LECTURE for MGU - MUTATIONS AND THEIR CONSEQUENCES.ppt
- Количество слайдов: 143
 MUTATIONS and their consequences
MUTATIONS and their consequences
 Mutation: definition • Mutation – a structural change in genomic DNA which can be transmitted from a cell to its daughter cells • Germline mutation: present in the gamete • Somatic mutation: present in the somatic cell
Mutation: definition • Mutation – a structural change in genomic DNA which can be transmitted from a cell to its daughter cells • Germline mutation: present in the gamete • Somatic mutation: present in the somatic cell
 Germline and somatic mutations Somatic cells Population of mutant cells MITOSES mutation Germ line cells Sexual reproduction No cell carry mutation All cells carry mutation
Germline and somatic mutations Somatic cells Population of mutant cells MITOSES mutation Germ line cells Sexual reproduction No cell carry mutation All cells carry mutation
 • 1 b. Regulatory point mutations") Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2 a. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2 a. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
 Point mutations: Normal sequence of gene Base substitution GGG AGT GTA GAT CGT T GGG AGT G A GAT CGT T Base insertion GGG AGT GTT AGA TCG T T Base deletion GGG AGT GAG ATC GT
Point mutations: Normal sequence of gene Base substitution GGG AGT GTA GAT CGT T GGG AGT G A GAT CGT T Base insertion GGG AGT GTT AGA TCG T T Base deletion GGG AGT GAG ATC GT
 Example of mutation: Substitutions Normal gene DNA GGTCTCCTCACGCCA ↓ RNA CCAGAGGAGUGCGGU Codons ↓ Pro-Glu-Cys-Gly Amino acids Substitution mutation GGTCACCTCACGCCA ↓ CCAGUGGAGUGCGGU ↓ Pro-Arg-Glu-Cys-Gly Substitutions will only affect a single codon Their effects may not be serious unless they affect an amino acid that is essential for the structure and function of the finished protein molecule (e. g. sickle cell anaemia) DNA RNA
Example of mutation: Substitutions Normal gene DNA GGTCTCCTCACGCCA ↓ RNA CCAGAGGAGUGCGGU Codons ↓ Pro-Glu-Cys-Gly Amino acids Substitution mutation GGTCACCTCACGCCA ↓ CCAGUGGAGUGCGGU ↓ Pro-Arg-Glu-Cys-Gly Substitutions will only affect a single codon Their effects may not be serious unless they affect an amino acid that is essential for the structure and function of the finished protein molecule (e. g. sickle cell anaemia) DNA RNA
 Example of mutation: Silent mutation Normal gene DNA GGTCTCCTCACGCCA ↓ RNA CCAGAGGAGUGCGGU Codons ↓ Pro-Glu-Cys-Gly Amino acids Substitution mutation GGTCTTCTCACGCCA ↓ CCAGAAGAGUGCGGU ↓ Pro-Glu-Cys-Gly Silent mutation has no effect on the phenotype (many exclusions present) Changes in the third base of a codon often have no effect on the phenotype due to degenerate nature of the genetic code DNA RNA
Example of mutation: Silent mutation Normal gene DNA GGTCTCCTCACGCCA ↓ RNA CCAGAGGAGUGCGGU Codons ↓ Pro-Glu-Cys-Gly Amino acids Substitution mutation GGTCTTCTCACGCCA ↓ CCAGAAGAGUGCGGU ↓ Pro-Glu-Cys-Gly Silent mutation has no effect on the phenotype (many exclusions present) Changes in the third base of a codon often have no effect on the phenotype due to degenerate nature of the genetic code DNA RNA
 Nonsense mutation: Stop-codon appears Normal gene DNA GGTCTCCTCACGCCA ↓ RNA CCAGAGGAGUGCGGU Codons ↓ Pro-Glu-Cys-Gly Amino acids Substitution mutation GGTCTCCTCACTCCA ↓ CCAGAAGAGUGAGGU ↓ Pro-Glu-STOP DNA RNA
Nonsense mutation: Stop-codon appears Normal gene DNA GGTCTCCTCACGCCA ↓ RNA CCAGAGGAGUGCGGU Codons ↓ Pro-Glu-Cys-Gly Amino acids Substitution mutation GGTCTCCTCACTCCA ↓ CCAGAAGAGUGAGGU ↓ Pro-Glu-STOP DNA RNA
 Point mutations: No Mut Missense Nonsense Silent DNA … AGT … AAT … A TT … C … AG C … m. RNA … UCA … … UUA … … UAA … … TCG … Stop codon Protein SER LEU X SER Missense: VAL (valine): GUU, GUA, GUC, GUG
Point mutations: No Mut Missense Nonsense Silent DNA … AGT … AAT … A TT … C … AG C … m. RNA … UCA … … UUA … … UAA … … TCG … Stop codon Protein SER LEU X SER Missense: VAL (valine): GUU, GUA, GUC, GUG
 Missense mutation: sickle cell anemia The change in amino acid sequence causes hemoglobin molecules to crystallize when oxygen levels in the blood are low. As a result, red blood cells sickle and get stuck in small blood vessels
Missense mutation: sickle cell anemia The change in amino acid sequence causes hemoglobin molecules to crystallize when oxygen levels in the blood are low. As a result, red blood cells sickle and get stuck in small blood vessels
 Low Density Lipoprotein Receptor Nonsense mutation in LDLR Protein") Nonsense mutation: familial hypercholesterolemia (LDLR) Low Density Lipoprotein Receptor Nonsense mutation in LDLR Protein is too short. It can not fold properly. Because of that, LDLRs never make it into membrane and got degraded in ER
Nonsense mutation: familial hypercholesterolemia (LDLR) Low Density Lipoprotein Receptor Nonsense mutation in LDLR Protein is too short. It can not fold properly. Because of that, LDLRs never make it into membrane and got degraded in ER
 Frame-shift mutations: Frameshift mutation No Mutation DNA TACAACGTCACCATT TACAACGA TCACCATT g m. RNA AUGUUGCAGUGGUAA Protein AUGUUGCCAGUGGUAA MET PHE GLN TRP MET PHE PRO VAL … … … VAL X
Frame-shift mutations: Frameshift mutation No Mutation DNA TACAACGTCACCATT TACAACGA TCACCATT g m. RNA AUGUUGCAGUGGUAA Protein AUGUUGCCAGUGGUAA MET PHE GLN TRP MET PHE PRO VAL … … … VAL X
 Frame-shift mutations: cystic fibrosis Protein aminoacid sequence is shifted the function of CFTR protein is a Cl- channel. Plus it is too short due to unexpected stop-codon. It can not fold properly. Because of that, CFTR channel never make it into membrane and got degraded in ER
Frame-shift mutations: cystic fibrosis Protein aminoacid sequence is shifted the function of CFTR protein is a Cl- channel. Plus it is too short due to unexpected stop-codon. It can not fold properly. Because of that, CFTR channel never make it into membrane and got degraded in ER
 detects the presence of a premature") Nonsense mediated m. RNA decay Nonsense-mediated decay (NMD) detects the presence of a premature stop codon in the m. RNA and initiates m. RNA degradation Nonsense GGA CCT AAG GCT A TT … T CCT GGA TTC CGT UAA GGACTGA CCT GGA TTC CGT UAA … Stop codon
Nonsense mediated m. RNA decay Nonsense-mediated decay (NMD) detects the presence of a premature stop codon in the m. RNA and initiates m. RNA degradation Nonsense GGA CCT AAG GCT A TT … T CCT GGA TTC CGT UAA GGACTGA CCT GGA TTC CGT UAA … Stop codon
 Damage control: Two-level check-up of protein length • 1. m. RNAs with pre-mature stop codons got degraded no protein synthesized • 2. If truncated protein synthesized, it often degrades in ER before delivery to membrane WHY? Damage control: If the truncated protein is not going to function anyway, We better save resources on its synthesis and delivery to proper location. Also, it might possess novel, cell-damaging function. Why risk it?
Damage control: Two-level check-up of protein length • 1. m. RNAs with pre-mature stop codons got degraded no protein synthesized • 2. If truncated protein synthesized, it often degrades in ER before delivery to membrane WHY? Damage control: If the truncated protein is not going to function anyway, We better save resources on its synthesis and delivery to proper location. Also, it might possess novel, cell-damaging function. Why risk it?
