Патология обмена веществ.ppt
- Количество слайдов: 45



Нарушения обмена веществ профессор Т. Л. Настаушева


Моногенно обусловленные заболевания с патологией гена. Картировано более 933 заболеваний обмена в-в. Передача аутосомно-рецессивно или сцепл. с Х хром. Полигенные заболевания (наследственная предрасп-сть, ф-ры среды) n Только среда (ожоги, травмы, отравления) n

Символы родословных Мужчина, женщина Брак Родственный брак Дважды женат Близнецы Пол не установлен Выкидыш, аборт Больные Носители непостоянного рецессивного гена Обладатели рудиментарных признаков Умершие

Патологический ген Вирьирует по экспрессивности – не все клинические признаки выражены у больного. n Неполная пенетрантность – болезнь не проявляет себя, но при передаче детям – развивается. n

Болезни обмена веществ I. Нарушение обмена аминокислот II. Нарушение обмена углеводов III. Нарушение обмена липидов IV. Нарушение пигментного обмена V. Нарушение минерального обмена VI. Нарушение обмена витаминов VII. Нарушение обмена пуринов VIII. Другие дефекты обмена

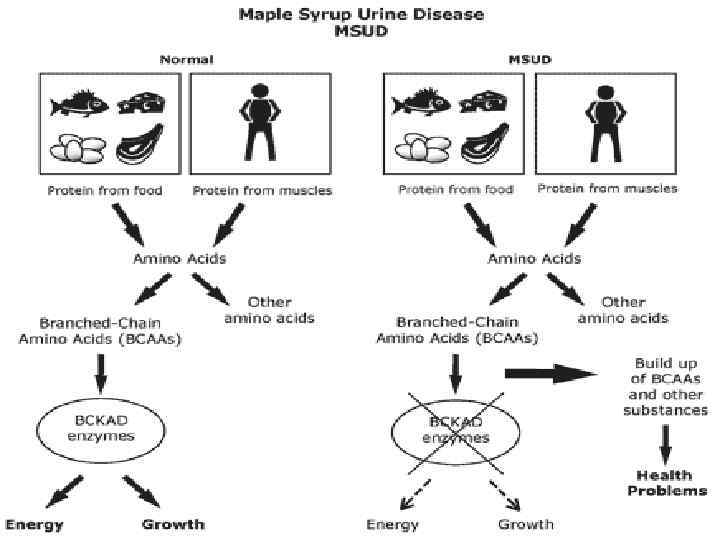
Клиника болезни обмена веществ Новорожденные диарея; u летаргия; u трудность кормления; u вздутие живота; u рвота; u поражение глаз; u t°; u поражение нервной системы; u судороги; u стигмы дизэмбриогенеза; u желтуха; u гепатоспленомегалия. u u Ацидоз с кетозом. u Запах мочи. u Гипогликемия.
")
Характер запаха при обменных заболеваниях (аминокислот)

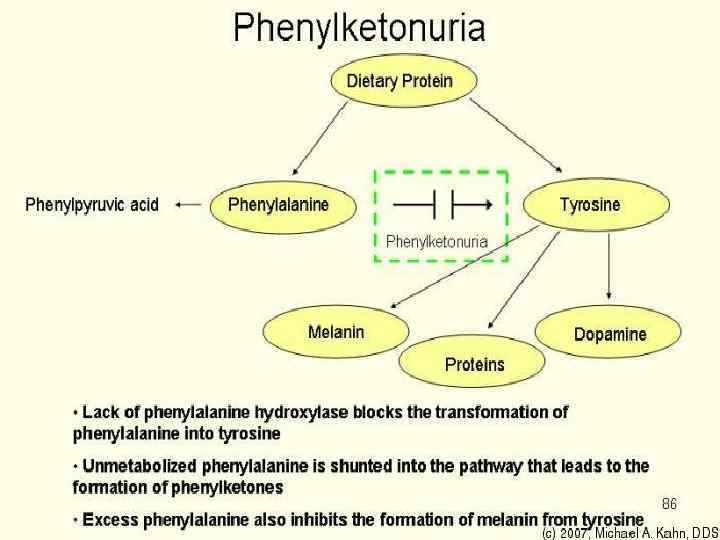
Нарушение обмена АК аутос. -рецесс. с ↑ в крови и моче II. с ↑ в мочи III. нарушение транспорта Большая часть АК внутри клеток, значит. концентр. в гол. мозге. ФКУ- ↓ ФА гидроксидазы → нарушение гидроксилирования ФА → <тирозина n >ФА до 4 мг% Умст. отсталость, судороги у25%, аллергическая сыпь, немотивированные вспышки гнева. I.

