НАРУШЕНИЯ ЛИПИДНОГО ОБМЕНА.ppt
- Количество слайдов: 27



НАРУШЕНИЯ ЛИПИДНОГО ОБМЕНА

ЛИПИДЫ - группа различных по химическому строению органических веществ биогенного происхождения, которые не растворимы в воде, а растворимы о органических растворителях – эфире, хлороформе, ацетоне, бензоле и др.
")
Триацилглицерины Холестерин Фосфолипиды Сфинголипиды Гликолипиды (ганглиозиды, цереброзиды)

ФУНКЦИИ ЛИПИДОВ Энергетическая Структурная, Эмульгирующая, Терморегулирующая, Механическая, Гормональная, Витаминная, Электроизолирующая
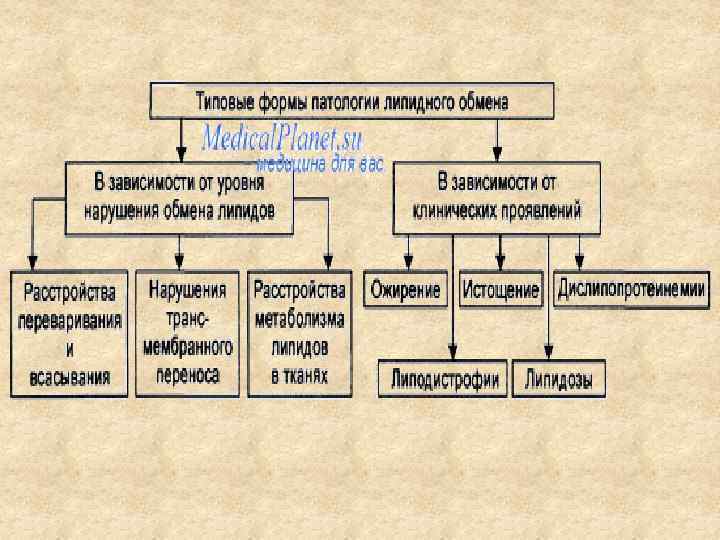

Переваривание и всасывание жиров Роль желчи 1. Наряду с панкреатическим соком нейтрализация кислого химуса, поступающего из желудка. 2. Усиливает перистальтику кишечника. 3. Обеспечивает эмульгирование для последующего воздействия липазой, 4. Активирует липазу. 5. Обеспечивает всасывание жирорастворимых витаминов. 6. Экскреция избытка ХС, желчных пигментов, креатинина, металлов Zn, Cu, Hg, лекарств.

Нарушения переваривания и всасывания жиров. Стеаторея Поступившие с пищей жиры, если они приняты в умеренном количестве (не более 100 150 г), усваиваются почти полностью, и при нормальном пищеварении кал содержит не более 5% жиров. При нарушениях переваривания и всасывания липидов наблюдается избыток липидов в кале – стеаторея (жирный стул). Различают 3 типа стеаторей: 1. Панкреатогенная – при дефиците панкреатической липазы; причины хронический панкреатит, врожденнная гипоплазия поджелудочной железы, врожденный или приобретенный дефицит панкреатической липазы, муковисцидоз. В кале желчные пигменты, понижено содержание свободных жирных кислот и повышено ТАГ. 2. Гепатогенная при закупорке желчных протоков (в результате сужения желчного протока желчными камнями или сдавления опухолью); В кале больных отсутствуют желчные пигменты, высоко содержание ТАГ, жирных кислот и мыл. 3. Энтерогенная – при снижении метаболической активности слизистой кишечника; Характерен сдвиг р. Н кала в кислую сторону, рост содержания в кале жирных кислот.