 • 1 b. Regulatory point mutations") Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
 Regulatory point mutations Affects promoters: or other regulatory sequences of the gene Typical effect: increase (or decrease) of the expression level for a certain gene
Regulatory point mutations Affects promoters: or other regulatory sequences of the gene Typical effect: increase (or decrease) of the expression level for a certain gene
 Mutation to abolish transcription factor binding site X In this example, promoter mutation leads to significant decrease in the expression of the liver-specific enzyme (HNF 3 beta – liver specific transcription factor)
Mutation to abolish transcription factor binding site X In this example, promoter mutation leads to significant decrease in the expression of the liver-specific enzyme (HNF 3 beta – liver specific transcription factor)
 an antigen 2) a receptor for") Duffy blood group protein serves 3 purposes: 1) an antigen 2) a receptor for chemokines 3) a receptor for Plasmodium vivax malaria parasites. Individuals with the Duffy-negative phenotype are resistant to P. vivax invasion. Despite P. vivax be widespread throughout the tropical and subtropical world, it is absent from West Africa, where more than 95% of the population is Duffy negative. -33 T/C mut The DARC (Duffy blood group, chemokine receptor) locus, showing the location of a mutation at -46 (red bar) that abolishes transcription in red blood cells
Duffy blood group protein serves 3 purposes: 1) an antigen 2) a receptor for chemokines 3) a receptor for Plasmodium vivax malaria parasites. Individuals with the Duffy-negative phenotype are resistant to P. vivax invasion. Despite P. vivax be widespread throughout the tropical and subtropical world, it is absent from West Africa, where more than 95% of the population is Duffy negative. -33 T/C mut The DARC (Duffy blood group, chemokine receptor) locus, showing the location of a mutation at -46 (red bar) that abolishes transcription in red blood cells
 C/T-13910 LCT gene: Normally – downregulated in") LCT gene: lactose tolerance in adults (mutation) C/T-13910 LCT gene: Normally – downregulated in adulthood. Adults do not tolerate milk (diarrhea). Promoter mutation C/T-13910 located in the position of -13 910 (bp) upstream of the lactase gene has occurred that has made part of mankind tolerate milk (lactase persistence). All 4 mutations influencing lactose production are actually found within an intron of the MCM 6 gene – way upstream. Wray Nature Reviews Genetics 8, 206– 216 (March 2007) | doi: 10. 1038/nrg 2063
LCT gene: lactose tolerance in adults (mutation) C/T-13910 LCT gene: Normally – downregulated in adulthood. Adults do not tolerate milk (diarrhea). Promoter mutation C/T-13910 located in the position of -13 910 (bp) upstream of the lactase gene has occurred that has made part of mankind tolerate milk (lactase persistence). All 4 mutations influencing lactose production are actually found within an intron of the MCM 6 gene – way upstream. Wray Nature Reviews Genetics 8, 206– 216 (March 2007) | doi: 10. 1038/nrg 2063
 locus: evil cousin of enkephalin and endorphin Encodes opioid neuropeptide precursor") The PDYN (prodynorphin) locus: evil cousin of enkephalin and endorphin Encodes opioid neuropeptide precursor for endogenous ligands for opiate receptor; balances “pleasure” genes A 68 bp tandem repeat Mutation downregulates “pleasure” controls; provides for addiction Influences: Schizophrenia cocaine addiction epilepsy -mediate the anticipation and experience of pain -influence social attachment and bonding behaviour -affect learning and memory Wray Nature Reviews Genetics 8, 206– 216 (March 2007) | doi: 10. 1038/nrg 2063
The PDYN (prodynorphin) locus: evil cousin of enkephalin and endorphin Encodes opioid neuropeptide precursor for endogenous ligands for opiate receptor; balances “pleasure” genes A 68 bp tandem repeat Mutation downregulates “pleasure” controls; provides for addiction Influences: Schizophrenia cocaine addiction epilepsy -mediate the anticipation and experience of pain -influence social attachment and bonding behaviour -affect learning and memory Wray Nature Reviews Genetics 8, 206– 216 (March 2007) | doi: 10. 1038/nrg 2063
 Regulatory mutations with interesting phenotypic consequences Gene Function of product Phenotype AVPR 1 A Vasopressin receptor Creative dance performance DARC Chemokine receptor Resistance to infection with malaria HTR 2 A Serotonin receptor Obsessive-compulsive behaviour IL 10 Interleukin 10 Outcome of infection with HIV and infection with leprosy IL 10 Interleukin 10 Susceptibility to schizophrenia LCT Digestive enzyme Lactose persistence MAOA Neurotransmitter turnover Aggressive behaviour MMP 3 Matrix metalloprotease Risk of heart disease PDYN Neuropeptide Memory, emotional status SLC 6 A 4 Serotonin transporter Depression, creativity, anxiety
Regulatory mutations with interesting phenotypic consequences Gene Function of product Phenotype AVPR 1 A Vasopressin receptor Creative dance performance DARC Chemokine receptor Resistance to infection with malaria HTR 2 A Serotonin receptor Obsessive-compulsive behaviour IL 10 Interleukin 10 Outcome of infection with HIV and infection with leprosy IL 10 Interleukin 10 Susceptibility to schizophrenia LCT Digestive enzyme Lactose persistence MAOA Neurotransmitter turnover Aggressive behaviour MMP 3 Matrix metalloprotease Risk of heart disease PDYN Neuropeptide Memory, emotional status SLC 6 A 4 Serotonin transporter Depression, creativity, anxiety
 • 1 b. Regulatory point mutations") Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
 RNA processing mutations / splicing site mutations Don’t you EVER forget that human genes contain introns!!!!! Biochemistry, Stryer, 4 th ed.
RNA processing mutations / splicing site mutations Don’t you EVER forget that human genes contain introns!!!!! Biochemistry, Stryer, 4 th ed.
 Splicing site mutations: Phenylketonuria ex 11 ex 12 ex 13 Phenylalanin hydroxylase PAH ex 11 ex 12 ex 13 PAH m. RNA : normal ex 11 ex 12 AU ex 13 The most common phenylkenouria mutations in Europeans PAH m. RNA : Phenylketonuria ex 11 ex 13 This leads to a truncated PAH protein that lacks the last 52 amino acids at the C-terminus. This results in an unstable protein, without PAH activity.
Splicing site mutations: Phenylketonuria ex 11 ex 12 ex 13 Phenylalanin hydroxylase PAH ex 11 ex 12 ex 13 PAH m. RNA : normal ex 11 ex 12 AU ex 13 The most common phenylkenouria mutations in Europeans PAH m. RNA : Phenylketonuria ex 11 ex 13 This leads to a truncated PAH protein that lacks the last 52 amino acids at the C-terminus. This results in an unstable protein, without PAH activity.
 • 1 b. Regulatory point mutations") Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
 Deletions and insertions • A. Small deletions and insertions • B. Large deletions and insertions – Unequal crossing-over events – Retrotransposition Deletion Insertion
Deletions and insertions • A. Small deletions and insertions • B. Large deletions and insertions – Unequal crossing-over events – Retrotransposition Deletion Insertion
 A. Small deletions and insertions • Account for approximately 22% of all known mutations • Often results from a slippage of DNA replication (slippage tend to happen in the repetetive nucleotide tracts, e. g. AAAAAA) • Can be in-frame, if the number of deleted/inserted nucleotides is divisible by three
A. Small deletions and insertions • Account for approximately 22% of all known mutations • Often results from a slippage of DNA replication (slippage tend to happen in the repetetive nucleotide tracts, e. g. AAAAAA) • Can be in-frame, if the number of deleted/inserted nucleotides is divisible by three
 Slippage of DNA replication leads to deletion or insertions This will cause misalignment producing a loop in the nascent strand increase the repeat length. Alternatively, the same will take place but on the template strand (right) causing decrease of repeat length (Ellergan, 2004).
Slippage of DNA replication leads to deletion or insertions This will cause misalignment producing a loop in the nascent strand increase the repeat length. Alternatively, the same will take place but on the template strand (right) causing decrease of repeat length (Ellergan, 2004).
 In-frame deletions: two possible outcomes 5423 nt Protein is too short: It is unable to fold properly. Protein is too short: It still folds OK. Because of it, protein get degraded in ER. Example: ΔF 508 1 aa del in CFTR protein It functions, not so great, But it functions!!!! Hooray!
In-frame deletions: two possible outcomes 5423 nt Protein is too short: It is unable to fold properly. Protein is too short: It still folds OK. Because of it, protein get degraded in ER. Example: ΔF 508 1 aa del in CFTR protein It functions, not so great, But it functions!!!! Hooray!
: changes conformation of the protein. Proteins") Duchenne/Becker dystrophy DMD protein Missense mutation (1 bp): changes conformation of the protein. Proteins non-functional. Duchenne dystrophy (death at 12 -16 years old) In-frame deletion (-2000 bp) Middle part of the protein is gone. Proteins is functional. Becker dystrophy (mild symptoms, live till 60 th)
Duchenne/Becker dystrophy DMD protein Missense mutation (1 bp): changes conformation of the protein. Proteins non-functional. Duchenne dystrophy (death at 12 -16 years old) In-frame deletion (-2000 bp) Middle part of the protein is gone. Proteins is functional. Becker dystrophy (mild symptoms, live till 60 th)
 B. Large deletions and insertions • Range in size from 20 bp to 10 Mb (after that they classify as chromosomal abnormalities, as they become visible in light microscope) • Account for approximately 5 -6% of all known mutations • Often caused by unequal crossing over between homologous sequences • Sometimes are caused by retrotransposition
B. Large deletions and insertions • Range in size from 20 bp to 10 Mb (after that they classify as chromosomal abnormalities, as they become visible in light microscope) • Account for approximately 5 -6% of all known mutations • Often caused by unequal crossing over between homologous sequences • Sometimes are caused by retrotransposition
 Unequal crossing over between two chromosomes Unequal crossing over within the same chromosome May cause a disruption of entire gene, change gene structure, produce fusion of genes From: Lee M. Silver “Concepts of mouse genetics”
Unequal crossing over between two chromosomes Unequal crossing over within the same chromosome May cause a disruption of entire gene, change gene structure, produce fusion of genes From: Lee M. Silver “Concepts of mouse genetics”
 • 1 b. Regulatory point mutations") Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
 - LINEs (autonomous) 1)") Retrotransposition • Transposable elements in human genome: - SINEs (non-autonomous) - LINEs (autonomous) 1) May directly disrupt genes; 2) May provoke illegitemate recombination by homology 3) Contain regulatory sequences - Promoters - Enhancers - Splicing sites - Transcription factor binding sites These sites can be brought close to the gene and change its regulation