Нарушения обмена АК Тирозинемия-аутос. рец. ↓ ацетоуксусной гидроксилазы-> тирозина. С-мы в 6 мес. Печен. нед-ть, желтуха, рвота, гипотр. , кровоточивость, гипотрофия, смерть в 90%. Алкаптонурия- аутос. рец. ↓ β-оксидазы → >гомогентизиновой кислоты (черный цвет мочи). Дегенер. костей, хрящей, пигментация хрящей.
 дефект аланин/глиоксилат аминотрансферазы, аутос. -рец. n n Нефрокальциноз в раннем возрасте,")
Оксалоз (гипероксалурия первичная) дефект аланин/глиоксилат аминотрансферазы, аутос. -рец. n n Нефрокальциноз в раннем возрасте, ИМП, 50%больных –ХПН. Повыш. оксалатов в моче (выше 0, 5 ммоль/1, 73 м 2) Цистиноз –полиурия, полидипсия, с-м Фанкони на 1 -м году. Отложение кристал. цистина в орг. ХПН у многих детей.

Липидозы u β-галактозидаза u гексозаминидаза Тея-Сакса u сфингомиелиназа Нимана-Пика u β-глюкозидаза Гарольд-Финкель Гоше

Синдром Тея -Сакса
 накопление сфинголипидов в клетках u u Начало: первый год")
Клиника болезни Нимана-Пика (младенческий тип) накопление сфинголипидов в клетках u u Начало: первый год жизни. Смерть: в 2– 3 года. печень> селезенка > сетчатка – вишнево-красные пятна неврологические изменения – трудности вскармливания апатия глухота + слепота + костный мозг — пенистые клетки кровь — ↑ холестерина β- ЛП
 (3. 01. 90 г. – 4. 06. 00 г. )")
Adam Michael Ward (NPD) (3. 01. 90 г. – 4. 06. 00 г. ) Три месяца 2 года

3 года Возникают проблемы с речью 4 года Занятия с логопедом, перелом руки при падении с велосипеда

4 года Хорошо конструирует из кубиков, но появляются проблемы в обучении 6 лет Задержка роста и моторного развития, проблемы с поведением в детском саду, большой эпи-припадок госпитализация, диагноз: Болезнь Нимана-Пика Тип С

7 лет Переведен на специальное обучение, проблемы с фиксацией взора, утратил речевые навыки 7 лет Частота судорожных приступов увеличилась

8 лет Ходит только с поддержкой, внимание не удерживает, не говорит, появились проблемы с глотанием, питание через зонд 9 лет Перемещается только на коляске, не глотает, зондовое питание, энурез, энкопрез, полная атония мышц рук Адам умер 4 июня 2000 года в возрасте 10 лет

Болезнь Гоше Описана в 1882 г. 1 на 40000 до 1 на 450 n Анемия, >печени и селезенки, тромбоцитопения n Патология костей: атрофия, склероз, некроз n Задержка роста и полового развития n Диагностика –изм. фермента β глюкозидазы, молекулярногенетич. анализ n

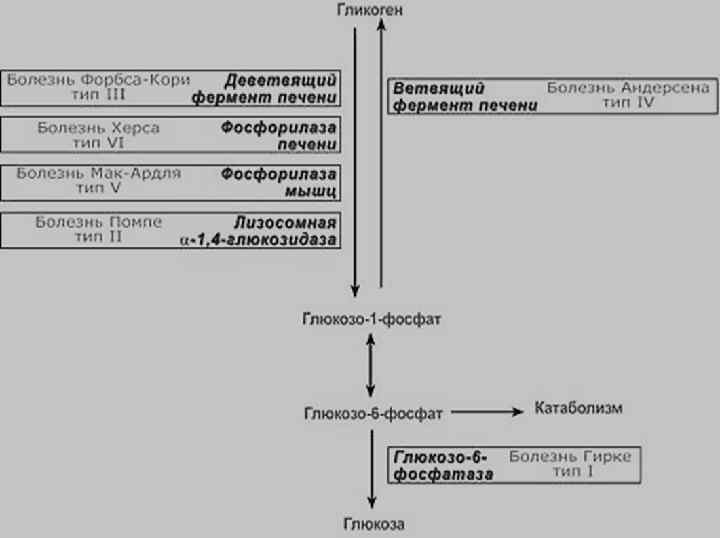
ГЛИКОГЕНОЗЫ n n n Наследственные болезни обмена полисахаридов Нарушение синтеза или распада гликогена на простые сахара Нормальный и аномальный гликоген одновременно накапливаются в клетках печени и других органах и тканях

