

ba1710ee88469e5041f6527473b24744.ppt
- Количество слайдов: 86
 Molecular diagnosis of heterogeneous genetic diseases: the example of muscular dystrophies Vincenzo Nigro Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli Telethon Institute of Genetics and Medicine (TIGEM)
Molecular diagnosis of heterogeneous genetic diseases: the example of muscular dystrophies Vincenzo Nigro Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli Telethon Institute of Genetics and Medicine (TIGEM)
 What is a mutation? A variation of the DNA sequence n that is only found in affected individuals n that is never found in non affected individuals n that accounts for the pathological process/status n that, when corrected in time, disease is rescued
What is a mutation? A variation of the DNA sequence n that is only found in affected individuals n that is never found in non affected individuals n that accounts for the pathological process/status n that, when corrected in time, disease is rescued
 . . that is only found in affected and that is never found in non affected incomplete penetrance that is more often found in affected than in non affected. . .
. . that is only found in affected and that is never found in non affected incomplete penetrance that is more often found in affected than in non affected. . .
 50. 000 private variants = innocuous differences belonging to one family CCCCAGCCTCCTTGCCAACGCCCCCTTTCCCTCTCCCCCTCCCGCTCGGCGCTGACC CCCCATCCCCACCCCCGTGGGAACACTGGGAGCCTGCACTCCACAGACCCTCTCCTT GCCTCTTCCCTCAGCCTCCGCTCCCCGCCCTCTTCCCGGCCCAGGGCGCCG GCCCACCCTTCCCTCCGCCGCCCCCCGGCCGCGGGGAGGACATGGCCGCGCACAG GCCGGTGGAATGGGTCCAGGCCGTGGTCAGCCGCTTCGACGAGCAGCTTCCAATAA AAACAGGACAGCAGAACACACATACCAAAGTCAGTACTGAGCACAACAAGGAATGTC TAATCAATATTTCCAAATACAAGTTTTCTTTGGTTATAAGCGGCCTCACTACTATTTTAA AGAATGTTAACAATATGAGAATATTTGGAGAAGCTGCTGAAAAAAATTTATATCTCTCT CAGTTGATTATATTGGATACACTGGAAAAATGTCTTGCTGGGCAACCAAAGGACACAA TGAGATTAGATGAAACGATGCTGGTCAAACAGTTGCTGCCAGAAATCTGCCATTTTCT TCACACCTGTCGTGAAGGAAACCAGCATGCAGCTGAACTTCGGAATTCTGCCTCTGG GGTTTTATTTTCTCTCAGCTGCAACAACTTCAATGCAGTCTTTAGTCGCATTTCTACCA GGTTACAGGAATTAACTGTTCAGAAGACAATGTTGATGTTCATGATATAGAATTG TTACAGTATATCAATGTGGATTGTGCAAAATTAAAACGACTCCTGAAGGAAACAGCAT TTAAATTTAAAGCCCTAAAGAAGGTTGCGCAGTTATAAATAGCCTGGAAAA GGCATTTTGGAACTGGGTAGAAAATTATCCAGATGAATTTACAAAACTGTACCAGATC CCACAGACTGATATGGCTGAATGTGCAGAAAAGCTATTTGACTTGGTGGATGGTTTTG CTGAAAGCACCAAACGTAAAGCAGCAGTTTGGCCACTACAAATCATTCTCCTTATCTT GTGTCCAGAAATAATCCAGGATATATCCAAAGACGTGGTTGATGAAAACAACATGAAT AAGAAGTTATTTCTGGACAGTCTACGAAAAGCTCTTGCTGGCCATGGAGGAAGTAGG CAGCTGACAGAAAGTGCTGCAATTGCCTGTGTCAAACTGTGTAAAGCAAGTACTTACA TCAATTGGGAAGATAACTCTGTCATTTTCCTACTTGTTCAGTCCATGGTGGTTGATCTT AAGAACCTGCTTTTTAATCCAAGTAAGCCATTCTCAAGAGGCAGTCAGCCTGCAGATG TGGATCTAATGATTGACTGCCTTGTTTCTTGCTTTCGTATAAGCCCTCACAACAACCAA CACTTTAAGATCTGCCTGGCTCAGAATTCACCTTCTACATTTCACTATGTGCTGGTAAA TTCACTCCATCGAATCATCACCAATTCCGCATTGGTGGCCTAAGATTGATGCT GTGTATTGTCACTCGGTTGAACTTCGAAATATGTTTGGTGAAACACTTCATAAAGCAGT GCAAGGTTGTGGAGCACACCCAGCAATACGAATGGCACCGAGTCTTACATTTAAAGA AAAAGTAACAAGCCTTAAATTTAAAGAAAAACCTACAGACCTGGAGACAAGAAGCTAT AAGTATCTTCTCTTGTCCATGGTGAAACTAATTCATGCAGATCCAAAGCTCTTGCTTTG TAATCCAAGAAAACAGGGGCCCGAAACCCAAGGCAGTACAGCAGAATTACAGG GCTCGTCCAACTGGTCCCTCAGTCACACATGCCAGAGATTGCTCAGGAAGCAATGGA