Retrotransposition • Transposable elements in human genome: - SINEs (non-autonomous) - LINEs (autonomous) 1) May directly disrupt genes; 2) May provoke illegitemate recombination by homology 3) Contain regulatory sequences - Promoters - Enhancers - Splicing sites - Transcription factor binding sites These sites can be brought close to the gene and change its regulation
 Troubles with Transposable Elements in the Human Genome 33 retrotransposition events identified result in human disease (hemophilia A and B, ßthalassemia, muscular dystrophy) Background rate of retrotransposition: 1/ 50 -100 germ cells Kazazian, H. , Science 289: 1152, 2000
Troubles with Transposable Elements in the Human Genome 33 retrotransposition events identified result in human disease (hemophilia A and B, ßthalassemia, muscular dystrophy) Background rate of retrotransposition: 1/ 50 -100 germ cells Kazazian, H. , Science 289: 1152, 2000
 How the insertion of the transposon may cause problems
How the insertion of the transposon may cause problems
 • 1 b. Regulatory point mutations") Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
Mutation types • 1 a. Point mutations (coding) • 1 b. Regulatory point mutations • 1 c. Alternative splicing point mutations • 2. Deletions/insertions • 3. Retrotransposition-related problems • 4. Unstable repeat expansion
 Unstable repeat expansion An increase in the size of triplet repeat, probably as a result of a slippage Expanded repeat : - Either suppresses expression of the gene (Fragile X) -Either results in the production of abnormal toxic product (Huntigton disease) S. Raskin et al. , 2000
Unstable repeat expansion An increase in the size of triplet repeat, probably as a result of a slippage Expanded repeat : - Either suppresses expression of the gene (Fragile X) -Either results in the production of abnormal toxic product (Huntigton disease) S. Raskin et al. , 2000
 Unstable repeat expansion: simple view
Unstable repeat expansion: simple view
 Tri-nucleotide repeat expansion disorders Disease Inheritance Repeat Normal range Premutation Full mutation Huntigton Autos. Dom CAG 10 -25 26 -35 36 -120 Myotonic dystrophy Autos. Dom CTG 5 -37 50 -80 (mildly aff) 80 -2000 Friedriech ataxia Autos. Rec GAA 6 -34 36 -100 100 -1700 Fragile X syndrome X-linked CGG 6 -54 55 -200 200 -1000 Spinal/bulbar muscl. atrophy CAG 11 -33 40 -62 Jacobsen syndrome CGG 11 100 -1000 Myoclonus epilepsy (U-L type) CCCCGCCC CGCG 2 -3 12 -13
Tri-nucleotide repeat expansion disorders Disease Inheritance Repeat Normal range Premutation Full mutation Huntigton Autos. Dom CAG 10 -25 26 -35 36 -120 Myotonic dystrophy Autos. Dom CTG 5 -37 50 -80 (mildly aff) 80 -2000 Friedriech ataxia Autos. Rec GAA 6 -34 36 -100 100 -1700 Fragile X syndrome X-linked CGG 6 -54 55 -200 200 -1000 Spinal/bulbar muscl. atrophy CAG 11 -33 40 -62 Jacobsen syndrome CGG 11 100 -1000 Myoclonus epilepsy (U-L type) CCCCGCCC CGCG 2 -3 12 -13
 Unstable repeat expansion causes suppression of FMR 1 gene and results in Fragile X syndrome
Unstable repeat expansion causes suppression of FMR 1 gene and results in Fragile X syndrome
 How do expanded repeats cause disease? Repeats in protein coding sequences = toxic proteins; Repeats in RNA coding regions = altered RNA function; Repeats in non-coding regions = reduced transcription or translation
How do expanded repeats cause disease? Repeats in protein coding sequences = toxic proteins; Repeats in RNA coding regions = altered RNA function; Repeats in non-coding regions = reduced transcription or translation
 FUNCTIONAL CLASSIFICATION OF MUTATIONS
FUNCTIONAL CLASSIFICATION OF MUTATIONS
 Functional classification of mutations • 1. Loss-of-Function mutations – Haploinsufficiency – Dominant negative mutations • 2. Gain-of-function mutations
Functional classification of mutations • 1. Loss-of-Function mutations – Haploinsufficiency – Dominant negative mutations • 2. Gain-of-function mutations
 Loss-of-Function mutations Loss-of-function mutations often cause recessive traits No gene expression Disease 50% of gene expression Enough to compensate 100% gene expression Normal
Loss-of-Function mutations Loss-of-function mutations often cause recessive traits No gene expression Disease 50% of gene expression Enough to compensate 100% gene expression Normal
 Loss of function causing dominant trait is an example of haploinsufficiency 50% of gene function is not enough to compensate Disease (heterozygote displays phenotypic effect) In most of the cases: 100% loss of gene function is either lethal either more severe phenotype
Loss of function causing dominant trait is an example of haploinsufficiency 50% of gene function is not enough to compensate Disease (heterozygote displays phenotypic effect) In most of the cases: 100% loss of gene function is either lethal either more severe phenotype
 1 per 20,") Loss-of-function mutations that leads to haploinsufficiency Osteogenesis imperfecta (brittle bone disease) 1 per 20, 000 live births a deficiency of collagen type I (30% of human weight) People with OI either have less collagen than normal or the quality is poorer than normal (4 different subtypes of disease) The condition has been found in an Egyptian mymmy from 1000 BC. The Norse king Ivar the Boneless may have had this condition as well. He was one of the Viking Lords that wreaked havoc on Europe and kept it firmly in the grips of darkness. he was unable to walk and he was carried around on a shield by his soldiers.
Loss-of-function mutations that leads to haploinsufficiency Osteogenesis imperfecta (brittle bone disease) 1 per 20, 000 live births a deficiency of collagen type I (30% of human weight) People with OI either have less collagen than normal or the quality is poorer than normal (4 different subtypes of disease) The condition has been found in an Egyptian mymmy from 1000 BC. The Norse king Ivar the Boneless may have had this condition as well. He was one of the Viking Lords that wreaked havoc on Europe and kept it firmly in the grips of darkness. he was unable to walk and he was carried around on a shield by his soldiers.
 Dominant negative mutations Some dominant loss-of-function mutations are called "dominant negative" or “antimorphic” mutations Mutant proteins often acts as competitive inhibitors Tumor suppressor gene TP 53, a guardian of genome, encodes a product p 53 that acts as a tetramer to regulate gene expression p 53
Dominant negative mutations Some dominant loss-of-function mutations are called "dominant negative" or “antimorphic” mutations Mutant proteins often acts as competitive inhibitors Tumor suppressor gene TP 53, a guardian of genome, encodes a product p 53 that acts as a tetramer to regulate gene expression p 53
 Dominant negative mutations: P 53 as an example Normally p 53 active as a tetramer ¼ of p 53 is mutant; Whole tetramer is inactive In the cell where one p 53 allele is mutated ½ of p 53 molecules are bad; No functional tetramers exist
Dominant negative mutations: P 53 as an example Normally p 53 active as a tetramer ¼ of p 53 is mutant; Whole tetramer is inactive In the cell where one p 53 allele is mutated ½ of p 53 molecules are bad; No functional tetramers exist
 Gain-of-Function mutations A mutation that confers new or enhanced activity on a protein. Less common than loss-of-function mutations Examples: Mutations in oncogenes Normal oncogene RAS mutation Huntington disease; mutated HD protein is able to suppress activity of transcription factors
Gain-of-Function mutations A mutation that confers new or enhanced activity on a protein. Less common than loss-of-function mutations Examples: Mutations in oncogenes Normal oncogene RAS mutation Huntington disease; mutated HD protein is able to suppress activity of transcription factors
") Spontaneous and Induced Mutation • Spontaneous mutation rate = 1 in 109 (a billion) replicated base pairs or 1 in 106 ( a million) replicated genes. Mistakes occur during DNA replication just before cell division. This is natural error rate of DNA polymerase. • Mutagens increase mistakes to 10– 5 (100 thousand) or 10– 3 (a thousand) per replicated gene. Mutations overload natural repair systems of the human genome
Spontaneous and Induced Mutation • Spontaneous mutation rate = 1 in 109 (a billion) replicated base pairs or 1 in 106 ( a million) replicated genes. Mistakes occur during DNA replication just before cell division. This is natural error rate of DNA polymerase. • Mutagens increase mistakes to 10– 5 (100 thousand) or 10– 3 (a thousand) per replicated gene. Mutations overload natural repair systems of the human genome
 • Chemical (different compounds) – Alkylating agents – Aromatic") Agents that cause mutations (mutagens) • Chemical (different compounds) – Alkylating agents – Aromatic hydrocarbons (e. g. benzo(a)pyrene) – Intercalating agents (e. g. fluorescent dyes) – Artificial derivatives of DNA bases • Physical (ionizing radiation, UV-radiation) • Biological (viruses, transposable elements)
Agents that cause mutations (mutagens) • Chemical (different compounds) – Alkylating agents – Aromatic hydrocarbons (e. g. benzo(a)pyrene) – Intercalating agents (e. g. fluorescent dyes) – Artificial derivatives of DNA bases • Physical (ionizing radiation, UV-radiation) • Biological (viruses, transposable elements)
 What is point mutation from the biochemistry point of view?
What is point mutation from the biochemistry point of view?
pyren Polycyclic aromatic hydrocarbon with confirmed carcinogenic activity Metabolic conversion A") Example of mutagen: benz(a)pyren Polycyclic aromatic hydrocarbon with confirmed carcinogenic activity Metabolic conversion A product of combustion processes (e. g. tobacco smoking, french fries) binds to DNA Converted by the body from promutagen to mutagen (addition of epoxide group and two OH-groups). A metabolic product binds to DNA → forms an adduct (a compound that results from addition). Ba. P – guanin adduct Presence of adduct change structure of DNA = cause mutation.
Example of mutagen: benz(a)pyren Polycyclic aromatic hydrocarbon with confirmed carcinogenic activity Metabolic conversion A product of combustion processes (e. g. tobacco smoking, french fries) binds to DNA Converted by the body from promutagen to mutagen (addition of epoxide group and two OH-groups). A metabolic product binds to DNA → forms an adduct (a compound that results from addition). Ba. P – guanin adduct Presence of adduct change structure of DNA = cause mutation.
 Many Mutagens Intercalate into DNA AT GC TA GC CG AT GC CG TA GC CG Carboplatin (anti-cancer drug) Benz(a)pyrene in cigarette smoke Daunarubicin (anti-cancer drug) Many anti-cancer drugs are MUTAGENS Aflatoxin from Aspergillus fungus growing on corn Bleomycin (anti-cancer drug produced by Streptomyces) (they treat cancer, but they also cause cancer)
Many Mutagens Intercalate into DNA AT GC TA GC CG AT GC CG TA GC CG Carboplatin (anti-cancer drug) Benz(a)pyrene in cigarette smoke Daunarubicin (anti-cancer drug) Many anti-cancer drugs are MUTAGENS Aflatoxin from Aspergillus fungus growing on corn Bleomycin (anti-cancer drug produced by Streptomyces) (they treat cancer, but they also cause cancer)
 UV exposure causes very specific damage: Thymine diners UV radiation causes formation of the thymine dimers, which block replication.
UV exposure causes very specific damage: Thymine diners UV radiation causes formation of the thymine dimers, which block replication.
