

Наследственные заболевания2.ppt
- Количество слайдов: 51
 Муковисцидоз Докладчик: Студентка 4 курса Педиатрического факультета. Иванова Алеся
Муковисцидоз Докладчик: Студентка 4 курса Педиатрического факультета. Иванова Алеся
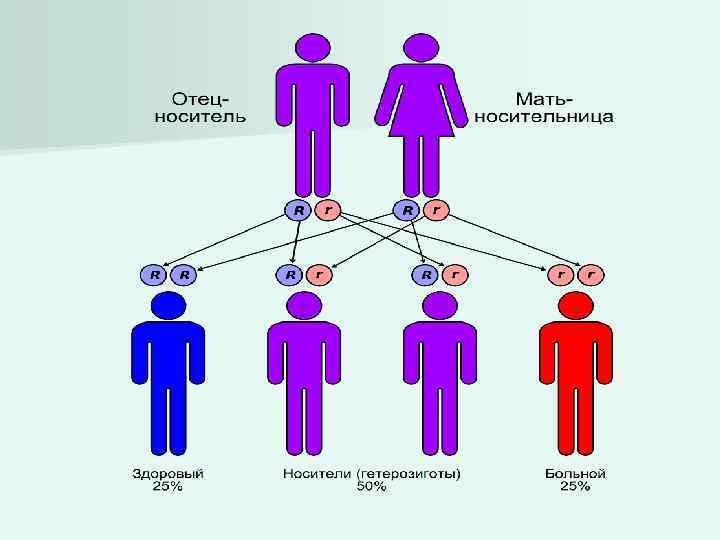
 – аутосомнорецессивное моногенное заболевание, обусловленное мутацией гена МВТР (муковисцидозный трансмембранный") n Муковисцидоз (кистозный фиброз) – аутосомнорецессивное моногенное заболевание, обусловленное мутацией гена МВТР (муковисцидозный трансмембранный регулятор проводимости), характеризующееся поражением экзокринных желез, жизненно важных органов и систем, имеющее тяжелое течение и прогноз.
n Муковисцидоз (кистозный фиброз) – аутосомнорецессивное моногенное заболевание, обусловленное мутацией гена МВТР (муковисцидозный трансмембранный регулятор проводимости), характеризующееся поражением экзокринных желез, жизненно важных органов и систем, имеющее тяжелое течение и прогноз.
 Развитие муковисцидоза связано с мутацией гена, расположенного на 7 -й хромосоме
Развитие муковисцидоза связано с мутацией гена, расположенного на 7 -й хромосоме
 Эпидемиология n n n Россия 1: 10480 Италия 1: 2500 США 1: 5000 Частота носительства мутаций гена CFTR среди белых европейцев– 5 человек из 100 Средняя продолжительность жизни больного МВ в Москве и Санкт-Петербурге равняется 35, 2 года.
Эпидемиология n n n Россия 1: 10480 Италия 1: 2500 США 1: 5000 Частота носительства мутаций гена CFTR среди белых европейцев– 5 человек из 100 Средняя продолжительность жизни больного МВ в Москве и Санкт-Петербурге равняется 35, 2 года.
 (80% всех случаев); — с преимущественно легочными проявлениями") Классификация Формы МВ — смешанная (легочно-кишечная) (80% всех случаев); — с преимущественно легочными проявлениями (15%); — с преимущественно кишечными проявлениями (5%). n
Классификация Формы МВ — смешанная (легочно-кишечная) (80% всех случаев); — с преимущественно легочными проявлениями (15%); — с преимущественно кишечными проявлениями (5%). n
 Патогенез Мутация гена. Дисфункция кодируемого белка. Нарушение транспорта и секреции ионов хлора, нарушения электрического потенциала. Сгущение секрета в клетках поджелудочной железы, эпителии бронхов, слизистой оболочки ЖКТ, семенных канальцев. Формирование вторичных изменений.
Патогенез Мутация гена. Дисфункция кодируемого белка. Нарушение транспорта и секреции ионов хлора, нарушения электрического потенциала. Сгущение секрета в клетках поджелудочной железы, эпителии бронхов, слизистой оболочки ЖКТ, семенных канальцев. Формирование вторичных изменений.
 Бронхолегочная система n Вязкий бронхиальный секрет способствует застою мокроты, ее инфицированию. Нарушается механизм самоочищения бронхов и развивается гнойное воспаление. Разрушению легочной ткани способствует и чрезмерный иммунный ответ организма. Прогрессирующая обструкция бронхов и задержка воздуха может сопровождаться образованием ателектазов и эмфиземы. По мере нарастания тяжести заболевания выявляются распространенные бронхоэктатические изменения и признаки разрушения паренхимы легких, нарастает гипоксемия, развивается легочная гипертензия и легочное сердце.