GGCTCTGCTGGTTCTTCATCAGTTAGATAGCATTGATTTGTGGAATCCTGATGCTCCT GTAGAAACATTTTGGGAGATTAGCTCACAAATGCTTTTTTACATCTGCAAGAAATTAAC TAGTCATCAAATGCTTAGTAGCACAGAAATTCTCAAGTGGTTGCGGGAAATATTGATC TGCAGGAATAAATTTCTTCTTAAAAATAAGCAGATAGAAGTTCCTGTCACTTTC CCCCAGCCTCCTTGCCAACGCCCCCTTTCCCTCTCCCCCTCCCGCTCGGCGCTGACC CCCCATCCCCACCCCCGTGGGAACACTGGGAGCCTGCACTCCACAGACCCTCTCCTT GCCTCTTCCCTCAGCCTCCGCTCCCCGCCCTCTTCCCGGCCCAGGGCGCCG GCCCACCCTTCCCTCCGCCGCCCCCCGGCCGCGGGGAGGACATGGCCGCGCACAG GCCGGTGGAATGGGTCCAGGCCGTGGTCAGCCGCTTCGACGAGCAGCTTCCAATAA AAACAGGACAGCAGAACACACATACCAAAGTCAGTACTGAGCACAACAAGGAATGTC TAATCAATATTTCCAAATACAAGTTTTCTTTGGTTATAAGCGGCCTCACTACTATTTTAA AGAATGTTAACTATATGAGAATATTTGGAGAAGCTGCTGAAAAAAATTTATATCTCTCT CAGTTGATTATATTGGATACACTGGAAAAATGTCTTGCTGGGCAACCAAAGGACACAA TGAGATTAGATGAAACGATGCTGGTCAAACAGTTGCTGCCAGAAATCTGCCATTTTCT TCACACCTGTCGTGAAGGAAACCAGCATGCAGCTGAACTTCGGAATTCTGCCTCTGG GGTTTTATTTTCTCTCAGCTGCAACAACTTCAATGCAGTCTTTAGTCGCATTTCTACCA GGTTACAGGAATTAACTGTTCAGAAGACAATGTTGATGTTCATGATATAGAATTG TTACAGTATATCAATGTGGATTGTGCAAAATTAAAACGACTCCTGAAGGAAACAGCAT TTAAATTTAAAGCCCTAAAGAAGGTTGCGCAGTTATAAATAGCCTGGAAAA GGCATTTTGGAACTGGGTAGAAAATTATCCAGATGAATTTACAAAACTGTACCAGATC CCACAGACTGATATGGCTGAATGTGCAGAAAAGCTATTTGACTTGGTGGATGGTTTTG CTGAAAGCACCAAACGTAAAGCAGCAGTTTGGCCACTACAAATCATTCTCCTTATCTT GTGTCCAGAAATAATCCAGGATATATCCAAAGACGTGGTTGATGAAAACAACATGAAT AAGAAGTTATTTCTGGACAGTCTACGAAAAGCTCTTGCTGGCCATGGAGGAAGTAGG CAGCTGACAGAAAGTGCTGCAATTGCCTGTGTCAAACTGTGTAAAGCAAGTACTTACA TCAATTGGGAAGATAACTCTGTCATTTTCCTACTTGTTCAGTCCATGGTGGTTGATCTT AAGAACCTGCTTTTTAATCCAAGTAAGCCATTCTCAAGAGGCAGTCAGCCTGCAGATG TGGATCTAATGATTGACTGCCTTGTTTCTTGCTTTCGTATAAGCCCTCACAACAACCAA CACTTTAAGATCTGCCTGGCTCAGAATTCACCTTCTACATTTCACTATGTGCTGGTAAA TTCACTCCATCGAATCATCACCAATTCCGCATTGGTGGCCTAAGATTGATGCT GTGTATTGTCACTCGGTTGAACTTCGAAATATGTTTGGTGAAACACTTCATAAAGCAGT GCAAGGTTGTGGAGCACACCCAGCAATACGAATGGCACCGAGTCTTACATTTAAAGA AAAAGTAACAAGCCTTAAATTTAAAGAAAAACCTACAGACCTGGAGACAAGAAGCTAT AAGTATCTTCTCTTGTCCATGGTGAAACTAATTCATGCAGCTCCAAAGCTCTTGCTTTG TAATCCAAGAAAACAGGGGCCCGAAACCCAAGGCAGTACAGCAGAATTACAGG GCTCGTCCAACTGGTCCCTCAGTCACACATGCCAGAGATTGCTCAGGAAGCAATGGA GGCTCTGCTGGTTCTTCATCAGTTAGATAGCATTGATTTGTGGAATCCTGATGCTCCT GTAGAAACATTTTGGGAGATTAGCTCACAAATGCTTTTTTACATCTGCAAGAAATTAAC TAGTCATCAAATGCTTAGTAGCACAGAAATTCTCAAGTGGTTGCGGGAAATATTGATC TGCAGGAATAAATTTCTTCTTAAAAATAAGCAGATAGAAGTTCCTGTCACTTTC
50. 000 private variants = innocuous differences belonging to one family CCCCAGCCTCCTTGCCAACGCCCCCTTTCCCTCTCCCCCTCCCGCTCGGCGCTGACC CCCCATCCCCACCCCCGTGGGAACACTGGGAGCCTGCACTCCACAGACCCTCTCCTT GCCTCTTCCCTCAGCCTCCGCTCCCCGCCCTCTTCCCGGCCCAGGGCGCCG GCCCACCCTTCCCTCCGCCGCCCCCCGGCCGCGGGGAGGACATGGCCGCGCACAG GCCGGTGGAATGGGTCCAGGCCGTGGTCAGCCGCTTCGACGAGCAGCTTCCAATAA AAACAGGACAGCAGAACACACATACCAAAGTCAGTACTGAGCACAACAAGGAATGTC TAATCAATATTTCCAAATACAAGTTTTCTTTGGTTATAAGCGGCCTCACTACTATTTTAA AGAATGTTAACAATATGAGAATATTTGGAGAAGCTGCTGAAAAAAATTTATATCTCTCT CAGTTGATTATATTGGATACACTGGAAAAATGTCTTGCTGGGCAACCAAAGGACACAA TGAGATTAGATGAAACGATGCTGGTCAAACAGTTGCTGCCAGAAATCTGCCATTTTCT TCACACCTGTCGTGAAGGAAACCAGCATGCAGCTGAACTTCGGAATTCTGCCTCTGG GGTTTTATTTTCTCTCAGCTGCAACAACTTCAATGCAGTCTTTAGTCGCATTTCTACCA GGTTACAGGAATTAACTGTTCAGAAGACAATGTTGATGTTCATGATATAGAATTG TTACAGTATATCAATGTGGATTGTGCAAAATTAAAACGACTCCTGAAGGAAACAGCAT