 Look at this picture one more time: that is how UV light damages DNA
Look at this picture one more time: that is how UV light damages DNA
 Now see how it may be fixed
Now see how it may be fixed
 If we are getting lots of sunlight exposure, repair system might get overloaded MELANOMA: EW! The 6 risk factors are: History of blistering sunburns as a teen Red or blond hair Marked freckling of the upper back Family history of melanoma History of actinic keratoses Outdoor summer jobs for 3 or more years as a teen
If we are getting lots of sunlight exposure, repair system might get overloaded MELANOMA: EW! The 6 risk factors are: History of blistering sunburns as a teen Red or blond hair Marked freckling of the upper back Family history of melanoma History of actinic keratoses Outdoor summer jobs for 3 or more years as a teen
 Sunny Queensland Simply, too many sunny days! Why more melanomas in Queensland? Better whether ; more sunny days
Sunny Queensland Simply, too many sunny days! Why more melanomas in Queensland? Better whether ; more sunny days
 Best prevention for melanoma
Best prevention for melanoma
 Sometimes the “repair job” introduces the wrong nucleotide, leading to a point mutation. There are three possible outcomes 1) If mutation is in the germline, it may be passed to offspring (if gamete is not lost) - worst 2) If mutation is in the somatic cells, it might lead to cancer – not good 3) In most cases, mutations in the somatic cells do not produce any effect – e. g. cell may die, but we will never fell it (the best case scenario)
Sometimes the “repair job” introduces the wrong nucleotide, leading to a point mutation. There are three possible outcomes 1) If mutation is in the germline, it may be passed to offspring (if gamete is not lost) - worst 2) If mutation is in the somatic cells, it might lead to cancer – not good 3) In most cases, mutations in the somatic cells do not produce any effect – e. g. cell may die, but we will never fell it (the best case scenario)
 Instability of the human genome
Instability of the human genome
 De novo mutations in human genes in germplasm spontaneous abortions or babies with genetic syndromes or just nothing when Inherited serve as material for evolution In somatic cells spontaneous cell death or improperly functioning cells (never going to be detected) or just nothing not inherited: lost in evolution or CANCER
De novo mutations in human genes in germplasm spontaneous abortions or babies with genetic syndromes or just nothing when Inherited serve as material for evolution In somatic cells spontaneous cell death or improperly functioning cells (never going to be detected) or just nothing not inherited: lost in evolution or CANCER
 In fact, mutation rarely happens in the egg or sperm itself Embryo with germ-line cells Hametes Zygote MUTATIONS usually happen in the germ-line cells – precursors of sperms and eggs (it means within the mother/father of embryo)
In fact, mutation rarely happens in the egg or sperm itself Embryo with germ-line cells Hametes Zygote MUTATIONS usually happen in the germ-line cells – precursors of sperms and eggs (it means within the mother/father of embryo)
 NO DEFECT") Mutation resides only in germ-plasm derivatives (small proportion of the body’s cell) NO DEFECT IS VISIBLE Mutation de novo Sick baby (mutation in every cell) NORMAL
Mutation resides only in germ-plasm derivatives (small proportion of the body’s cell) NO DEFECT IS VISIBLE Mutation de novo Sick baby (mutation in every cell) NORMAL
") Are spontaneous mutations a health risk? The human mutation rate for base substitutions 1) is much higher in males than in females 2) increases with paternal age Risch et al. , 1987 Apert syndrome (N = 111) Achondroplasia, n = 152 Neurofibromatosis, n = 243. Relative frequency of affected children of normal parents (ordinate) as a function of paternal age (abscissa).
Are spontaneous mutations a health risk? The human mutation rate for base substitutions 1) is much higher in males than in females 2) increases with paternal age Risch et al. , 1987 Apert syndrome (N = 111) Achondroplasia, n = 152 Neurofibromatosis, n = 243. Relative frequency of affected children of normal parents (ordinate) as a function of paternal age (abscissa).
 Why aging males? Females: 22 cell divisions before meiosis and + meiosis = 23. All cell divisions are completed before birth. No increase with postnatal age. J. Crow, 2000
Why aging males? Females: 22 cell divisions before meiosis and + meiosis = 23. All cell divisions are completed before birth. No increase with postnatal age. J. Crow, 2000
 de novo mutations in humans • Humans have an exceptionally high spontaneous mutation rate of between 7. 6 × 10− 9 and 2. 2 × 10− 8 /bp /generation • An average newborn is calculated to have acquired 50 to 100 new mutations in their genome – -> 0. 86 novel non-synonymous mutations • The high-frequency of de novo mutations may explain the high frequency of conditions that cause reduced fecundity (autism, schizophrenia, etc).
de novo mutations in humans • Humans have an exceptionally high spontaneous mutation rate of between 7. 6 × 10− 9 and 2. 2 × 10− 8 /bp /generation • An average newborn is calculated to have acquired 50 to 100 new mutations in their genome – -> 0. 86 novel non-synonymous mutations • The high-frequency of de novo mutations may explain the high frequency of conditions that cause reduced fecundity (autism, schizophrenia, etc).
 “Soft” conditions are associated with decreased fertility, to maintain these prevalences, the diseases must be replenished by new mutations Prevalence Age onset (%) Mortality Fertility Paternal age (fraction of Heritability effect normal) 0. 05 0. 90 1. 4 Autism 0. 30 1 2. 0 Anorexia nervosa 0. 60 15 6. 2 0. 33 0. 56 — Schizophrenia 0. 70 22 2. 6 0. 40 0. 81 1. 4 Bipolar affective disorder 1. 25 25 2. 0 0. 65 0. 85 1. 2 Unipolar depression 10. 22 32 1. 8 0. 90 0. 37 1 Anxiety disorders 28. 80 11 1. 2 0. 90 0. 32 — The role of genetic variation in the causation of mental illness: an evolution-informed framework Uher, R. Molecular Psychiatry (2009) Dec; 14(12): 1072 -82, “
“Soft” conditions are associated with decreased fertility, to maintain these prevalences, the diseases must be replenished by new mutations Prevalence Age onset (%) Mortality Fertility Paternal age (fraction of Heritability effect normal) 0. 05 0. 90 1. 4 Autism 0. 30 1 2. 0 Anorexia nervosa 0. 60 15 6. 2 0. 33 0. 56 — Schizophrenia 0. 70 22 2. 6 0. 40 0. 81 1. 4 Bipolar affective disorder 1. 25 25 2. 0 0. 65 0. 85 1. 2 Unipolar depression 10. 22 32 1. 8 0. 90 0. 37 1 Anxiety disorders 28. 80 11 1. 2 0. 90 0. 32 — The role of genetic variation in the causation of mental illness: an evolution-informed framework Uher, R. Molecular Psychiatry (2009) Dec; 14(12): 1072 -82, “
 Paternal age is strong contributor to a number of de novo mutations per genome Age = 33 70 new mutations Kong et al. , 2012
Paternal age is strong contributor to a number of de novo mutations per genome Age = 33 70 new mutations Kong et al. , 2012
 disorders: A small group of disorders, including Apert syndrome") “paternal age effect” (PAE) disorders: A small group of disorders, including Apert syndrome (caused by FGFR 2 mutations), achondroplasia (FGFR 3), Costello syndrome (HRAS), PTPN 11 (Noonan syndrome) Direct quantification of PAE mutations in sperm and testes: these mutations provide selective advantage for mutated cells through dysregulation of spermatogonial cell behavior by growth factor receptor-RAS signal transduction pathway !!! (same way as pre-malignant close outcompetes normal cells)
“paternal age effect” (PAE) disorders: A small group of disorders, including Apert syndrome (caused by FGFR 2 mutations), achondroplasia (FGFR 3), Costello syndrome (HRAS), PTPN 11 (Noonan syndrome) Direct quantification of PAE mutations in sperm and testes: these mutations provide selective advantage for mutated cells through dysregulation of spermatogonial cell behavior by growth factor receptor-RAS signal transduction pathway !!! (same way as pre-malignant close outcompetes normal cells)
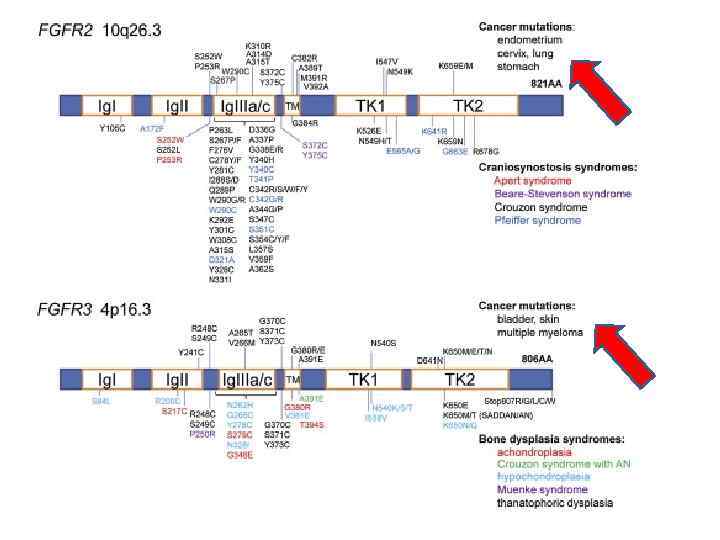
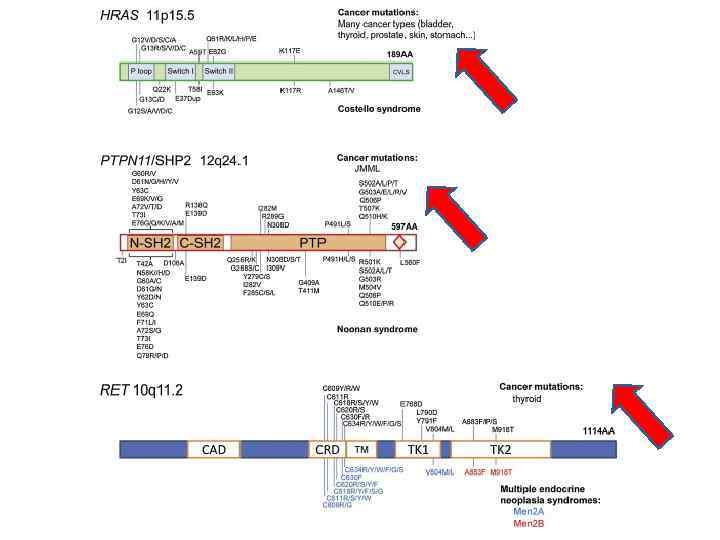
-RAS Signaling Pathway. Signaling downstream") PAE Disorders Cluster within the Receptor Tyrosine Kinase (RTK)-RAS Signaling Pathway. Signaling downstream of RAS involves the RAF/MEK/ERK (MAPK-ERK branch) (in red) and the PI 3 K-AKT-m. TOR (in green) pathways. The MEK 1/2 inhibitor (in red box) specifically blocks the phosphorylation of ERK 1/2.