Бронхолегочная система n Вязкий бронхиальный секрет способствует застою мокроты, ее инфицированию. Нарушается механизм самоочищения бронхов и развивается гнойное воспаление. Разрушению легочной ткани способствует и чрезмерный иммунный ответ организма. Прогрессирующая обструкция бронхов и задержка воздуха может сопровождаться образованием ателектазов и эмфиземы. По мере нарастания тяжести заболевания выявляются распространенные бронхоэктатические изменения и признаки разрушения паренхимы легких, нарастает гипоксемия, развивается легочная гипертензия и легочное сердце.
 Поджелудочная железа и гепатобилиарная система n Застой вязкого секрета поджелудочной железы ведет к нарушению ее внешнесекреторной функции и фиброзированию. Нарушаются процессы переваривания и всасывания жиров и белков, развивается дефицит жирорастворимых витаминов (А, D, Е, К). Нарушение оттока желчи приводит к фиброзу печени и билиарному циррозу.
Поджелудочная железа и гепатобилиарная система n Застой вязкого секрета поджелудочной железы ведет к нарушению ее внешнесекреторной функции и фиброзированию. Нарушаются процессы переваривания и всасывания жиров и белков, развивается дефицит жирорастворимых витаминов (А, D, Е, К). Нарушение оттока желчи приводит к фиброзу печени и билиарному циррозу.
 Желудочно-кишечный тракт n Из-за клейкого секрета слизистой и плотных каловых масс развивается хроническая обструкция дистальных отделов тонкой и проксимальных отделов толстых кишок. Мекониальный илеус — закупорка дистальных отделов тонкой кишки густым и вязким меконием у новорожденных, опасное осложнение — мекониальный перитонит. Гастроэзофагеальный рефлюкс (ГЭР) встречается часто, обусловлен задержкой эвакуации содержимого желудка и нарушением его перистальтики, повышенной продукцией соляной кислоты.
Желудочно-кишечный тракт n Из-за клейкого секрета слизистой и плотных каловых масс развивается хроническая обструкция дистальных отделов тонкой и проксимальных отделов толстых кишок. Мекониальный илеус — закупорка дистальных отделов тонкой кишки густым и вязким меконием у новорожденных, опасное осложнение — мекониальный перитонит. Гастроэзофагеальный рефлюкс (ГЭР) встречается часто, обусловлен задержкой эвакуации содержимого желудка и нарушением его перистальтики, повышенной продукцией соляной кислоты.
 Кожные покровы n Содержание Na. Cl в поте превышает норму в 3– 5 раз. В жару чрезмерная потеря солей приводит к нарушению электролитного баланса и метаболическому алкалозу, возникает тепловой коллапс, рвота, потеря сознания.
Кожные покровы n Содержание Na. Cl в поте превышает норму в 3– 5 раз. В жару чрезмерная потеря солей приводит к нарушению электролитного баланса и метаболическому алкалозу, возникает тепловой коллапс, рвота, потеря сознания.