Желчекаменная болезнь — заболевание, связанное с нарушением обменных процессов, проявляющееся образованием камней в желчном пузыре и желчных протоках. Факторы, предрасполагающие к желчнокаменной болезни — ожирение, сахарный диабет, беременность, наследственная предрасположенность, воспалительные процессы в желчном пузыре и желчных путях. Механизм образования камней связан с выпадением в осадок составных частей желчи в результате изменения ее состава и застоя в желчном пузыре. Желчные камни по своему составу разделяются на 3 группы: однородные камни (холестериновые, билирубиновые, известковые), смешанные камни (наиболее часто встречающиеся), состоящие из холестерина, билирубина и солей кальция, сложные камни. Биохимический анализ крови показывает повышение содержания билирубина (связанной фракции), при камнях общего желчного протока с желтухой, повышение активности щелочной фосфатазы, АЛТ, ГГТП и уровня холестерина в крови.

Последствия нарушения всасывания жира При отсутствии их в пище у животных в эксперименте развиваются хронические заболевания кожи (в виде некротических очагов). Полное отсутствие ненасыщенных высших жирных кислот в пище человека также может быть причиной более или менее серьезных расстройств обмена. Незаменимые жирные кислоты: линолевая, линоленовая, арахидоновая кислоты Выделение жира с мочой — липурия — может возникнуть после приема с пищей очень больших количеств жира, при переломах трубчатых костей, сопровождающихся размозжением костного мозга, травме обширных участков жировой ткани, при липоидном нефрозе.

Нарушение перехода жира из крови в ткани Общие липиды 3, 5 ‑ 8, 0 г/л повышаются при эссенциальной гиперлипемии, ожирении, атеросклерозе, сахарном диабете, гипотиреозе, панкреатите, злоупотреблении алкоголем, билиарном циррозе печени, остром гепатите, при остром и хроническом нефрите. Триацилглицерины 0, 55 2, 1 ммоль/л Первичные гипертриглицеринемии обнаруживаются при гиперлипопротеинемиях типа, при генетическом дефиците фермента липопротеинлипазы. Вторичное повышение наблюдается при нефротическом синдроме, переломах костей, подагре, сахарном диабете (транспортная липемия), острых и хронических панкреатитах, беременности, билиарном циррозе, беременности, алкоголизме. Снижение при гипотиреозе, гиперпаратиреозе, синдроме мальабсорбции.

Холестерин 3, 0 ‑ 6, 0 ммоль/л повышение содержания холестерина отмечается при гиперлипопротеинемии, заболеваниях печени (внутри‑ и внепеченочный холестаз), заболеваниях почек, злокачественных опухолях поджелудочной железы, гипотиреозе, заболеваниях сердечно‑сосудистой системы, беременности, сахарном диабете, болезни Иценко Кушинга, ожирении. Снижение гипертиреозе, циррозе печени, злокачественных опухолях печени, гипопротеинемии и аb‑липопротеинемии. Спинномозговая жидкость Накопление холестерина выявляется при менингите, опухоли или абсцессе мозга, кровоизлияниях в мозг, при рассеянном склерозе. Снижение при церебральной и кортикальной атрофии.

Фосфолипиды 1, 98 ‑ 4, 71 ммоль/л повышение при сахарном диабете, механической желтухе, нефрозе, хроническом нефрите, гиперлипемии. Снижение истощении, лихорадочных состояниях, гипертиреозе. При повышенном поступлении жира с пищей алиментарная гиперлипемия. Гиперлипемия может быть результатом усиления мобилизации жира из депо — транспортная гиперлипемия Ретенционная гиперлипемия (retentio — задерживать) — результат задержки перехода нейтральных жиров из крови в ткани, возникает преимущественно при уменьшении в крови содержания альбумина и фактора просветления (ФП), специфической липопротеидлипазы.
![Дислипопротеинемии (греч. dys + липопротеин [ы] + греч. haima кровь) количественные и качественные нарушения](https://present5.com/presentation/-44807045_133383175/image-13.jpg "Дислипопротеинемии (греч. dys + липопротеин [ы] + греч. haima кровь) количественные и качественные нарушения")
Дислипопротеинемии (греч. dys + липопротеин [ы] + греч. haima кровь) количественные и качественные нарушения состава липопротеинов крови. Основными Д. являются гипер и гиполипопротеинемии — соответственно повышенное или пониженное содержание в плазме крови липопротеинов одного или нескольких классов; алипопротеинемии — отсутствие липопротеинов одного из классов в плазме крови. Чаще встречаются гиперлипопротеинемии. Прогноз особенно неблагоприятен при гиперлипопротеинемиях II—V типа в случае поздней диагностики и развития сосудистых осложнений, а также при алипопротеинемиях.