PAE Disorders Cluster within the Receptor Tyrosine Kinase (RTK)-RAS Signaling Pathway. Signaling downstream of RAS involves the RAF/MEK/ERK (MAPK-ERK branch) (in red) and the PI 3 K-AKT-m. TOR (in green) pathways. The MEK 1/2 inhibitor (in red box) specifically blocks the phosphorylation of ERK 1/2.
 Long-Term Consequences of Selfish Selection in the Testis. Blue ovals represent the testis at three ages from puberty (left) to senescence (right). During the recurrent rounds of replication required for spermatogenesis and SSC self-renewal, stochastic mutations (represented by X) occur randomly in the testis.
Long-Term Consequences of Selfish Selection in the Testis. Blue ovals represent the testis at three ages from puberty (left) to senescence (right). During the recurrent rounds of replication required for spermatogenesis and SSC self-renewal, stochastic mutations (represented by X) occur randomly in the testis.
 Three scenarios for spermatogonial mutations Functionally neutral mutations do not accumulate and are associated with a very low risk of individual transmission (∼ 10− 8, the background rate of nucleotide substitution in the genome). Typical PAE mutations confer a strong selective advantage to the mutant cell clone, leading over time to the formation of large “mole-like” clones (ovals) and an increased risk of transmission in older men (up to 1, 000 -fold higher than the background mutation rate). In rare cases, these mutations are associated with spermatocytic seminoma (SPS). An intermediate scenario: milder selective advantage (i. e. , weak gain-of-function, change in copy number or regulation of expression) lead to enrichment over time to a lesser extent (>1 - to 100 -fold). Although strongly activating mutations associated with classical PAE disorders are deleterious and will be rapidly eliminated because of low reproductive fitness, neutral and mildly pathogenic mutations are potentially transmissible over many generations, contributing to genetic heterogeneity. Some mildly pathogenic mutations, although associated with deleterious phenotypes, might be maintained in the population either by recurrent mutation or because they provide a beneficial fitness trait during spermatogenesis.
Three scenarios for spermatogonial mutations Functionally neutral mutations do not accumulate and are associated with a very low risk of individual transmission (∼ 10− 8, the background rate of nucleotide substitution in the genome). Typical PAE mutations confer a strong selective advantage to the mutant cell clone, leading over time to the formation of large “mole-like” clones (ovals) and an increased risk of transmission in older men (up to 1, 000 -fold higher than the background mutation rate). In rare cases, these mutations are associated with spermatocytic seminoma (SPS). An intermediate scenario: milder selective advantage (i. e. , weak gain-of-function, change in copy number or regulation of expression) lead to enrichment over time to a lesser extent (>1 - to 100 -fold). Although strongly activating mutations associated with classical PAE disorders are deleterious and will be rapidly eliminated because of low reproductive fitness, neutral and mildly pathogenic mutations are potentially transmissible over many generations, contributing to genetic heterogeneity. Some mildly pathogenic mutations, although associated with deleterious phenotypes, might be maintained in the population either by recurrent mutation or because they provide a beneficial fitness trait during spermatogenesis.
 Prof. John Edwards Professor of Genetics at Oxford University and Fellow of the Royal College of Physicians QUOTE: “A normal male may well produce a whole of the gene mutation catalogue in every ejaculate”
Prof. John Edwards Professor of Genetics at Oxford University and Fellow of the Royal College of Physicians QUOTE: “A normal male may well produce a whole of the gene mutation catalogue in every ejaculate”
 , who has spent his entire career at") Victor Mc. Kusick (1921 – 2008) , who has spent his entire career at Johns Hopkins, is widely lauded as the 'father of genetic medicine. ' Mendelian Inheritance in Man: A Catalog of Human Genes and Genetic Disorders, first published in 1966
Victor Mc. Kusick (1921 – 2008) , who has spent his entire career at Johns Hopkins, is widely lauded as the 'father of genetic medicine. ' Mendelian Inheritance in Man: A Catalog of Human Genes and Genetic Disorders, first published in 1966
 How do we identify the de novo mutation responsible? • Compared to the Human genome reference sequence, which is itself constructed from 13 individuals 1000 Genomes project: A map of human genome variation from population-scale sequencing, Nature 467: 1061– 1073
How do we identify the de novo mutation responsible? • Compared to the Human genome reference sequence, which is itself constructed from 13 individuals 1000 Genomes project: A map of human genome variation from population-scale sequencing, Nature 467: 1061– 1073
 Identifying a causative de novo mutation Veltman and colleagues - Nat Genet. 2010 Dec; 42(12): 1109 -12 (1) Sequence genome Patient with idiopathic disorder (3) Exclude known variants seen in healthy people (2) Select only coding mutations ~22, 000 variants (exome re-sequencing) MSGTCASTTR MSGTNASTTR ~5, 640 coding variants (4) Sequence parents and exclude their private variants ~143 novel coding variants For 6/9 patients, they were able to identify a single likely-causative mutation (5) Look at affected gene function and mutational impact ~5 de novo novel coding variants
Identifying a causative de novo mutation Veltman and colleagues - Nat Genet. 2010 Dec; 42(12): 1109 -12 (1) Sequence genome Patient with idiopathic disorder (3) Exclude known variants seen in healthy people (2) Select only coding mutations ~22, 000 variants (exome re-sequencing) MSGTCASTTR MSGTNASTTR ~5, 640 coding variants (4) Sequence parents and exclude their private variants ~143 novel coding variants For 6/9 patients, they were able to identify a single likely-causative mutation (5) Look at affected gene function and mutational impact ~5 de novo novel coding variants
 showed that mut rate in humans is appr") By pseudogene resequencing (no selection) showed that mut rate in humans is appr 2. 5 x 10 -8 mut per nucl 175 mutations per diploid genome per generation Transitions are more common than transversions Cp. G mutate more often than non-Cp. G
By pseudogene resequencing (no selection) showed that mut rate in humans is appr 2. 5 x 10 -8 mut per nucl 175 mutations per diploid genome per generation Transitions are more common than transversions Cp. G mutate more often than non-Cp. G
 Mutations vs. chromosomal changes Event Point mutations Intragenic deletions Non-disjunction events Gender and paternal age bias Substantially increase with paternal age Slightly increase with both ages Substantially increase with maternal age Examples Apert syndrome, dwarfism Duchenne myotrophy (other large genes) Down syndrome, 0. 3% liveborn are aneuploid
Mutations vs. chromosomal changes Event Point mutations Intragenic deletions Non-disjunction events Gender and paternal age bias Substantially increase with paternal age Slightly increase with both ages Substantially increase with maternal age Examples Apert syndrome, dwarfism Duchenne myotrophy (other large genes) Down syndrome, 0. 3% liveborn are aneuploid
 is a very large gene 93% of point mutations paternal, 87%") Duchenne myotropy (DM) is a very large gene 93% of point mutations paternal, 87% of deletions maternal
Duchenne myotropy (DM) is a very large gene 93% of point mutations paternal, 87% of deletions maternal
 Relative frequency of all trisomies for different maternal ages. The ordinate is the percentage of trisomy occurring among recognized pregnancies
Relative frequency of all trisomies for different maternal ages. The ordinate is the percentage of trisomy occurring among recognized pregnancies
 From an evolutionary standpoint, an increase in mutation rate at later reproductive ages is not surprising. In our remote ancestry, probably very few males lived to reproduce in their 40 s. So, there would be very little selection pressure to reduce the harmful mutation rates late in the reproductive period. The luxury of reproducing at older ages is a bonus that contemporary men receive from higher living standards, medical advances and other environmental improvements.
From an evolutionary standpoint, an increase in mutation rate at later reproductive ages is not surprising. In our remote ancestry, probably very few males lived to reproduce in their 40 s. So, there would be very little selection pressure to reduce the harmful mutation rates late in the reproductive period. The luxury of reproducing at older ages is a bonus that contemporary men receive from higher living standards, medical advances and other environmental improvements.
 J. F. Crow; public lecture at the National Academy of Sciences, November 14, 1996 Truncation selection: eliminates these with mildly deleterious mutations quite efficiently Well. Not anymore. Most of the mutations have a very small effect and are to a large extent compensated for by environmental improvements. Who worries about having to wear spectacles? But can we continue to improve the environment indefinitely? Will a time come, especially if there is some sort of catatastrophe (war, epidemic or famine), when we are forced to return to the life of our ancestors? Under those circumstances we would surely see an increase in human misery, for all the mutations that have accumulated would be expressed in full force.
J. F. Crow; public lecture at the National Academy of Sciences, November 14, 1996 Truncation selection: eliminates these with mildly deleterious mutations quite efficiently Well. Not anymore. Most of the mutations have a very small effect and are to a large extent compensated for by environmental improvements. Who worries about having to wear spectacles? But can we continue to improve the environment indefinitely? Will a time come, especially if there is some sort of catatastrophe (war, epidemic or famine), when we are forced to return to the life of our ancestors? Under those circumstances we would surely see an increase in human misery, for all the mutations that have accumulated would be expressed in full force.
 J. F. Crow; public lecture at the National Academy of Sciences, November 14, 1996 "greatest mutational health hazard to the human genome is fertile older males".
J. F. Crow; public lecture at the National Academy of Sciences, November 14, 1996 "greatest mutational health hazard to the human genome is fertile older males".