Гликогенозы -12 типов u Глюкозо-6 -фосфат u Кислая мальтаза u Амило-1, 6 -глюкозидаза тип I (Гирке) тип II (Помпе) тип III (Кори)
. Гипогликемия у новорожденных может привести")
Клиника гликогенозов § гипогликемия (рвота, судороги, потеря сознания, кома). Гипогликемия у новорожденных может привести к синдрому внезапной смерти § Для дифференциальной д-ки с гиперинсулинемией – проба с глюкагоном § Гипотрофия, лицо «китайской куклы» , увеличение печени Течение болезни зависит от места депонирования гликогена: печень (цирроз печени), почки (почечная недостаточность), мышечная ткань (гипотония, сердечная недостаточность). Прогноз зависит от типа болезни

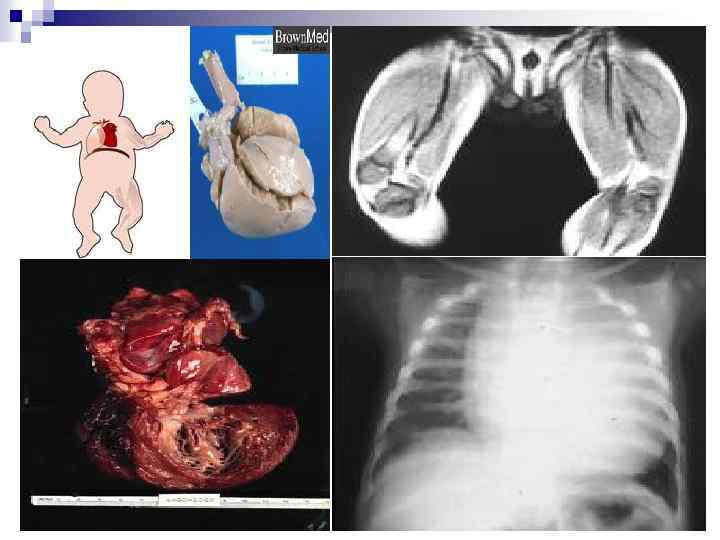
БОЛЕЗНЬ ПОМПЕ

§ § § § § Аутосомно-рецессивный тип наследования 1: 40 -300 тыс. Всего в мире 10 000 тыс. больных Симптомы проявляются с первых дней жизни- 6 месяцев Дефект фермента найден в печени, почках, селезенке, мышцах, нервной ткани, лейкоцитах Клиника: расстройство дыхания, беспокойство или адинамия, отсутствие аппетита, задержка роста, мышечная гипотония Объективно: увеличение размеров ♥, печени, почек, селезенки, ♥ шаровидной формы, в связи с гипертрофией миокарда, изменения ЭКГ Часто возникают гипостатические пневмонии, бронхиты, ателектазы легких, миодистрофия, гипорефлексия, спастические параличи Мышечная форма гликогеноза II типа возникает только в мышцах при дефиците кислой α-1, 4 -глюкозидазы, болезнь проявляется в более поздние сроки и по клинической картине напоминает миопатию
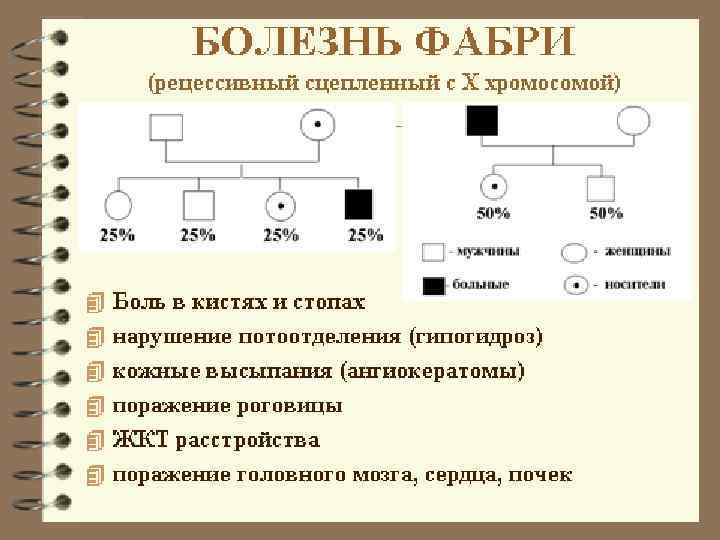

Болезнь Фабри n Женщины –носители, но м. б. клинические проявления. Накопление гликосфинголипидов (полисахариды и липиды) в эндотелиоцитах. С-мы заболевания с 10 лет в среднем. Смерть от инсультов, инфарктов, ХПН.