 развивается азооспермия, связанная с") Репродуктивная система Почти у всех больных муковисцидозом мужского пола (97%) развивается азооспермия, связанная с врожденным отсутствием, атрофией или обструкцией семенного канатика. Соответственно большинство мужчин, больных муковисцидозом, не способно иметь потомство. n У пациентов женского пола МВ сопровождается снижением фертильности: повышенная вязкость отделяемого цервикального канала матки затрудняет миграцию сперматозоидов. Однако многие женщины, страдающие МВ, сохраняют детородную функцию. n
Репродуктивная система Почти у всех больных муковисцидозом мужского пола (97%) развивается азооспермия, связанная с врожденным отсутствием, атрофией или обструкцией семенного канатика. Соответственно большинство мужчин, больных муковисцидозом, не способно иметь потомство. n У пациентов женского пола МВ сопровождается снижением фертильности: повышенная вязкость отделяемого цервикального канала матки затрудняет миграцию сперматозоидов. Однако многие женщины, страдающие МВ, сохраняют детородную функцию. n
 Клинические проявления В грудном возрасте. n n n n n Рецидивирующие или хронические респираторные симптомы, такие как кашель или одышка Повторные пневмонии Мекониальный илеус Отставание в физическом развитие Неоформленный, обильный, маслянистый и зловонный стул Хроническая диарея Выпадение прямой кишки Затяжная неонатальная желтуха Соленый вкус кожи Тепловой удар или дегидратация при жаркой погоде
Клинические проявления В грудном возрасте. n n n n n Рецидивирующие или хронические респираторные симптомы, такие как кашель или одышка Повторные пневмонии Мекониальный илеус Отставание в физическом развитие Неоформленный, обильный, маслянистый и зловонный стул Хроническая диарея Выпадение прямой кишки Затяжная неонатальная желтуха Соленый вкус кожи Тепловой удар или дегидратация при жаркой погоде
 Клинические проявления У детей дошкольного возраста n n n n n Стойкий кашель с или без гнойной мокроты Диагностически неясная рецидивирующая или хроническая одышка Отставание в весе и росте Выпадение прямой кишки, инвагинация Хроническая диарея Симптом «барабанных палочек» Кристаллы соли на коже Гипотоническая дегидратация Гипоэлектролитемия и метаболический алкалоз Гепатомегалия или диагностически неясное нарушение функции печени
Клинические проявления У детей дошкольного возраста n n n n n Стойкий кашель с или без гнойной мокроты Диагностически неясная рецидивирующая или хроническая одышка Отставание в весе и росте Выпадение прямой кишки, инвагинация Хроническая диарея Симптом «барабанных палочек» Кристаллы соли на коже Гипотоническая дегидратация Гипоэлектролитемия и метаболический алкалоз Гепатомегалия или диагностически неясное нарушение функции печени
 Клинические проявления У детей школьного возраста n n n Хронические респираторные симптомы неясной этиологии Pseudomonas aeruginosa в мокроте Хронический синусит Назальный полипоз Бронхоэктазы Симптом “барабанных палочек” Хроническая диарея Панкреатит Выпадение прямой кишки Сахарный диабет в сочетании с респираторными симптомами Гепатомегалия Заболевание печени неясной этиологии
Клинические проявления У детей школьного возраста n n n Хронические респираторные симптомы неясной этиологии Pseudomonas aeruginosa в мокроте Хронический синусит Назальный полипоз Бронхоэктазы Симптом “барабанных палочек” Хроническая диарея Панкреатит Выпадение прямой кишки Сахарный диабет в сочетании с респираторными симптомами Гепатомегалия Заболевание печени неясной этиологии
 Клинические проявления. У подростков и взрослых n n n n n Гнойное заболевание легких неясной этиологии Симптом “барабанных палочек” Панкреатит Синдром дистальной интестинальной обструкции Сахарный диабет в сочетании с респираторными симптомами Признаки цирроза печени и портальной гипертензии Отставание в росте Задержка полового развития Стерильность с азооспермией у лиц мужского пола Снижение фертильности у лиц женского пола