Нарушения b-окисления жирных кислот Ackee «ямайская рвотная болезнь» Нарушения b-окисления жирных кислот - более 20 генетических болезней: üДефект переносчика длинноцепочечных жирных кислот üНарушения b-окисления длинноцепочечных жирных кислот. üНарушения окисления среднецепочечных жирных кислот. üНарушения окисления кроткоцепочечных жирных кислот üНарушения кетогенеза. üЗамедление окисления жирных кислот с нечетным числом атомов углерода, накопление токсичного метилмалоната – наблюдается при дефиците В 12 Биохимические методы диагностики Определение карнитина, ацилкарнитинов, жирных кислот плазмы

ПАТОЛОГИЧЕСКАЯ АКТИВАЦИЯ КЕТОГЕНЕЗА Кетогенез нормальный метаболический процесс, происходящий в митохондриях печени и производящий альтернативные глюкозе субстраты окисления. а) В норме суммарная концентрация кетоновых тел 0, 2 0, 6 м. М/л, б) Основные причины кетоза: сахарный диабет, голодание, несбалансированное питание, токсикозы беременности, желудочно кишечные расстройства у детей, почечная глюкозурия. в) Механизм развития кетоза нарушение принципа жиры сгорают в пламени углеводов). г) Опасность кетоза: кетоацидоз, мембранотропный эффект.

Избыточное накопление жира в жировой ткани Ожире ние — отложение жира, увеличение массы тела за счёт жировой ткани. Жировая ткань может отлагаться как в местах физиологических отложений, так и в области молочных желёз, бёдер, живота. В настоящее время ожирение рассматривается как хроническое обменное заболевание, возникающее в любом возрасте, проявляющееся избыточным увеличением массы тела преимущественно за счёт чрезмерного накопления жировой ткани, сопровождающееся увеличением случаев общей заболеваемости и смертности населения.

Жировая инфильтрация печени Жировая дистрофия печени — хроническое заболевание печени, характеризующееся жировой дистрофией печеночных клеток. Развивается под воздействием алкоголя, токсических веществ (медикаментов), при сахарном диабете, анемиях, заболеваниях легких, тяжелых панкреатитах и энтеритах, неполноценном питании, ожирении. • Избыточное поступление в печень свободных жирных кислот (СЖК); • Снижение скорости b–окисления СЖК в митохондриях гепатоцитов; • Избыточное образование и всасывание СЖК в кишечнике; • Снижение синтеза липопротеинов разной плотности в самой печени; • Функциональной печеночной недостаточности При жировом гепатозе отмечается увеличение содержания в сыворотке крови ГГТП (возможно связано с употреблением алкоголя). Активность сывороточных трансаминаз и ЩФ повышена, уровень билирубина, альбумина и протромбина обычно нормальные.
 — патологическое состояние, характеризующееся зачастую общим отсутствием объёма жировой ткани в")
Липодистрофия (жировая дистрофия) — патологическое состояние, характеризующееся зачастую общим отсутствием объёма жировой ткани в подкожной клетчатке. Врождённые липодистрофии Приобретённые липодистрофии Инсулиновые липодистрофии; ВИЧ ассоциированные липодистрофии;
 наследственные заболевания нервной системы, обусловленные расстройством обмена липидов. В основе патогенеза липидозов")
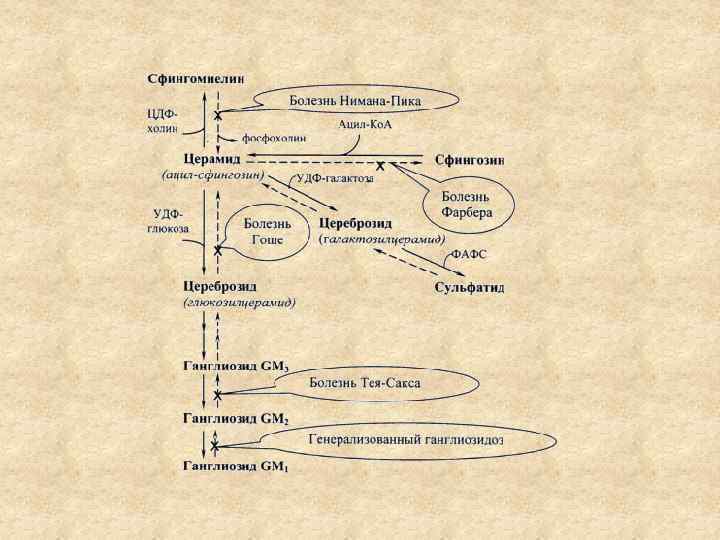
Липидозы (липоидозы) наследственные заболевания нервной системы, обусловленные расстройством обмена липидов. В основе патогенеза липидозов генетически детерминированные разнообразные ферментативные дефекты. К болезням накопления (внутриклеточным липидозам) относятся амавротическая идиотия, болезнь Ниманна Пика, болезнь Гоше и болезнь Рефсума Обширный класс наследственных ферментопатий, или, иначе говоря, лизосомные болезни накопления (ЛБН), включает около 40 нозологических единиц и характеризуется генетической гетерогенностью и выраженным клиническим полиморфизмом. Практически все ЛБН имеют приводят к ранней инвалидизации и преждевременной смерти.