TTAAATTTAAAGCCCTAAAGAAGGTTGCGCAGTTATAAATAGCCTGGAAAA GGCATTTTGGAACTGGGTAGAAAATTATCCAGATGAATTTACAAAACTGTACCAGATC CCACAGACTGATATGGCTGAATGTGCAGAAAAGCTATTTGACTTGGTGGATGGTTTTG CTGAAAGCACCAAACGTAAAGCAGCAGTTTGGCCACTACAAATCATTCTCCTTATCTT GTGTCCAGAAATAATCCAGGATATATCCAAAGACGTGGTTGATGAAAACAACATGAAT AAGAAGTTATTTCTGGACAGTCTACGAAAAGCTCTTGCTGGCCATGGAGGAAGTAGG CAGCTGACAGAAAGTGCTGCAATTGCCTGTGTCAAACTGTGTAAAGCAAGTACTTACA TCAATTGGGAAGATAACTCTGTCATTTTCCTACTTGTTCAGTCCATGGTGGTTGATCTT AAGAACCTGCTTTTTAATCCAAGTAAGCCATTCTCAAGAGGCAGTCAGCCTGCAGATG TGGATCTAATGATTGACTGCCTTGTTTCTTGCTTTCGTATAAGCCCTCACAACAACCAA CACTTTAAGATCTGCCTGGCTCAGAATTCACCTTCTACATTTCACTATGTGCTGGTAAA TTCACTCCATCGAATCATCACCAATTCCGCATTGGTGGCCTAAGATTGATGCT GTGTATTGTCACTCGGTTGAACTTCGAAATATGTTTGGTGAAACACTTCATAAAGCAGT GCAAGGTTGTGGAGCACACCCAGCAATACGAATGGCACCGAGTCTTACATTTAAAGA AAAAGTAACAAGCCTTAAATTTAAAGAAAAACCTACAGACCTGGAGACAAGAAGCTAT AAGTATCTTCTCTTGTCCATGGTGAAACTAATTCATGCAGATCCAAAGCTCTTGCTTTG TAATCCAAGAAAACAGGGGCCCGAAACCCAAGGCAGTACAGCAGAATTACAGG GCTCGTCCAACTGGTCCCTCAGTCACACATGCCAGAGATTGCTCAGGAAGCAATGGA GGCTCTGCTGGTTCTTCATCAGTTAGATAGCATTGATTTGTGGAATCCTGATGCTCCT GTAGAAACATTTTGGGAGATTAGCTCACAAATGCTTTTTTACATCTGCAAGAAATTAAC TAGTCATCAAATGCTTAGTAGCACAGAAATTCTCAAGTGGTTGCGGGAAATATTGATC TGCAGGAATAAATTTCTTCTTAAAAATAAGCAGATAGAAGTTCCTGTCACTTTC CCCCAGCCTCCTTGCCAACGCCCCCTTTCCCTCTCCCCCTCCCGCTCGGCGCTGACC CCCCATCCCCACCCCCGTGGGAACACTGGGAGCCTGCACTCCACAGACCCTCTCCTT GCCTCTTCCCTCAGCCTCCGCTCCCCGCCCTCTTCCCGGCCCAGGGCGCCG GCCCACCCTTCCCTCCGCCGCCCCCCGGCCGCGGGGAGGACATGGCCGCGCACAG GCCGGTGGAATGGGTCCAGGCCGTGGTCAGCCGCTTCGACGAGCAGCTTCCAATAA AAACAGGACAGCAGAACACACATACCAAAGTCAGTACTGAGCACAACAAGGAATGTC TAATCAATATTTCCAAATACAAGTTTTCTTTGGTTATAAGCGGCCTCACTACTATTTTAA AGAATGTTAACTATATGAGAATATTTGGAGAAGCTGCTGAAAAAAATTTATATCTCTCT CAGTTGATTATATTGGATACACTGGAAAAATGTCTTGCTGGGCAACCAAAGGACACAA TGAGATTAGATGAAACGATGCTGGTCAAACAGTTGCTGCCAGAAATCTGCCATTTTCT TCACACCTGTCGTGAAGGAAACCAGCATGCAGCTGAACTTCGGAATTCTGCCTCTGG GGTTTTATTTTCTCTCAGCTGCAACAACTTCAATGCAGTCTTTAGTCGCATTTCTACCA GGTTACAGGAATTAACTGTTCAGAAGACAATGTTGATGTTCATGATATAGAATTG TTACAGTATATCAATGTGGATTGTGCAAAATTAAAACGACTCCTGAAGGAAACAGCAT TTAAATTTAAAGCCCTAAAGAAGGTTGCGCAGTTATAAATAGCCTGGAAAA GGCATTTTGGAACTGGGTAGAAAATTATCCAGATGAATTTACAAAACTGTACCAGATC CCACAGACTGATATGGCTGAATGTGCAGAAAAGCTATTTGACTTGGTGGATGGTTTTG CTGAAAGCACCAAACGTAAAGCAGCAGTTTGGCCACTACAAATCATTCTCCTTATCTT GTGTCCAGAAATAATCCAGGATATATCCAAAGACGTGGTTGATGAAAACAACATGAAT AAGAAGTTATTTCTGGACAGTCTACGAAAAGCTCTTGCTGGCCATGGAGGAAGTAGG CAGCTGACAGAAAGTGCTGCAATTGCCTGTGTCAAACTGTGTAAAGCAAGTACTTACA TCAATTGGGAAGATAACTCTGTCATTTTCCTACTTGTTCAGTCCATGGTGGTTGATCTT AAGAACCTGCTTTTTAATCCAAGTAAGCCATTCTCAAGAGGCAGTCAGCCTGCAGATG TGGATCTAATGATTGACTGCCTTGTTTCTTGCTTTCGTATAAGCCCTCACAACAACCAA CACTTTAAGATCTGCCTGGCTCAGAATTCACCTTCTACATTTCACTATGTGCTGGTAAA TTCACTCCATCGAATCATCACCAATTCCGCATTGGTGGCCTAAGATTGATGCT GTGTATTGTCACTCGGTTGAACTTCGAAATATGTTTGGTGAAACACTTCATAAAGCAGT GCAAGGTTGTGGAGCACACCCAGCAATACGAATGGCACCGAGTCTTACATTTAAAGA AAAAGTAACAAGCCTTAAATTTAAAGAAAAACCTACAGACCTGGAGACAAGAAGCTAT AAGTATCTTCTCTTGTCCATGGTGAAACTAATTCATGCAGCTCCAAAGCTCTTGCTTTG TAATCCAAGAAAACAGGGGCCCGAAACCCAAGGCAGTACAGCAGAATTACAGG GCTCGTCCAACTGGTCCCTCAGTCACACATGCCAGAGATTGCTCAGGAAGCAATGGA GGCTCTGCTGGTTCTTCATCAGTTAGATAGCATTGATTTGTGGAATCCTGATGCTCCT GTAGAAACATTTTGGGAGATTAGCTCACAAATGCTTTTTTACATCTGCAAGAAATTAAC TAGTCATCAAATGCTTAGTAGCACAGAAATTCTCAAGTGGTTGCGGGAAATATTGATC TGCAGGAATAAATTTCTTCTTAAAAATAAGCAGATAGAAGTTCCTGTCACTTTC