 COMMON TYPES OF POINT MUTATIONS
COMMON TYPES OF POINT MUTATIONS
 Sources of point mutations in DNA Spontaneous or assisted chemical changes in the DNA Oxidation (red arrows) Hydrolysis (blue arrows) Methylation (green arrows)
Sources of point mutations in DNA Spontaneous or assisted chemical changes in the DNA Oxidation (red arrows) Hydrolysis (blue arrows) Methylation (green arrows)
 Spontaneous or assisted chemical changes in the") Sources of point mutations in DNA 1) Spontaneous or assisted chemical changes in the DNA Xantine; Uracil; pairs with T No deamination Hypoxantine; pairs with C Deamination as an example
Sources of point mutations in DNA 1) Spontaneous or assisted chemical changes in the DNA Xantine; Uracil; pairs with T No deamination Hypoxantine; pairs with C Deamination as an example
 PURINE or TRANSITIONS MOST COMMON PYRIMIDINE") Transitions and transversions Point mutations (single nucleotide changes) PURINE or TRANSITIONS MOST COMMON PYRIMIDINE PIRIMIDINE Pairing is possible due to tautomeric shifts or ionizing that allows mispairing TRANSVERSIONS purine pyrimidine or pyrimidine purine Pairing is energetically infavourable, but Pur-Pur pairs are possible (G-A)
Transitions and transversions Point mutations (single nucleotide changes) PURINE or TRANSITIONS MOST COMMON PYRIMIDINE PIRIMIDINE Pairing is possible due to tautomeric shifts or ionizing that allows mispairing TRANSVERSIONS purine pyrimidine or pyrimidine purine Pairing is energetically infavourable, but Pur-Pur pairs are possible (G-A)
 Ultraviolet Radiation Cause Thymine Dimers 260 nanometer wavelength Disrupts synthesis; good for sterilization of bacteria, bad for skin cancer.
Ultraviolet Radiation Cause Thymine Dimers 260 nanometer wavelength Disrupts synthesis; good for sterilization of bacteria, bad for skin cancer.
 Other types of mutations that can occur 2. Small insertions/ deletions of 1 -5 bp By Failure to repair abasic sites 3. Large chromosomal deletions 4. Translocations Do not forget about them ! When people talk about carcinogenic mutations, most often they talk about point mutations. Point mutations much more tolerable for cell reparation system, and unlikely to awaken apoptosis pathway.
Other types of mutations that can occur 2. Small insertions/ deletions of 1 -5 bp By Failure to repair abasic sites 3. Large chromosomal deletions 4. Translocations Do not forget about them ! When people talk about carcinogenic mutations, most often they talk about point mutations. Point mutations much more tolerable for cell reparation system, and unlikely to awaken apoptosis pathway.
 Rate of mutations in human RATES of molecular events in human genes cells Random gain of mutations (low-level; natural cause) Early stages of natural cancer in elderly; spontaneous mutations in germ plasm Forced gain of mutations (median level; X-ray, chemical carcinogens) Early stages of cancer and germplasm in exposed people; Very high rate of mutations (in cells that lost one or more major mechanisms of DNA repair) Certain genetic syndromes; late stages of almost any cancer
Rate of mutations in human RATES of molecular events in human genes cells Random gain of mutations (low-level; natural cause) Early stages of natural cancer in elderly; spontaneous mutations in germ plasm Forced gain of mutations (median level; X-ray, chemical carcinogens) Early stages of cancer and germplasm in exposed people; Very high rate of mutations (in cells that lost one or more major mechanisms of DNA repair) Certain genetic syndromes; late stages of almost any cancer
 HOW TO CALCULATE RATE OF MUTATIONS IN HUMAN CELLS ? By HPRT assay
HOW TO CALCULATE RATE OF MUTATIONS IN HUMAN CELLS ? By HPRT assay
 Human HPRT gene IN VIVO : complete deficiency of HPRT activity = too much purunes = Lesch-Nyhan. Syndrome (urate crystals + self-Injuring) Partial deficiency = nephrolithiasis, gouty arthritis, & some neurological manifestations -- Located on chromosome X -- Encodes the enzyme hypoxanthine phospho ribosyl transferase -- Normal function of HPRT is metabolic salvage of the purines (hypoxanthine and guanine) into nucleotides, inosinic acid, and guanylic acid Human lymphocytes survive treatment with 6 -thioguanine (poison) ONLY if HPRT gene is mutated 6 -thioguanine
Human HPRT gene IN VIVO : complete deficiency of HPRT activity = too much purunes = Lesch-Nyhan. Syndrome (urate crystals + self-Injuring) Partial deficiency = nephrolithiasis, gouty arthritis, & some neurological manifestations -- Located on chromosome X -- Encodes the enzyme hypoxanthine phospho ribosyl transferase -- Normal function of HPRT is metabolic salvage of the purines (hypoxanthine and guanine) into nucleotides, inosinic acid, and guanylic acid Human lymphocytes survive treatment with 6 -thioguanine (poison) ONLY if HPRT gene is mutated 6 -thioguanine
 HPRT ASSAY The Hypoxanthine Phosphoribsyltransferase Assay. HPRT +/+ 6 -thioguanine = Poison So, if cell acquire mutation in HPRT, it become resistant to 6 -thioguanine compound HPRT -/ 6 -thioguanine = OK We can directly count mutant colonies and compare this number with number of cell seeded on plate
HPRT ASSAY The Hypoxanthine Phosphoribsyltransferase Assay. HPRT +/+ 6 -thioguanine = Poison So, if cell acquire mutation in HPRT, it become resistant to 6 -thioguanine compound HPRT -/ 6 -thioguanine = OK We can directly count mutant colonies and compare this number with number of cell seeded on plate
 HPRT mutation frequency In peripheral T cells In 49 healthy, non-smoking adults: rates varied 0. 25 up to 9. 64 x 10 -6 bp. REMEMBER!!! The real life mutation rates are somewhere in between PURELY SPONTANEOUS RATE 1 per 109 bp As measured by HPRT 0. 25 up to 9. 64 per 106 bp MUTAGEN-ENHANCED 1 per 105 up to 1 per 103 bp
HPRT mutation frequency In peripheral T cells In 49 healthy, non-smoking adults: rates varied 0. 25 up to 9. 64 x 10 -6 bp. REMEMBER!!! The real life mutation rates are somewhere in between PURELY SPONTANEOUS RATE 1 per 109 bp As measured by HPRT 0. 25 up to 9. 64 per 106 bp MUTAGEN-ENHANCED 1 per 105 up to 1 per 103 bp
 That means: we are under constant pressure by the everyday life mutagens that come from everywhere!! But anyway, what makes individual mutation frequencies so different (>15 times)? ? ? Going hypothesis: 1. Polymorphisms of genes metabolizing carcinogens; 2. Polymorphisms of genes responsible for DNA repair; 3. Alcohol consumption and smoking; 4. Exposure to environmental carcinogens; 5. Exposure to radiation CYP 1 A 1, GSTM 1 and NAT 2 polymorphisms have no influence on HPRT mutation frequencies; Mismatch repair genes – no mutations;
That means: we are under constant pressure by the everyday life mutagens that come from everywhere!! But anyway, what makes individual mutation frequencies so different (>15 times)? ? ? Going hypothesis: 1. Polymorphisms of genes metabolizing carcinogens; 2. Polymorphisms of genes responsible for DNA repair; 3. Alcohol consumption and smoking; 4. Exposure to environmental carcinogens; 5. Exposure to radiation CYP 1 A 1, GSTM 1 and NAT 2 polymorphisms have no influence on HPRT mutation frequencies; Mismatch repair genes – no mutations;
 Less test the hypothesis that the frequency of mutations is influenced by one’s genes Correlation between maternal alcohol consumption during pregnancy and results of HPRT assay on T-lymphocytes from newborns? Early pregnancy alcohol: RR high mutation status = 1. 84 Through pregnancy alcohol: RR high mutation status = 2. 99 Smoking during pregnancy have an influence on mutational spectrum, but not on mutation frequency Mutat Res 1999 Dec 17; 431(2): 279 -89
Less test the hypothesis that the frequency of mutations is influenced by one’s genes Correlation between maternal alcohol consumption during pregnancy and results of HPRT assay on T-lymphocytes from newborns? Early pregnancy alcohol: RR high mutation status = 1. 84 Through pregnancy alcohol: RR high mutation status = 2. 99 Smoking during pregnancy have an influence on mutational spectrum, but not on mutation frequency Mutat Res 1999 Dec 17; 431(2): 279 -89
 Mutation frequency and smoking Yes, smoking increases mutation frequency No, smoke does not have any influence NON-cancerous LUNGS of SMOKERS AND NON-SMOKERS checked for mutations in K-ras, p 53 and HPRT genes In the normal lung tissue of smokers and non-smokers the rate of mutation in smokers > non-smokers (~1. 6 fold )
Mutation frequency and smoking Yes, smoking increases mutation frequency No, smoke does not have any influence NON-cancerous LUNGS of SMOKERS AND NON-SMOKERS checked for mutations in K-ras, p 53 and HPRT genes In the normal lung tissue of smokers and non-smokers the rate of mutation in smokers > non-smokers (~1. 6 fold )
 Exposure to envinronmental carcinogens and frequency of mutations ethylene oxide ; 1, 3 -butadiene ; benzene Marginal increase in HPRT mutability when measured as in vivo exposure (on plant workers populations) Cell line-based or mice/ rat-based HPRT assays after direct addition of carcinogen show strong increase in HPRT mutability
Exposure to envinronmental carcinogens and frequency of mutations ethylene oxide ; 1, 3 -butadiene ; benzene Marginal increase in HPRT mutability when measured as in vivo exposure (on plant workers populations) Cell line-based or mice/ rat-based HPRT assays after direct addition of carcinogen show strong increase in HPRT mutability
 HPRT rates : 1") Exposure to radiation Among Hiroshima-Nagasaki survivors (43 Rad in average) HPRT rates : 1 mut per 10 -8 per base pair per generation Lower than that of Japanese controls Chernobyl clean-up workers: 40% increase in mutation rate in the first year after accident, …. then it declined…. Conclusion: classical HPRT assays in lymphocytes do not support any hypothesis explaining population variance of the mutational load
