается у 0, 05 % населения, в основном у детей. В 3 раза чаще у мужчин (из них 95 % в возрасте 2 -5 лет). Также может наблюдаться у людей в возрасте от 15 до 30 лет. Непроизвольные движения людей страдающих синдромом Туретта однотипны в своих проявлениях (резкие, быстрые, порывистые). Наряду с двигательным тиками проявляются также и звуковые симптомы: произнесение отдельных звуков и нечленораздельных слов является характерной особенностью синдрома. В некоторых случаях может отмечаться так называемая эхолалия, то есть навязчивое повторение слов, слогов или звуков. В половине случаев при синдроме Туретта возможны вокальные тики с неприличными ругательными словами, а также неприличные жесты. Больные могут нанести себе травмы, поскольку не способны контролировать внезапные движения.

Заключение остановки иагноза бщего линического п д о к обследования любого больного: ü четко поставлен диагноз ненаследственного заболевания; ü четко поставлен диагноз наследственного заболевания; ü имеется подозрение, что основная или сопутствующая болезнь является наследственной.
 диагностика гетерозиготного носительства;")
Можно выделить несколько организационных форм профилактики врожденных и наследственных болезней: 1) диагностика гетерозиготного носительства; 2) пренатальная (дородовая) диагностика и внутриутробная коррекция патологии плода; 3) преклиническая (досимптоматическая) диагностика; 4) медико-генетическое консультирование; 5) ранняя постнатальная диагностика (идентификация) наследственных болезней, поддающихся лечению; 6) диспансеризация семей с наследственной патологией; 7) контроль мутагенности факторов окружающей среды и гигиеническая регламентация (профилактика новых мутаций); 8) пропаганда медико-генетических знаний.

Каждый человек ответственен за наследственное благополучие своих детей, при этом важным фактором является его биологическое образование, так как знания в области аномалии, физиологии, генетики предостерегут человека от совершения ошибок. Нужно вести здоровый образ жизни, чтобы за наши ошибки не страдали наши дети…

Список используемой литературы: http: //ru. wikipedia. org www. Krugosvet. ru http: //www. lechenieblog. ru/archives/24 Ю. Я. Керкис. Лечение и предупреждение некоторых наследственных болезней человека. А. О. Рувинский. Наследственная изменчивость человека. Лильин Е. Т. и др. Генетика для врачей М. , “Медицина”, 1990; Фогель Ф. Генетика человека, М. , “Мир”, 1989.

СПАСИБО ЗА ВНИМАНИЕ
ГЕНЕТИКА))).pptx