 1 -allele diseases monoallelic mutations may be responsible for dominant or X-linked disorders n new random mutations are the rule with an unpredictable pattern of distribution n
1 -allele diseases monoallelic mutations may be responsible for dominant or X-linked disorders n new random mutations are the rule with an unpredictable pattern of distribution n
 Gender effect in mutations n n For mutations other than point mutations, sex biases in the mutation rate are very variable Small deletions are more frequent in females Germline base substitution mutations occur more frequently in males than in females, especially in older males Point mutations at some loci occur almost exclusively in males, whereas others occur ten times more than in females
Gender effect in mutations n n For mutations other than point mutations, sex biases in the mutation rate are very variable Small deletions are more frequent in females Germline base substitution mutations occur more frequently in males than in females, especially in older males Point mutations at some loci occur almost exclusively in males, whereas others occur ten times more than in females
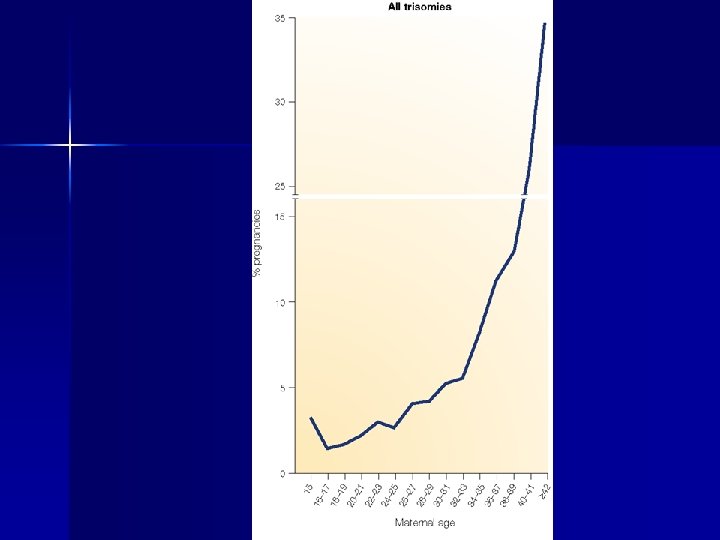
 Relative frequency of de novo achondroplasia for different paternal ages
Relative frequency of de novo achondroplasia for different paternal ages
 Relative frequency of de novo neurofibromatosis for different paternal ages
Relative frequency of de novo neurofibromatosis for different paternal ages
 the number of male germ-cell divisions
the number of male germ-cell divisions
 2 -allele diseases n novel mutations are rare, usually mutations have a long history (100 -1000 generations) n mutations have an ethnical signature with a predictable pattern of distribution and frequency n biallelic mutations may be responsible for autosomal recessive disorders n polymorphisms and private variants are more easily discriminated vs true mutations
2 -allele diseases n novel mutations are rare, usually mutations have a long history (100 -1000 generations) n mutations have an ethnical signature with a predictable pattern of distribution and frequency n biallelic mutations may be responsible for autosomal recessive disorders n polymorphisms and private variants are more easily discriminated vs true mutations
 2 -allele diseases n n consanguineity is a risk factor for homozygosity high carrier frequency is a risk factor for compound heterozygosity
2 -allele diseases n n consanguineity is a risk factor for homozygosity high carrier frequency is a risk factor for compound heterozygosity
 The effect of an allele n null or amorph = no product n hypomorph = reduced amount / activity n hypermorph = increased amount / activity n neomorph = novel product / activity n antimorph = antagonistic product / activity
The effect of an allele n null or amorph = no product n hypomorph = reduced amount / activity n hypermorph = increased amount / activity n neomorph = novel product / activity n antimorph = antagonistic product / activity
 Dominant or recessive phenotype?