Клинические проявления. У подростков и взрослых n n n n n Гнойное заболевание легких неясной этиологии Симптом “барабанных палочек” Панкреатит Синдром дистальной интестинальной обструкции Сахарный диабет в сочетании с респираторными симптомами Признаки цирроза печени и портальной гипертензии Отставание в росте Задержка полового развития Стерильность с азооспермией у лиц мужского пола Снижение фертильности у лиц женского пола
 Потовая проба Б) Копрологическое исследование 3.") Диагностика 1. Анамнез. n 2. Лабораторные исследования. А) Потовая проба Б) Копрологическое исследование 3. Инструментальные исследования. А) Рентгенография грудной клетки Б) Исследование ФВД В) Измерение разницы назальных потенциалов n 4. Генетический анализ. n 5. Пренатальная диагностика. n 6. Неонатальная диагностика. • первое определение концентрации иммунореактивного трипсина (на первой неделе жизни); • повторное определение концентрации иммунореактивного трипсина (3– 4 -я неделя жизни); • проведение потового теста; • ДНК-диагностика. n
Диагностика 1. Анамнез. n 2. Лабораторные исследования. А) Потовая проба Б) Копрологическое исследование 3. Инструментальные исследования. А) Рентгенография грудной клетки Б) Исследование ФВД В) Измерение разницы назальных потенциалов n 4. Генетический анализ. n 5. Пренатальная диагностика. n 6. Неонатальная диагностика. • первое определение концентрации иммунореактивного трипсина (на первой неделе жизни); • повторное определение концентрации иммунореактивного трипсина (3– 4 -я неделя жизни); • проведение потового теста; • ДНК-диагностика. n
 Лечение n n n n 1. Кинезитерапия. 2. Муколитическая терапия. 3. Антибактериальная терапия. 4. Противовоспалительная терапия. 5. Заместительная ферментотерапия. 6. Витамины (А, Д, Е, К) 7. Диетотерапия 8. Генная терапия
Лечение n n n n 1. Кинезитерапия. 2. Муколитическая терапия. 3. Антибактериальная терапия. 4. Противовоспалительная терапия. 5. Заместительная ферментотерапия. 6. Витамины (А, Д, Е, К) 7. Диетотерапия 8. Генная терапия
 Недостаточность функции") Дифференциальная диагностика n n n n n n Синдром приобретенного иммунодефицита (СПИД) Недостаточность функции надпочечников Псевдогипоальдостеронизм Адреногенитальный синдром Синдром Дауна Синдром Клайнфелтера Атопический дерматит Эктодермальная дисплазия Семейный холестатический синдром Фукозидоз Гликогеноз, тип II Недостаточность глюкозо-6 -фосфатазы Гипотиреоз Гипопаратиреоз Резко выраженная гипотрофия (кахексия) Нервная анорексия Синдром Мориака Мукополисахаридоз Нефрогенный несахарный диабет Хронический панкреатит Гипогаммаглобулинемия Целиакия
Дифференциальная диагностика n n n n n n Синдром приобретенного иммунодефицита (СПИД) Недостаточность функции надпочечников Псевдогипоальдостеронизм Адреногенитальный синдром Синдром Дауна Синдром Клайнфелтера Атопический дерматит Эктодермальная дисплазия Семейный холестатический синдром Фукозидоз Гликогеноз, тип II Недостаточность глюкозо-6 -фосфатазы Гипотиреоз Гипопаратиреоз Резко выраженная гипотрофия (кахексия) Нервная анорексия Синдром Мориака Мукополисахаридоз Нефрогенный несахарный диабет Хронический панкреатит Гипогаммаглобулинемия Целиакия
 Осложнения муковисцидоза n n n n n Дыхательная и сердечная недостаточность Обструкция дистальных отделов тонкой кишки. Назальный полипоз. Холелитиаз. Сахарный диабет. Фиброз и цирроз печени. Бесплодие. Легочные и желудочные кровотечения. Ателектазы. Пневмоторакс.
Осложнения муковисцидоза n n n n n Дыхательная и сердечная недостаточность Обструкция дистальных отделов тонкой кишки. Назальный полипоз. Холелитиаз. Сахарный диабет. Фиброз и цирроз печени. Бесплодие. Легочные и желудочные кровотечения. Ателектазы. Пневмоторакс.
 Фенилкетонурия
Фенилкетонурия
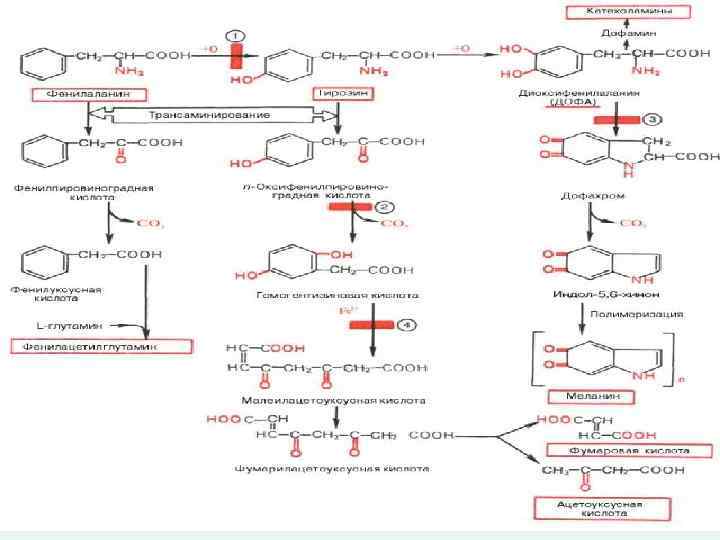
 n Фенилкетонурия – группа аутосомнорецессивных заболеваний, связанных с нарушением аминокислотного метаболизма и приводящие к поражению главным образом ЦНС.
n Фенилкетонурия – группа аутосомнорецессивных заболеваний, связанных с нарушением аминокислотного метаболизма и приводящие к поражению главным образом ЦНС.