Семейная амавротическая идиотия системное заболе вание, которое сопровождается типичными изменениями со стороны глаз, особенно в области желтого пятна, ведущими к слепоте; умствен ной отсталостью и прогрессирующей слабостью мускулатуры. Различают ганглиозидозы и цероид липофусцинозы. Диагноз необходимы офтальмологические и биохимические исследования. Выявление на глазном дне ярко красного участка в области макулы, имеющего округлую форму и слегка выступающего над поверхностью сетчатки (симптом «вишневой косточки» ) При биохимическом исследовании • диагноз ганглиозидозов устанавливается на основании выявления дефекта галактозидаз в лейкоцитах крови, фибробластах кожи. • цероид липофусцинозы диагностируют на основании наличия в крови вакуолизированных лейкоцитов.
 дефиците фермента b галактозидазы в")
Ганглиозидозы Болезнь Нормана — Ландинга (CM 1 генерализованный ганглиозидоз) дефиците фермента b галактозидазы в головном мозге и во внутренних Смерть наступает обычно на втором году жизни. Амавротическая идиотия Тея — Сакса (GM 2 ганглиозидоз, тип I). недостаточность фермента гексозаминидазы А и увеличения активности гексозаминидазы В. Смерть наступает через 1— 2 года после начала заболевания. Болезнь Дерри (ювенильный GM 1 ганглиозидоз), снижение активности b галактозидазы и головном мозге начинается на 2 м году жизни с развития атаксии, деградации интеллекта, спастического тетрапареза, косоглазия. Болезнь Сандхоффа (GM 2 ганглиозидоз, типа III, вариант 0) частичный дефицита гексозаминидазы А. Начинается в возрасте 2— 6 лет с атаксии. Смерть наступает в возрасте до 3 лет. Врожденная амавротическая идиотия Нормана — Вуда (GM 3 ганглиозидоз) развивается при дефиците фермента GM 3 N ацетилгалактозаминилтрансферазы.