Dominant or recessive phenotype?
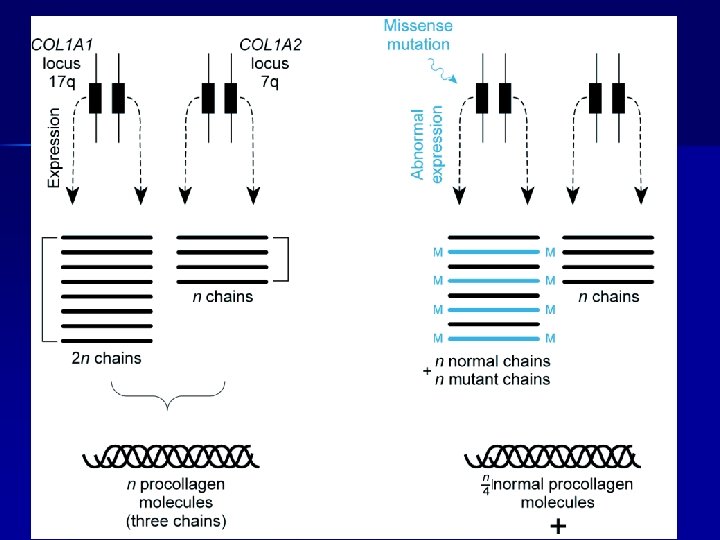
 haploinsufficiency") Loss of function mutations in the PAX 3 gene (Waardenburg syndrome) haploinsufficiency
Loss of function mutations in the PAX 3 gene (Waardenburg syndrome) haploinsufficiency
 n deletion – the entire gene – part of the") amorph / hypomorph (1) n deletion – the entire gene – part of the gene n disruption of the gene structure – by insertion, inversion, translocation n promoter inactivation m. RNA destabilization splicing mutation – inactivating donor/acceptor – activating criptic splice sites
amorph / hypomorph (1) n deletion – the entire gene – part of the gene n disruption of the gene structure – by insertion, inversion, translocation n promoter inactivation m. RNA destabilization splicing mutation – inactivating donor/acceptor – activating criptic splice sites
 n frame-shift in translation – by insertion of n+1 or") amorph / hypomorph (2) n frame-shift in translation – by insertion of n+1 or n+2 bases into the coding sequence – by deletion of n+1 or n+2 bases into the coding sequence n n nonsense mutation missense mutation / aa deletion – – – essential / conserved amino acid defect in post-transcriptional processing defect in cellular localization
amorph / hypomorph (2) n frame-shift in translation – by insertion of n+1 or n+2 bases into the coding sequence – by deletion of n+1 or n+2 bases into the coding sequence n n nonsense mutation missense mutation / aa deletion – – – essential / conserved amino acid defect in post-transcriptional processing defect in cellular localization
 n chromatin derepression (FSH) n trasposition under") hypermorph trisomia n duplication n amplification (cancer) n chromatin derepression (FSH) n trasposition under a strong promoter n – leukemia n overactivity of an abnormal protein
hypermorph trisomia n duplication n amplification (cancer) n chromatin derepression (FSH) n trasposition under a strong promoter n – leukemia n overactivity of an abnormal protein
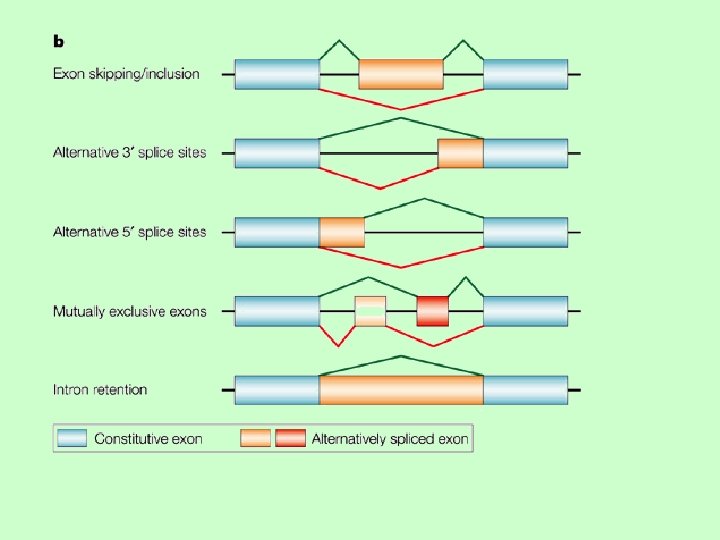
 missense mutations") neomorph n n n n generation of chimeric proteins duplication amplification (cancer) missense mutations inclusion of coding cryptic exons usage of alternative ORFs overactivity of an abnormal protein
neomorph n n n n generation of chimeric proteins duplication amplification (cancer) missense mutations inclusion of coding cryptic exons usage of alternative ORFs overactivity of an abnormal protein
 antimorph n n n missense mutations inclusion of coding cryptic exons usage of alternative ORFs
antimorph n n n missense mutations inclusion of coding cryptic exons usage of alternative ORFs
 Mutation detection n mutation scanning – or resequencing methods for identifying previously unknown mutations n genotyping – methods for scoring previously known mutations or single nucleotide polymorphisms (SNPs)
Mutation detection n mutation scanning – or resequencing methods for identifying previously unknown mutations n genotyping – methods for scoring previously known mutations or single nucleotide polymorphisms (SNPs)