 n Фенилкетонурия III") Классификация n Фенилкетонурия I типа(классическая фенилкетонурия) n Фенилкетонурия III
Классификация n Фенилкетонурия I типа(классическая фенилкетонурия) n Фенилкетонурия III
 - это заболевание с аутосомно-рецессивным типом наследования. В") n Классическая фенилкетонурия (фенилкетонурия I типа) - это заболевание с аутосомно-рецессивным типом наследования. В основе болезни лежит дефицит фермента фенилаланин-4 -гидроксилаза, обеспечивающего превращения фенилаланина в тирозин. Россия 1: 9000 Ирландия 1: 4560 Япония 1: 100000
n Классическая фенилкетонурия (фенилкетонурия I типа) - это заболевание с аутосомно-рецессивным типом наследования. В основе болезни лежит дефицит фермента фенилаланин-4 -гидроксилаза, обеспечивающего превращения фенилаланина в тирозин. Россия 1: 9000 Ирландия 1: 4560 Япония 1: 100000
 Клиническая картина n n n n Фенилкетонурия проявляется на первом году жизни. отсутствие интереса к окружающему; повышенная раздражительность; беспокойство; срыгивания; мышечные судороги; дерматит, экзема появляется характерный «мышиный» запах ; признаки аллергического дерматита ; задержка статикомоторного и психоречевого развития гипопигментация кожи, волос, радужной оболочки глаз ; атаксия ; гиперкинезы ; тремор ; эпилептические припадки ; микроцефалия ; умственная отсталость ;
Клиническая картина n n n n Фенилкетонурия проявляется на первом году жизни. отсутствие интереса к окружающему; повышенная раздражительность; беспокойство; срыгивания; мышечные судороги; дерматит, экзема появляется характерный «мышиный» запах ; признаки аллергического дерматита ; задержка статикомоторного и психоречевого развития гипопигментация кожи, волос, радужной оболочки глаз ; атаксия ; гиперкинезы ; тремор ; эпилептические припадки ; микроцефалия ; умственная отсталость ;
 n Фенилкетонурия II- это заболевание с аутосомнорецессивным типом наследования. При этом состоянии был обнаружен дефект дигидроптеридинредуктазы. Вследствие которого развиваются метаболические блоки на путях превращения фенилаланина в тирозин, а также образования предшественников нейромедиаторов катехоламинового и серотонинового ряда.
n Фенилкетонурия II- это заболевание с аутосомнорецессивным типом наследования. При этом состоянии был обнаружен дефект дигидроптеридинредуктазы. Вследствие которого развиваются метаболические блоки на путях превращения фенилаланина в тирозин, а также образования предшественников нейромедиаторов катехоламинового и серотонинового ряда.
 Клиническая картина n Тяжелая умственная отсталость n Судороги n Признаки повышенной возбудимости n Сухожильная гиперрефлексия n Мышечная дистония n Спастический тетрапарез Течение болезни прогрессирующее и нередко приводит к смерти в возрасте 2 -3 лет
Клиническая картина n Тяжелая умственная отсталость n Судороги n Признаки повышенной возбудимости n Сухожильная гиперрефлексия n Мышечная дистония n Спастический тетрапарез Течение болезни прогрессирующее и нередко приводит к смерти в возрасте 2 -3 лет
 n Фенилкетонурия III - это заболевание с аутосомнорецессивным типом наследования и связано с недостаточностью 6 -пирувоилтетрагидроптеринсинтазы, участвующей в процессе синтеза тетрогидроптерина из дигидроптерин трифосфат.
n Фенилкетонурия III - это заболевание с аутосомнорецессивным типом наследования и связано с недостаточностью 6 -пирувоилтетрагидроптеринсинтазы, участвующей в процессе синтеза тетрогидроптерина из дигидроптерин трифосфат.