Exposure to radiation Among Hiroshima-Nagasaki survivors (43 Rad in average) HPRT rates : 1 mut per 10 -8 per base pair per generation Lower than that of Japanese controls Chernobyl clean-up workers: 40% increase in mutation rate in the first year after accident, …. then it declined…. Conclusion: classical HPRT assays in lymphocytes do not support any hypothesis explaining population variance of the mutational load
 In kidney epithelium Mutational load is 0. 5") Mutations in Epithelial tissues (carcinoma progenitors) In kidney epithelium Mutational load is 0. 5 - 4. 2 x 10 -4 – much higher than in lymphocytes (10 - 100 times higher!) This rate is sufficient to account for a large proportion of human cancers without the need of increased frequency of mutations At least in kidney, tumor may arise by themselves without any additional environmental/genetic causes
Mutations in Epithelial tissues (carcinoma progenitors) In kidney epithelium Mutational load is 0. 5 - 4. 2 x 10 -4 – much higher than in lymphocytes (10 - 100 times higher!) This rate is sufficient to account for a large proportion of human cancers without the need of increased frequency of mutations At least in kidney, tumor may arise by themselves without any additional environmental/genetic causes
 Mind that somatic mutation rates and germline mutation rates are different • Mitochondrial DNA has been estimated to have mutation rates of ~3× 10− 6 or ~2. 7× 10− 5 per base per 20 year generation • whole genome sequencing in families showed the human genome mutation rate is ~1. 1× 10− 8 per site per generation MIT > Somatic nuclear > Germline nuclear
Mind that somatic mutation rates and germline mutation rates are different • Mitochondrial DNA has been estimated to have mutation rates of ~3× 10− 6 or ~2. 7× 10− 5 per base per 20 year generation • whole genome sequencing in families showed the human genome mutation rate is ~1. 1× 10− 8 per site per generation MIT > Somatic nuclear > Germline nuclear
 Rate of mutations in human RATES of molecular events in human genes cells Random gain of mutations (low-level; natural cause) Early stages of natural cancer in elderly; spontaneous mutations in germ cells Forced gain of mutations (median level; X-ray, chemical carcinogens) Early stages of cancer and germ cells in exposed people; Very high rate of mutations (cell lost one or more major mechanisms of DNA repair) Certain genetic syndromes; late stages of almost any cancer
Rate of mutations in human RATES of molecular events in human genes cells Random gain of mutations (low-level; natural cause) Early stages of natural cancer in elderly; spontaneous mutations in germ cells Forced gain of mutations (median level; X-ray, chemical carcinogens) Early stages of cancer and germ cells in exposed people; Very high rate of mutations (cell lost one or more major mechanisms of DNA repair) Certain genetic syndromes; late stages of almost any cancer
 Another test for mutagenesis: comet assay “Comet assay” uncomplicated and sensitive technique for the detection of DNA damage at the level of the individual eukaryotic cell. Most often, non-invasive sample of blood lymphocytes is used. a standard technique for evaluation of DNA damage/repair in biomonitoring and genotoxicity testing
Another test for mutagenesis: comet assay “Comet assay” uncomplicated and sensitive technique for the detection of DNA damage at the level of the individual eukaryotic cell. Most often, non-invasive sample of blood lymphocytes is used. a standard technique for evaluation of DNA damage/repair in biomonitoring and genotoxicity testing

 The length of the tail is quantified automatically in every cell The extent of DNA damage is measured by the size and intensity of the tail produced from a single nucleous following electrophoreisis
The length of the tail is quantified automatically in every cell The extent of DNA damage is measured by the size and intensity of the tail produced from a single nucleous following electrophoreisis
, norepinephrine (NE) and cortisol (Cort) murine 3 T") 20 min treatment by Epinephrine (E), norepinephrine (NE) and cortisol (Cort) murine 3 T 3 cell Cadmium induced stress in Euglena gracilis
20 min treatment by Epinephrine (E), norepinephrine (NE) and cortisol (Cort) murine 3 T 3 cell Cadmium induced stress in Euglena gracilis
 Oxidative DNA damage occur in airway cells following exposure to whole smoke. SMOKE (AIR DILUTION) the lesion specific enzyme formamidopyrimidine N-glycosylase (FPG) was used to access oxidative DNA base damage (it detects alkylation, 8 -OH guanine and other oxidatively damaged purines, instead of Physical DNA breaks. )
Oxidative DNA damage occur in airway cells following exposure to whole smoke. SMOKE (AIR DILUTION) the lesion specific enzyme formamidopyrimidine N-glycosylase (FPG) was used to access oxidative DNA base damage (it detects alkylation, 8 -OH guanine and other oxidatively damaged purines, instead of Physical DNA breaks. )
 Amount of breaks depend on given time point on life of individual
Amount of breaks depend on given time point on life of individual
 Strong envinronmental carcinogens ethylene oxide ; 1, 3 -butadiene ; benzene JUST marginal increase in HPRT mutability when measured as in vivo exposure (on plant workers populations) In the same time cell line-based or mice/rat-based assays with direct addition of carcinogen show strong increase in HPRT mutability
Strong envinronmental carcinogens ethylene oxide ; 1, 3 -butadiene ; benzene JUST marginal increase in HPRT mutability when measured as in vivo exposure (on plant workers populations) In the same time cell line-based or mice/rat-based assays with direct addition of carcinogen show strong increase in HPRT mutability
 derivates can bind and damage DNA In the Liver Oxidized") polycyclic aromatic hydrocarbons (PAHs) derivates can bind and damage DNA In the Liver Oxidized form of bp
polycyclic aromatic hydrocarbons (PAHs) derivates can bind and damage DNA In the Liver Oxidized form of bp
 Aflatoxin B 1 certain strains of the fungi Aspergillus flavus and A. parasiticus Prevention trial in China oltipraz, an inducer of Phase 2 metabolizing enzymes, significantly increased biomarkers of aflatoxin detoxification
Aflatoxin B 1 certain strains of the fungi Aspergillus flavus and A. parasiticus Prevention trial in China oltipraz, an inducer of Phase 2 metabolizing enzymes, significantly increased biomarkers of aflatoxin detoxification
 EGFR mutation in lung cancer: LUNG CANCER IN NON-SMOKERS Smokers are LESS likely to have this mutation Even second-hand smokers (ETS, environmental tobacco smoke) are more likely to have non-EFGR cancer than EGFR IRESSA treats EGFR mutated cancers (more common in Japan and in smokers) J. Paez et al. , 2004
EGFR mutation in lung cancer: LUNG CANCER IN NON-SMOKERS Smokers are LESS likely to have this mutation Even second-hand smokers (ETS, environmental tobacco smoke) are more likely to have non-EFGR cancer than EGFR IRESSA treats EGFR mutated cancers (more common in Japan and in smokers) J. Paez et al. , 2004
 Rate of mutations in human RATES of molecular events in human genes cells Random gain of mutations (low-level; natural cause) Early stages of natural cancer in elderly; spontaneous mutations in germplasm Forced gain of mutations (median level; X-ray, chemical carcinogens) Early stages of cancer and germplasm in exposed people; Very high rate of mutations (cell lost one or more major mechanisms of DNA repair) Certain genetic syndromes; late stages of almost any cancer
Rate of mutations in human RATES of molecular events in human genes cells Random gain of mutations (low-level; natural cause) Early stages of natural cancer in elderly; spontaneous mutations in germplasm Forced gain of mutations (median level; X-ray, chemical carcinogens) Early stages of cancer and germplasm in exposed people; Very high rate of mutations (cell lost one or more major mechanisms of DNA repair) Certain genetic syndromes; late stages of almost any cancer
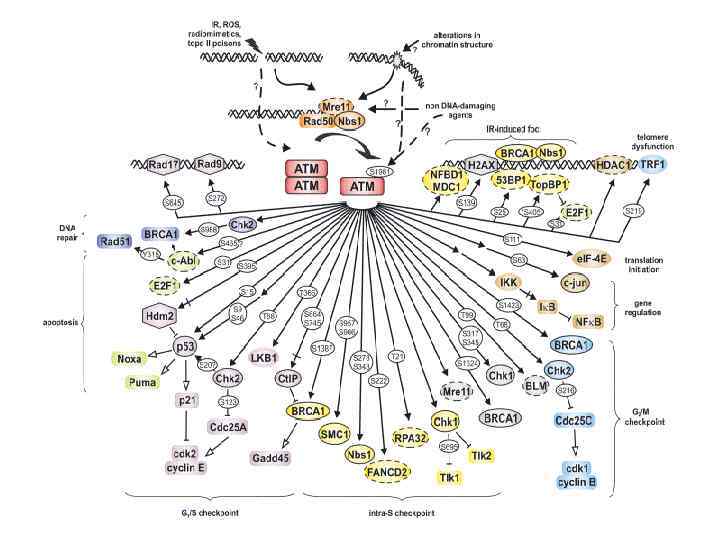
 Amount and type of damage can be handled Damage is excessive and/or irreparable Activation of the apoptosis Activation of the survival response network Low fidelity repair Repair CELL SURVIVAL Give up CANCER Nature Reviews Cancer 3; 155 -168 (2003); doi: 10. 1038/nrc 1011 ATM AND RELATED PROTEIN KINASES: SAFEGUARDING GENOME INTEGRITY CELL DEATH
Amount and type of damage can be handled Damage is excessive and/or irreparable Activation of the apoptosis Activation of the survival response network Low fidelity repair Repair CELL SURVIVAL Give up CANCER Nature Reviews Cancer 3; 155 -168 (2003); doi: 10. 1038/nrc 1011 ATM AND RELATED PROTEIN KINASES: SAFEGUARDING GENOME INTEGRITY CELL DEATH
 DNA repair 1/1000 bp of newly synthesized DNA is incorrect but most of mutations are fixed on spot Only 1/ 1 mln bp are actually miscopied (thanks to the help of DNA repair) Loss of DNA repair mechanisms results in ‘genomic instability’, resulting in massive amount of genetic mutations
DNA repair 1/1000 bp of newly synthesized DNA is incorrect but most of mutations are fixed on spot Only 1/ 1 mln bp are actually miscopied (thanks to the help of DNA repair) Loss of DNA repair mechanisms results in ‘genomic instability’, resulting in massive amount of genetic mutations
 As seen by the graph there is a direct correlation between DNA repair rate and life span.