 Key questions for mutation detection strategy n n n n expected mutations are monoallelic or biallelic? is the gene well recognized for that disease? is the mutation pattern known? (deletion, dup, small mutations, etc. ) which is the complexity of the gene? how many patients must be examined? how many controls should be examined? how many mutations and how many variations have already been identified in this gene? are there more members of the same gene family (or pseudogenes) in the genome?
Key questions for mutation detection strategy n n n n expected mutations are monoallelic or biallelic? is the gene well recognized for that disease? is the mutation pattern known? (deletion, dup, small mutations, etc. ) which is the complexity of the gene? how many patients must be examined? how many controls should be examined? how many mutations and how many variations have already been identified in this gene? are there more members of the same gene family (or pseudogenes) in the genome?
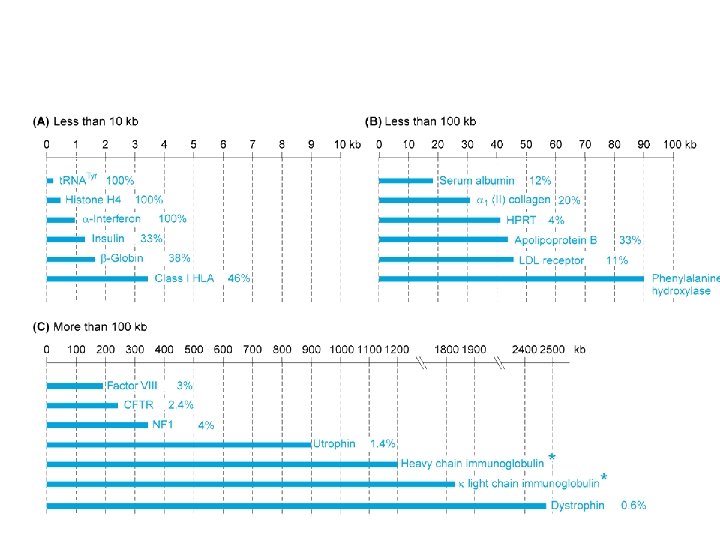
 Dimension of the mutation detection study Number of patients Gene size X Number of controls
Dimension of the mutation detection study Number of patients Gene size X Number of controls
 General strategy for mutation detection screening of recurrent mutations frequent mutations are known? YES mutations are identified? NO NO YES SEQUENCING mutation scanning
General strategy for mutation detection screening of recurrent mutations frequent mutations are known? YES mutations are identified? NO NO YES SEQUENCING mutation scanning
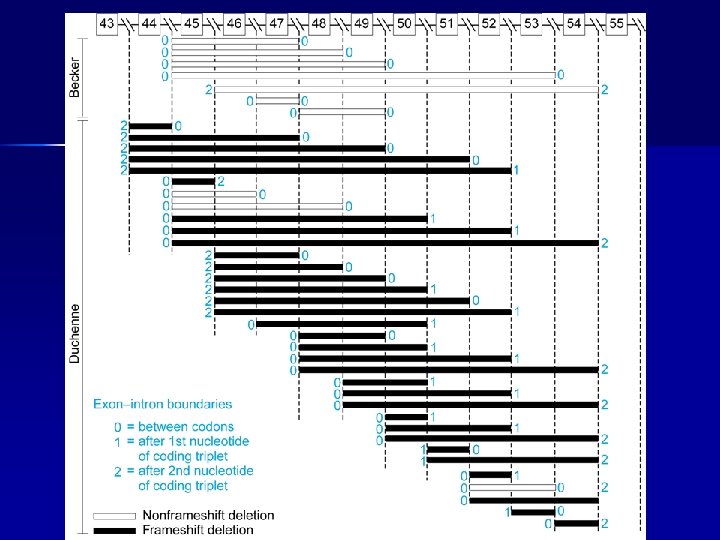
 n DMD Duchenne Muscular Dystrophy - 1/3, 500 boys Onset -- Early childhood - about 2 to 6 years – Laboratory -- CK (50 x to 1. 000 x), LDH 5, ALT, AST, aldolase increase Symptoms -- Generalized weakness and muscle wasting affecting proximal limb muscles first. Calves often enlarged. Heart involvement Progression -- Disease progresses slowly but will affect all voluntary muscles. Survival possible beyond late twenties n BMD Becker Muscular Dystrophy - 1/10, 000 boys Onset -- Adolescence or adulthood Symptoms -- Almost identical to Duchenne but often much less severe. Heart involvement Progression -- Slower and more variable than DMD with survival well into mid to late adulthood
n DMD Duchenne Muscular Dystrophy - 1/3, 500 boys Onset -- Early childhood - about 2 to 6 years – Laboratory -- CK (50 x to 1. 000 x), LDH 5, ALT, AST, aldolase increase Symptoms -- Generalized weakness and muscle wasting affecting proximal limb muscles first. Calves often enlarged. Heart involvement Progression -- Disease progresses slowly but will affect all voluntary muscles. Survival possible beyond late twenties n BMD Becker Muscular Dystrophy - 1/10, 000 boys Onset -- Adolescence or adulthood Symptoms -- Almost identical to Duchenne but often much less severe. Heart involvement Progression -- Slower and more variable than DMD with survival well into mid to late adulthood
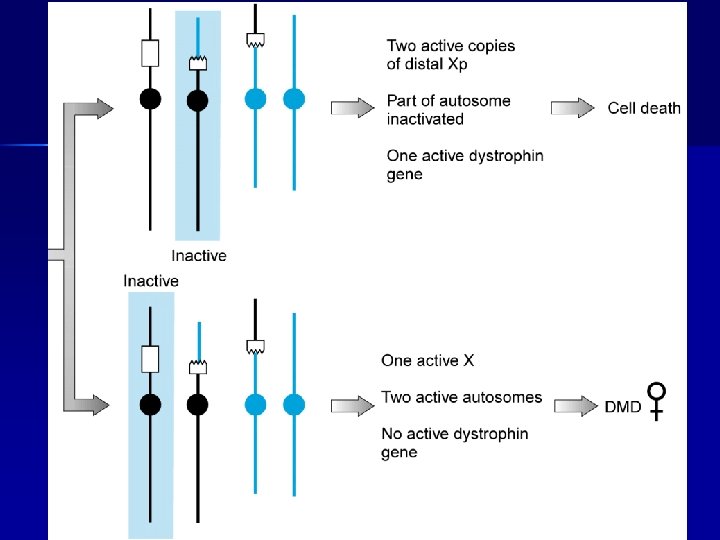
 Carrier of a balanced reciprocal X-autosome translocation