 Клиническая картина n n n Тяжелая умственная отсталость Признаки повышенной возбудимости Сухожильная гиперрефлексия Мышечная дистония Спастический тетрапарез Микроцефалия
Клиническая картина n n n Тяжелая умственная отсталость Признаки повышенной возбудимости Сухожильная гиперрефлексия Мышечная дистония Спастический тетрапарез Микроцефалия
 Транзиторная ГФА Умеренно повышение ФА при глубокой недоношенности или функциональной незрелости. n После дозревания системы окисления тирозина у ребенка нормализуется уровень ФА и тирозина. n
Транзиторная ГФА Умеренно повышение ФА при глубокой недоношенности или функциональной незрелости. n После дозревания системы окисления тирозина у ребенка нормализуется уровень ФА и тирозина. n
 Материнская ФКУ У женщин больных ФКУ, рождались дети с умственной отсталостью. n Тяжесть поражения плода коррелирует с концентрацией ФА в крови матери. ФА проникает через ГЭБ и накапливается в плаценте. n Адекватная диетотерапия может предупредить большинство проявлений материнской ФКУ. n
Материнская ФКУ У женщин больных ФКУ, рождались дети с умственной отсталостью. n Тяжесть поражения плода коррелирует с концентрацией ФА в крови матери. ФА проникает через ГЭБ и накапливается в плаценте. n Адекватная диетотерапия может предупредить большинство проявлений материнской ФКУ. n
 Диагностика. 1 этап-массовый скрининг новорожденных. n 2 этап- тонкослойная хроматография, молекулярногенетический метод. n
Диагностика. 1 этап-массовый скрининг новорожденных. n 2 этап- тонкослойная хроматография, молекулярногенетический метод. n
 Лечение n Диетотерапия – патогенетически обоснованный и наиболее эффективный метод лечения классической ФКУ; ее основной целью является предупреждение развития повреждения ЦНС, нарушений физического и интеллектуального развития. Диетотерапия должна быть начата не позднее первых недель жизни ребенка.
Лечение n Диетотерапия – патогенетически обоснованный и наиболее эффективный метод лечения классической ФКУ; ее основной целью является предупреждение развития повреждения ЦНС, нарушений физического и интеллектуального развития. Диетотерапия должна быть начата не позднее первых недель жизни ребенка.
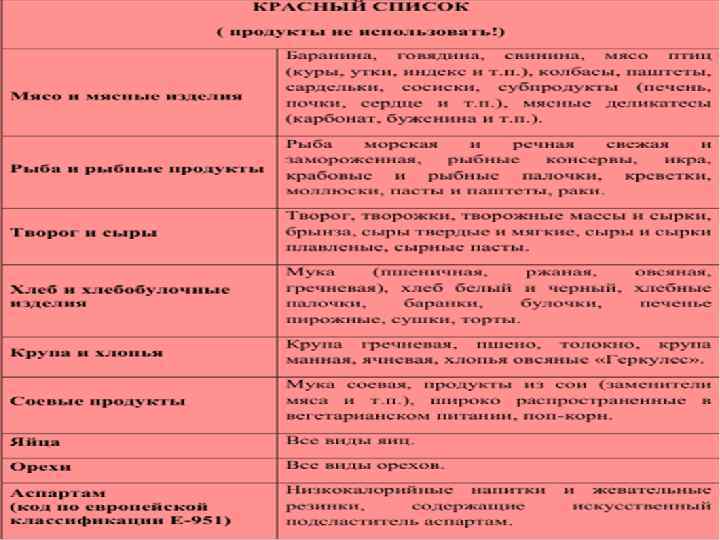
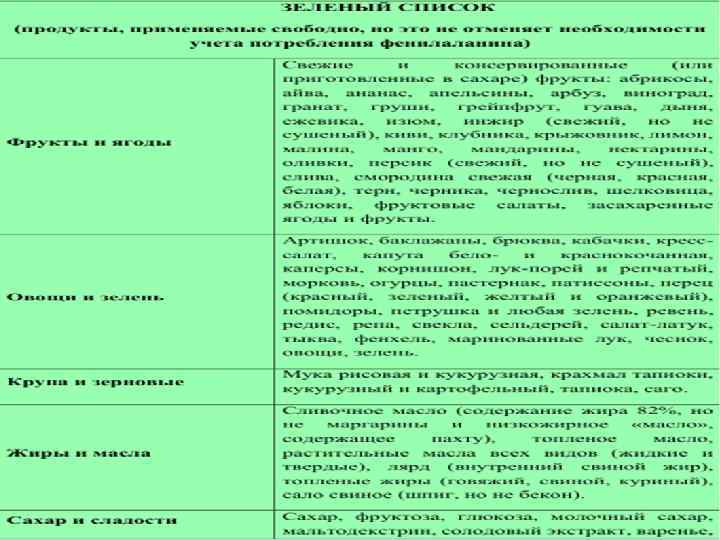
 n Меню вегетарианского типа с использованием малобелковых продуктов питания и ежедневное употребление лечебного продукта в виде смеси аминокислот или гидролизата белка, которые не содержат ФА с добавлением витаминов и микроэлементов. Для детей до 1 года – « Афенилак» , «Лофеналак» . После года- «Тетрафен» , «Фенил-фри» , «Максамаид ХР» .
n Меню вегетарианского типа с использованием малобелковых продуктов питания и ежедневное употребление лечебного продукта в виде смеси аминокислот или гидролизата белка, которые не содержат ФА с добавлением витаминов и микроэлементов. Для детей до 1 года – « Афенилак» , «Лофеналак» . После года- «Тетрафен» , «Фенил-фри» , «Максамаид ХР» .