Липофусцинозы Липофусцин — содержит гетерогенную смесь сшитых поперечными связями полимеров липидов и недопереваренных в лизосомах белков. Липофусцин накапливается в лизосомах. Содержание липофусцина обычно выше в неделящихся клетках (например, нейронах, клетках скелетной и сердечной мышечной ткани). Цероид липофусцинозы — группа наследственных аутосомно рецессивных заболеваний, характеризующихся накоплением пигментов. • Хроническая ювенильная (болезнь Баттена) — медленное прогрессирование поведенческих и зрительных симптомов. • Острая поздняя детская (болезнь Янского Бильшовского). • Хроническая взрослая (болезнь Куфса). • Острая детская болезнь Santavuori Haltia) — быстрое развитие двигательных и умственных расстройств в сочетании с миоклоническими судорогами и прогрессирующей слепотой. • Описано множество более редких форм
 наследственное нарушение обмена сфингомиелина, • происходит накопление сфингомиелина в")
• Болезнь Ниманна-Пика (сфингомиелиноз) наследственное нарушение обмена сфингомиелина, • происходит накопление сфингомиелина в мозге, печени, селезенке, ретикулоэндотелиальной системе. • Тип наследования аутосомно рецессивный. • Дефицит сфингомиелиназы. • Различают раннюю тяжелую форму заболевания с первых дней жизни отказ от пищи, периодическая рвота, обезвоживание организма, увеличиваются печень и селезенка, задержка психического развития, развиваются спастические парезы, нарушается координация движений, возникают глухота и слепота. На глазном дне атрофия сосков зрительных нервов, Гибель наступает через 2 3 года. • При поздней хронической форме заболевания, возникающей в подростковом возрасте или у взрослых, болезнь течет длительно, нервная система обычно не поражается или вовлекается в патологический процесс в позднем периоде.
 наследственно обусловленное нарушение обмена глюкоцереброзидов, • Цереброзиды накапливаются в")
• Болезнь Гоше (глюкоцереброзидоз) наследственно обусловленное нарушение обмена глюкоцереброзидов, • Цереброзиды накапливаются в клетках ретикулоэндотелиальной системы. • Тип наследования аутосомно рецессивный • Дефицит фермента бета глюкоцереброзидазы, • Развитие спленомегалического синдрома, анемии и поражения костной системы, сопровождается задержкой общего и психического развития, эпилептические припадки, слепота. • Смерть наступает через 1, 5 2 года. • У взрослых заболевание отличается хроническим течением и протекает более доброкачественно.
 обусловлено блокадой нормального альфа окисления фитановой кислоты")
• Болезнь Рефсума (полиневритоподобная гемералопическая гередоатаксия) обусловлено блокадой нормального альфа окисления фитановой кислоты (присутствующей в пище как компонент растительных и животных жиров) в альфа гидроксифитановую. • Наследуется по аутосомно рецессивному типу. • Измененный фермент: Оксидаза фитановой кислоты Накопление фитановой кислоты в крови и тканях, в центральной и периферической нервной системе. • Клинические симптомы: Пигментный ретинит, глухота, ихтиоз, мозжечковая атаксия, множественная эпифизарная дисплазия • Течение заболевания медленно прогрессирующее. • Дифференциальный базируется на биохимических показателях высоком содержании фитановой кислоты в сыворотке крови и повышенном выделении с мочой жирных кислот. CH 3·CH CH 2 CH 2 CH·CH 3 CH 2 COOH Фитановая кислота
 — тяжелая наследственная патология с аутосомно рецессивным типом наследования,")
Ø Болезнь Вольмана (генерализованный ксантоматоз) — тяжелая наследственная патология с аутосомно рецессивным типом наследования, х Ø арактеризуется накоплением холестеринэстеров в печени, селезенке, лимфатических узлах и надпочечниках Ø Недостаточность лизосомной холестеринэстергидролазы и кислой липазы. Симптомы появляются в первые недели жизни ребенка: неукротимая рвота, диарея, дистрофия и обезвоживание, признаки поражения нервной системы — атаксия, деменция, гепатоспленомегалия, развивается ксантоматоз кожи. Ø Прогноз неблагоприятный, дети умирают в первом полугодии жизни. Болезнь накопления холестеринэстеров Лизосомный дефектом α нафтилацетатэстеразы приводит к накоплению холестеринэстеров в печени. гепатомегалия, развивается цирроз печени с осложнениями, повышено содержание атерогенных липопротеидов. • прогноз зависит от особенностей течения цирроза печени. Специфическая терапия отсутствует. • •
НАРУШЕНИЯ ЛИПИДНОГО ОБМЕНА.ppt