Carrier of a balanced reciprocal X-autosome translocation

 Dystrophin gene: page 1/185
Dystrophin gene: page 1/185
 Dystrophin gene: page 2/185
Dystrophin gene: page 2/185
 Dystrophin gene: page 3/185
Dystrophin gene: page 3/185
 Dystrophin gene: page 185/185
Dystrophin gene: page 185/185
 Telethon-UILDM 250/300 DMD/BMD Qualitative test rejected Quantitative test more DNA 80 plex-PCR Deletions duplications Point mutations m. RNA study Family tests
Telethon-UILDM 250/300 DMD/BMD Qualitative test rejected Quantitative test more DNA 80 plex-PCR Deletions duplications Point mutations m. RNA study Family tests
 with uniform spacing and gel") Log-PCR = 4 multiplex-PCR (2 x 20+2 x 18) with uniform spacing and gel position according to chromosomal position A DMD patient : groups A, B BMD patient : groups C, D Deletion ex 17 -43 Duplication ex 13 -23 C B DMD BMD 1: del ex 43 2: del ex 11, 17, 19, 21 3: del ex 17, 19, 21 4: del ex 50, 52 5: del ex 7, 11, 17, 19 6: del ex 61 1 2 3 4 5 6 1: no del 2: del ex 8, 12, 18, 20, 22 3: del ex 12, 18, 20, 22 4: del ex 46, 51 5: del ex 6, 8, 12, 18 6: del ex 62 D
Log-PCR = 4 multiplex-PCR (2 x 20+2 x 18) with uniform spacing and gel position according to chromosomal position A DMD patient : groups A, B BMD patient : groups C, D Deletion ex 17 -43 Duplication ex 13 -23 C B DMD BMD 1: del ex 43 2: del ex 11, 17, 19, 21 3: del ex 17, 19, 21 4: del ex 50, 52 5: del ex 7, 11, 17, 19 6: del ex 61 1 2 3 4 5 6 1: no del 2: del ex 8, 12, 18, 20, 22 3: del ex 12, 18, 20, 22 4: del ex 46, 51 5: del ex 6, 8, 12, 18 6: del ex 62 D
 large deletions in 377/506 DMD/BMD 74. 5%
large deletions in 377/506 DMD/BMD 74. 5%
 large duplications in 51/506 patients 10. 1%
large duplications in 51/506 patients 10. 1%
 SALSA MLPA probes
SALSA MLPA probes
 Hybridysation 1. 2. The MLPA probemix is added to denatured genomic DNA The two parts of each probe hybridise to adjacent target sequences
Hybridysation 1. 2. The MLPA probemix is added to denatured genomic DNA The two parts of each probe hybridise to adjacent target sequences
 Ligation 3. Probes are ligated by a thermostable ligase
Ligation 3. Probes are ligated by a thermostable ligase
 PCR amplification 4. A universal primer pair is used to amplify all ligated probes The PCR product of each probe has a unique length (130 480 bp)
PCR amplification 4. A universal primer pair is used to amplify all ligated probes The PCR product of each probe has a unique length (130 480 bp)
 Separation and quantification by capillary electrophoresis Each peak is the amplification product of a specific probe. Samples are compared to a control sample. A difference in relative peak height or peak area indicates a copy number change of the probe target sequence
Separation and quantification by capillary electrophoresis Each peak is the amplification product of a specific probe. Samples are compared to a control sample. A difference in relative peak height or peak area indicates a copy number change of the probe target sequence
 Detection of Chr X copy number X Male Female Triple X 283 bp 346 bp MRC-Holland b. v.