 Лечение птерин-зависимых форм ФКУ n Препарат L-дофы n 5 -окситриптофан n Тетрагидроптерин
Лечение птерин-зависимых форм ФКУ n Препарат L-дофы n 5 -окситриптофан n Тетрагидроптерин
 Критерий эффективности лечения n n n в возрасте до 3 -х месяцев - 1 раз в неделю (до получения стабильных результатов) и далее не менее 2 -х раз в месяц от 3 -х месяцев до 6 лет – 1 раз в месяц, при необходимости -2 раза в месяц с 7 до 12 лет - не менее 1 раза в 2 месяца, после 12 лет - 1 раз в 3 месяца, у беременных с ФКУ 1 раз в 7 - 10 дней.
Критерий эффективности лечения n n n в возрасте до 3 -х месяцев - 1 раз в неделю (до получения стабильных результатов) и далее не менее 2 -х раз в месяц от 3 -х месяцев до 6 лет – 1 раз в месяц, при необходимости -2 раза в месяц с 7 до 12 лет - не менее 1 раза в 2 месяца, после 12 лет - 1 раз в 3 месяца, у беременных с ФКУ 1 раз в 7 - 10 дней.
 Галактоземия
Галактоземия
 n Галактоземия- наследственное аутосомнорецессивное нарушение обмена углеводов, при котором накапливается избыток галактозы и ее метаболитов, что обуславливает клиническую картину заболевания и формирует отсроченных осложнений.
n Галактоземия- наследственное аутосомнорецессивное нарушение обмена углеводов, при котором накапливается избыток галактозы и ее метаболитов, что обуславливает клиническую картину заболевания и формирует отсроченных осложнений.
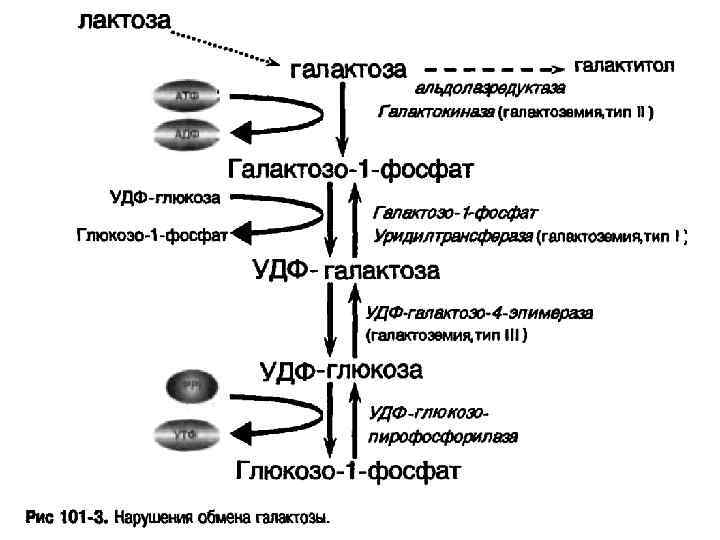
 Эпидемиология Галактоземия, тип I – панэтническое заболеание, частота странах Европы составляет в среднем 1: 40000 живых новорожденных. n Галактоземия, тип I I-в странах Европы частота заболевания составляет 1: 150000 живых новорожденных. Более высокая заболеваемость отмечена в Румынии и Болгарии. С высокой частотой встречается у цыган 1: 2500. n Галактоземия, тип I I I – редкая наследственная болезнь обмена веществ. Доброкачественная форма широко распространена в Японии(1: 23000 новорожденных). n
Эпидемиология Галактоземия, тип I – панэтническое заболеание, частота странах Европы составляет в среднем 1: 40000 живых новорожденных. n Галактоземия, тип I I-в странах Европы частота заболевания составляет 1: 150000 живых новорожденных. Более высокая заболеваемость отмечена в Румынии и Болгарии. С высокой частотой встречается у цыган 1: 2500. n Галактоземия, тип I I I – редкая наследственная болезнь обмена веществ. Доброкачественная форма широко распространена в Японии(1: 23000 новорожденных). n
. n Галактоземия") Классификация Классическая - галактоземия I типа, обусловленная дефицитом фермента галактозо-1 -фосфатуридилтрансферазы (ГАЛТ). n Галактоземия I I-недостаточность галактокиназы (ГАЛК). n Галактоземия I I I-дефицит уридиндифосфат-галактозо -4 -эпимеразы. n
Классификация Классическая - галактоземия I типа, обусловленная дефицитом фермента галактозо-1 -фосфатуридилтрансферазы (ГАЛТ). n Галактоземия I I-недостаточность галактокиназы (ГАЛК). n Галактоземия I I I-дефицит уридиндифосфат-галактозо -4 -эпимеразы. n
 Клиническая картина Галактоземия I тип. n Манифестация на 1 -2 неделе n Частые срыгивания n Диарея n Гепатоспленомегалия n Гипербилирубинемия n Гипоальбунемия n Асцит n Катаракта n Острая печеночная недостаточность n Сепсис n Затруднена организация речевых движений n Гипергонадотропный гипогонанизм