Болезнь Фабри
 -8 типов.")
Мукополисахаридозы Дефект гидролазы, нерасщ. полимерных углеводов (ГАГгепарансульфат, дерматансульфат, хонд роитинсульф. кератансульф. ) -8 типов. n 1 тип-Гурлера, 2 -Гунтера, 3 -Санфилиппо n 4, 6, 7 типы –нет отставания умств. развития n
 Задержка умственного развития u Грубые черты лица u Дизостоз u ↑ печени,")
Мукополисахаридоз (МПС) Задержка умственного развития u Грубые черты лица u Дизостоз u ↑ печени, селезенки u Нарушение сердечно-сосудистой системы u поражение глаз u глухота u продолжительность жизни 5 -50 лет u ↑ ГАГ в моче u

1 год 2 года 4 года Ребенок с мукополисахаридозом IIтипа 6 лет
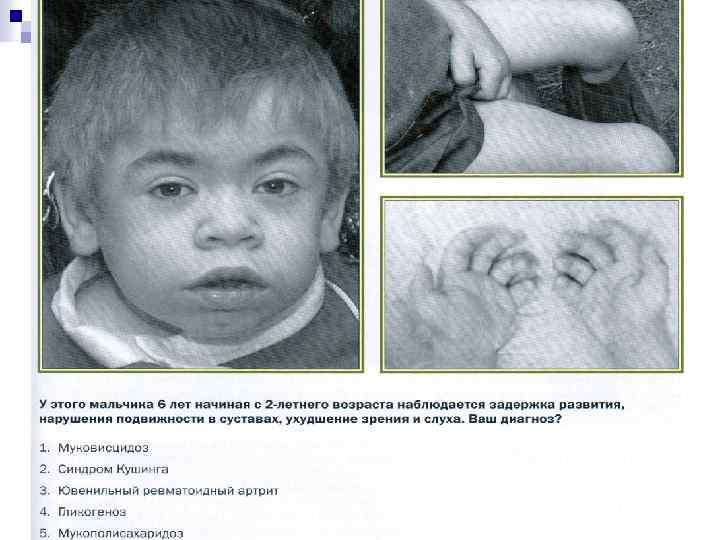

n Hurler's Syndrome n Morquio's Syndrome

Диагностика болезней обмена веществ Антенатальная – α-фетопротеин, УЗИ, генотип, биопсия кожи, кровь. II. Неонатальная – скрининг массовый и селективный. III. Постнатальная – родственные браки, аномалии, стигмы, частые пневмонии, осмотр окулиста, невролога. I. u u Специфические клетки, Биохимические изменения, ↓ ферментов Нанобиочипы (ФКУ).

Лечение болезней обмена веществ Нарушение обмена АК n 1 Симптоматическое: диета, витамины, ми кроэл. n ФКУ-белковые гидролизаты, уменьш ФА берлофен, лофенолак, норфенолак, арфенолак, тетрафен. n Цистиноз: Н 20, ощелачивание, цистеамин (растворяет цистин) n Оксалоз: Н 20, фосфаты, Магний, В 6, диета орган. щавелевой к-ты)
 в/в")
Лечение липидозов Ограничение в диете жира n Болезнь Гоше: заместительная терапия –Церезим (илиглюцераза) в/в 30 -60 ед. 1 р/2 недели. Уменьшение клинич. проявлений, норм. гематологических измен. n

Лечение гликогенозов § Цель лечения — предупредить тяжелую гипогликемию § Диета - богатая белками и углеводами § Питание должно быть частым (каждые 4 ч) § Белки служат источником аминокислот — субстратов глюконеогенеза, уменьшают углеводную нагрузку, приводящую к лактатацидозу § Диета предотвращает гипогликемию и кетоацидоз натощак, способствует ускорению роста Предпринимаются попытки введения больным недостающих ферментов (Enzyme Replacement Therapy (ERT)
 Myozyme® Genzyme Corp. USA Цена курса: 300.")
n n n Болезнь Помпе (Лиозим, Миозим) Myozyme® Genzyme Corp. USA Цена курса: 300. 000$/год для взрослого и 100. 000$/год для ребенка Применение: «разрушает мышцы» и увеличивает постепенно размер камер сердца. Получают 900 больных в мире (США, Бельгия, Нидерланды) Разрешен FDA для лечение больных старше 8 лет По данным «Forbes» - 5 место в рейтинге самых дорогих лекарств мира
 n Мукополисахаридоз 1 типа Гунтер – альбуразим")
Заместительная терапия Болезнь Фабри – фабразим (fabrazyme) n Мукополисахаридоз 1 типа Гунтер – альбуразим (alburazyme) n

Спасибо за внимание.
Патология обмена веществ.ppt