As seen by the graph there is a direct correlation between DNA repair rate and life span.
 Most syndromes caused by mutations in DNA repair genes cause both progeria/ short life span and cancer
Most syndromes caused by mutations in DNA repair genes cause both progeria/ short life span and cancer
 Many syndromes connected to mutations in reparation-related genes are associated with increase in cancer incidence
Many syndromes connected to mutations in reparation-related genes are associated with increase in cancer incidence
 DNA damage response networks have been uncovered while studying human genetic disorders DNA repair Damage sensing and signaling DNA topology Xeroderma pigmentosum (XP) Cockayne syndrome (CS) Trichothiodystrophy (TTD) Severe combined immunodeficiency (SCID) Ligase IV Syndrome (LIG 4 S) Ataxia Oculomoter Apraxia 1 (AOA 1) Hereditary non-polyposis colon cancer (HNPCC) Fanconi anemia (FA) Ataxia-telangiectasia (A-T) Nijmegen breakage syndrome (NBS) Ataxia-telangiectasia-like disorder (ATLD) Seckel Syndrome Bloom syndrome (BS) Rothmund-Thomson syndrome (RTS) Werner syndrome (WS)
DNA damage response networks have been uncovered while studying human genetic disorders DNA repair Damage sensing and signaling DNA topology Xeroderma pigmentosum (XP) Cockayne syndrome (CS) Trichothiodystrophy (TTD) Severe combined immunodeficiency (SCID) Ligase IV Syndrome (LIG 4 S) Ataxia Oculomoter Apraxia 1 (AOA 1) Hereditary non-polyposis colon cancer (HNPCC) Fanconi anemia (FA) Ataxia-telangiectasia (A-T) Nijmegen breakage syndrome (NBS) Ataxia-telangiectasia-like disorder (ATLD) Seckel Syndrome Bloom syndrome (BS) Rothmund-Thomson syndrome (RTS) Werner syndrome (WS)
, examples of DNA lesions (middle), and the relevant repair mechanisms (bottom).") DNA-damaging agents (top), examples of DNA lesions (middle), and the relevant repair mechanisms (bottom).
DNA-damaging agents (top), examples of DNA lesions (middle), and the relevant repair mechanisms (bottom).
 DNA checkpoint A control mechanism that makes the each stage of the cell cycle dependent on the successful completion of an earlier stage
DNA checkpoint A control mechanism that makes the each stage of the cell cycle dependent on the successful completion of an earlier stage
 Cell Cycle Checkpoints Apoptosis Checkpoint G 2 DNA Damage Checkpoints assembly of components for division S chromosomes replicate Mitosis P M A T Spindle Assembly Checkpoint cytokinesis G 1 cytoplasm doubles DNA Damage Checkpoint
Cell Cycle Checkpoints Apoptosis Checkpoint G 2 DNA Damage Checkpoints assembly of components for division S chromosomes replicate Mitosis P M A T Spindle Assembly Checkpoint cytokinesis G 1 cytoplasm doubles DNA Damage Checkpoint
 Concepts of checkpoint 1. Historical DNA damage checkpoints is a NON-ESSENTIAL regulatory pathways that control the ability of cells to arrest the cell cycle in response to DNA damage, allowing time for repair. 2. Modern Several checkpoint genes (ATR, CHK 1) are essential for cell and organism survival Checkpoint pathways are not only surveyors of occasional damage, but are firmly integrated components of cellular physiology.
Concepts of checkpoint 1. Historical DNA damage checkpoints is a NON-ESSENTIAL regulatory pathways that control the ability of cells to arrest the cell cycle in response to DNA damage, allowing time for repair. 2. Modern Several checkpoint genes (ATR, CHK 1) are essential for cell and organism survival Checkpoint pathways are not only surveyors of occasional damage, but are firmly integrated components of cellular physiology.
 Damage checkpoint pathways Control cell-cycle arrest activate DNA repair pathways Control the movement of DNA repair proteins to sites of DNA damage Activate different transcriptional programms control telomere length induce cell death by apoptosis
Damage checkpoint pathways Control cell-cycle arrest activate DNA repair pathways Control the movement of DNA repair proteins to sites of DNA damage Activate different transcriptional programms control telomere length induce cell death by apoptosis
 as example ATAXIA telangiectasia") Physiological importance of checkpoint pathways • ATM (ataxia telangiectasia) as example ATAXIA telangiectasia
Physiological importance of checkpoint pathways • ATM (ataxia telangiectasia) as example ATAXIA telangiectasia
 1 in 40, 000 -300, 000 live births: Ataxia: loss of") ATM (ataxia telangiectasia) 1 in 40, 000 -300, 000 live births: Ataxia: loss of motor control owing to Purkinje cell loss, masked faces; oculomotor apraxia Telangectasias: Dilated small blood vessels; Skin & Ocular; Onset 4 to 6 years Immune deficiencies: Recurrent respiratory infections; T-cell function is reduced High frequencies of cancer: 14 q+ leukemia (T-CLL) B-cell lymphoma Overall, ~40 -50% of A-T patients will develop some kind of malignancy www. neuro. wustl. edu/neuromuscular/ataxia/dnarep. html
ATM (ataxia telangiectasia) 1 in 40, 000 -300, 000 live births: Ataxia: loss of motor control owing to Purkinje cell loss, masked faces; oculomotor apraxia Telangectasias: Dilated small blood vessels; Skin & Ocular; Onset 4 to 6 years Immune deficiencies: Recurrent respiratory infections; T-cell function is reduced High frequencies of cancer: 14 q+ leukemia (T-CLL) B-cell lymphoma Overall, ~40 -50% of A-T patients will develop some kind of malignancy www. neuro. wustl. edu/neuromuscular/ataxia/dnarep. html
 Why ATM defect leads not only to cancer, but also to organismal defects? Repair-requiring events are the part of the normal life of our body 1. 2. 3. Stalled replication forks needs to be removed during regular cell divisions DSBs in normal VDJ recombination and in meiosis also needs repair Cells under high-oxidative stress and long-living cells alos needs repair – e. g. Purkinje cells (ataxia)
Why ATM defect leads not only to cancer, but also to organismal defects? Repair-requiring events are the part of the normal life of our body 1. 2. 3. Stalled replication forks needs to be removed during regular cell divisions DSBs in normal VDJ recombination and in meiosis also needs repair Cells under high-oxidative stress and long-living cells alos needs repair – e. g. Purkinje cells (ataxia)
 ATM mutations in Ataxia-telangioectasia syndrome locus. umdnj. edu/nigms/ charmut/atmut. html
ATM mutations in Ataxia-telangioectasia syndrome locus. umdnj. edu/nigms/ charmut/atmut. html
 Consequences of standard radiation therapy in an undiagnosed A-T patient
Consequences of standard radiation therapy in an undiagnosed A-T patient
 Survival of ATM +/- and ATM-/- cells after IR HUMAN MICE Normal mice Normal lymphocytes ATM +/- mice +/- parents of ATM patient ATM -/- mice ATM patient Kevin Spring et al. , Nature genetics 2002
Survival of ATM +/- and ATM-/- cells after IR HUMAN MICE Normal mice Normal lymphocytes ATM +/- mice +/- parents of ATM patient ATM -/- mice ATM patient Kevin Spring et al. , Nature genetics 2002
 ATM carriers develop breast cancer in response to radiation (due to enchanced radiosensitivity of the cells) Tuberculosis screening and mammogramms are harmful for ATM carriers
ATM carriers develop breast cancer in response to radiation (due to enchanced radiosensitivity of the cells) Tuberculosis screening and mammogramms are harmful for ATM carriers
 ATM is a true Tumor Suppressor Gene ATM disease is found in 1 in 40, 000 -300, 000 live births (different for different nations) Heterozygotes in populations: 0. 35% to 1% ATM +/- are breast cancer susceptible (9 -16 -time increase in comparison to population) 60% of ATM +/- women develop cancer by the age of 70. Swift et al. NEJM 325: 1831 -1836, 1991 Incidence of female breast cancer in blood relatives of A-T patients
ATM is a true Tumor Suppressor Gene ATM disease is found in 1 in 40, 000 -300, 000 live births (different for different nations) Heterozygotes in populations: 0. 35% to 1% ATM +/- are breast cancer susceptible (9 -16 -time increase in comparison to population) 60% of ATM +/- women develop cancer by the age of 70. Swift et al. NEJM 325: 1831 -1836, 1991 Incidence of female breast cancer in blood relatives of A-T patients
 DNA damage response networks DNA repair is an endpoint of the response of the cell to DNA damaging event Damage Detection Signaling Effectors What proteins detect the damage? unclear cell cycle arrest ATM acts at the first stage of cellular response (after damage recognition) DNA repair apoptosis
DNA damage response networks DNA repair is an endpoint of the response of the cell to DNA damaging event Damage Detection Signaling Effectors What proteins detect the damage? unclear cell cycle arrest ATM acts at the first stage of cellular response (after damage recognition) DNA repair apoptosis
 SUBSTRATES Chk 2, which is involved") ATM acts like any other kinase (via phosphorylation) SUBSTRATES Chk 2, which is involved in control of both the G 1/S and G 2/M cell-cycle checkpoints BRCA 1 NBS 1 tumor suppressor p 53 BIN-BING S. ZHOU AND STEPHEN J. ELLEDGE
ATM acts like any other kinase (via phosphorylation) SUBSTRATES Chk 2, which is involved in control of both the G 1/S and G 2/M cell-cycle checkpoints BRCA 1 NBS 1 tumor suppressor p 53 BIN-BING S. ZHOU AND STEPHEN J. ELLEDGE
 ATM and its DNA-PK family of kinases ATM is activated by the presence of DSBs (Rapid, within 1 minute, response). ATR is activated primarily by stalled replication forks. DNA-PKs can autophosphorylate or be phosphorylated by ATM.
ATM and its DNA-PK family of kinases ATM is activated by the presence of DSBs (Rapid, within 1 minute, response). ATR is activated primarily by stalled replication forks. DNA-PKs can autophosphorylate or be phosphorylated by ATM.