Клиническая картина Галактоземия I тип. n Манифестация на 1 -2 неделе n Частые срыгивания n Диарея n Гепатоспленомегалия n Гипербилирубинемия n Гипоальбунемия n Асцит n Катаракта n Острая печеночная недостаточность n Сепсис n Затруднена организация речевых движений n Гипергонадотропный гипогонанизм
 Клиническая картина Галактоземия I I тип. n Единственное проявление заболевания-катаракта. Галактоземия I I I тип. Доброкачественная Тяжелая Дефицит фермента желтуха только в циркулирующих рвота клетках крови. задержка развития гепатоспленомегалия катаракта сепсис нейросенсорная глухота
Клиническая картина Галактоземия I I тип. n Единственное проявление заболевания-катаракта. Галактоземия I I I тип. Доброкачественная Тяжелая Дефицит фермента желтуха только в циркулирующих рвота клетках крови. задержка развития гепатоспленомегалия катаракта сепсис нейросенсорная глухота
 Лабораторная диагностика Галактоземия, тип I- биохимическое определение концентрации галактозы и галактозо- I-фосфата в крови и/или измерение активности фермента галактозо- I-фосфатуридилтрансферазы в эритроцитах. ДНК-диагностика. n Галактоземия, тип I I-заболевание диагностируется при проведение массового скрининга на галактоземию. Диагноз подтерждается путем определения активности фермента в эритроцитах или фибробластах. ДНК-диагностика. n Галактоземия, тип I I I- определение активности фермента в эритроцитах крови и содержание галактозы и галактозо-1 -фосфата. ДНК-диагностика. n
Лабораторная диагностика Галактоземия, тип I- биохимическое определение концентрации галактозы и галактозо- I-фосфата в крови и/или измерение активности фермента галактозо- I-фосфатуридилтрансферазы в эритроцитах. ДНК-диагностика. n Галактоземия, тип I I-заболевание диагностируется при проведение массового скрининга на галактоземию. Диагноз подтерждается путем определения активности фермента в эритроцитах или фибробластах. ДНК-диагностика. n Галактоземия, тип I I I- определение активности фермента в эритроцитах крови и содержание галактозы и галактозо-1 -фосфата. ДНК-диагностика. n
 Лечение n n n Диетотерапия Урацил-4 -карбоновая кислота Гепатопротекторы Антиоксиданты Хирургическое лечение катаракты
Лечение n n n Диетотерапия Урацил-4 -карбоновая кислота Гепатопротекторы Антиоксиданты Хирургическое лечение катаракты
 Список литературы n n n n Шабалов Н. П. Детские болезни. -Питер, 2012. Баранов А. А. Педиатрия. Национальное руководство. М: ГЭОТАР-Медиа, 2014 Федеральные клинические рекомендации по диагностике и лечению фенилкетонурии. http: //www. ncagip. ru/ Баранов А. А. Педиатрия. Национальное руководство. Том 2. -М: ГЭОТАР-Медиа, 2009. http: //mukoviscidoz. org/ Федеральные клинические рекомендации по оказанию медицинской помощи детям с муковисцидозом.
Список литературы n n n n Шабалов Н. П. Детские болезни. -Питер, 2012. Баранов А. А. Педиатрия. Национальное руководство. М: ГЭОТАР-Медиа, 2014 Федеральные клинические рекомендации по диагностике и лечению фенилкетонурии. http: //www. ncagip. ru/ Баранов А. А. Педиатрия. Национальное руководство. Том 2. -М: ГЭОТАР-Медиа, 2009. http: //mukoviscidoz. org/ Федеральные клинические рекомендации по оказанию медицинской помощи детям с муковисцидозом.