Detection of Chr X copy number X Male Female Triple X 283 bp 346 bp MRC-Holland b. v.
 MLPA discriminates sequences that differ in only a single nucleotide and can be used to detect known mutations. Mismatch at the probe ligation site No ligation, no amplification product Perfect match Ligation of the two probe oligonucleotides Amplification product
MLPA discriminates sequences that differ in only a single nucleotide and can be used to detect known mutations. Mismatch at the probe ligation site No ligation, no amplification product Perfect match Ligation of the two probe oligonucleotides Amplification product
 MS-MLPA M M Methylated Target Denaturation and Multiplex probe hybridization Ligation and Digestion with methylation sensitive endonucleases Unmethylated Target M M Only undigested (methylated) and ligated probes are exponentially amplified MRC-Holland b. v.
MS-MLPA M M Methylated Target Denaturation and Multiplex probe hybridization Ligation and Digestion with methylation sensitive endonucleases Unmethylated Target M M Only undigested (methylated) and ligated probes are exponentially amplified MRC-Holland b. v.
 Limb-girdle weakness proximal weakness: most common n Lower extremities – difficulty climbing stairs – arising from a low chair or toilet – getting up from a squatted position n Upper extremities – trouble lifting objects over their head – brushing their hair n distal weakness – difficulty opening jars, inability to turn a key in the ignition, or tripping due to foot drop n cranial weakness – dysarthria, dysphagia or ptosis
Limb-girdle weakness proximal weakness: most common n Lower extremities – difficulty climbing stairs – arising from a low chair or toilet – getting up from a squatted position n Upper extremities – trouble lifting objects over their head – brushing their hair n distal weakness – difficulty opening jars, inability to turn a key in the ignition, or tripping due to foot drop n cranial weakness – dysarthria, dysphagia or ptosis
 Genetics of limb-girdle muscular dystrophies autosomal dominant n n n n LGMD 1 A LGMD 1 B LGMD 1 C LGMD 1 D LGMD 1 E LGMD 1 F LGMD 1 G 5 q 31. 2 1 q 21 3 p 25. 3 6 q 22 7 q 35 7 q 31. 1 4 p 21 autosomal recessive n n n LGMD 2 A LGMD 2 B LGMD 2 C LGMD 2 D 1994) LGMD 2 E Lim, 1995) LGMD 2 F LGMD 2 G LGMD 2 H LGMD 2 I LGMD 2 J LGMD 2 K 15 q 15 2 p 13. 2 13 q 12 17 q 21. 33 4 q 12 5 q 33 17 q 12 9 q 33. 1 19 q 13. 3 2 q 24. 3 9 q 34. 1 myotilin (Hauser, 2000) lamin A/C (Bonne, 1999) caveolin 3 (Minetti, 1997) ? ? filamin C ? calpain 3 (Richard, 1995) dysferlin (Bashir, Liu, 1998) g-sarcoglycan (Noguchi, 1995) a-sarcoglycan (Roberds, b-sarcoglycan (Bonnemann, d-sarcoglycan (Nigro, 1996) telethonin (Moreira, 2000) TRIM 32 (Frosk, 2002) FKRP (Brockington, 2001) titin (Udd, 2002) POMT 1 (Balci, 2005)
Genetics of limb-girdle muscular dystrophies autosomal dominant n n n n LGMD 1 A LGMD 1 B LGMD 1 C LGMD 1 D LGMD 1 E LGMD 1 F LGMD 1 G 5 q 31. 2 1 q 21 3 p 25. 3 6 q 22 7 q 35 7 q 31. 1 4 p 21 autosomal recessive n n n LGMD 2 A LGMD 2 B LGMD 2 C LGMD 2 D 1994) LGMD 2 E Lim, 1995) LGMD 2 F LGMD 2 G LGMD 2 H LGMD 2 I LGMD 2 J LGMD 2 K 15 q 15 2 p 13. 2 13 q 12 17 q 21. 33 4 q 12 5 q 33 17 q 12 9 q 33. 1 19 q 13. 3 2 q 24. 3 9 q 34. 1 myotilin (Hauser, 2000) lamin A/C (Bonne, 1999) caveolin 3 (Minetti, 1997) ? ? filamin C ? calpain 3 (Richard, 1995) dysferlin (Bashir, Liu, 1998) g-sarcoglycan (Noguchi, 1995) a-sarcoglycan (Roberds, b-sarcoglycan (Bonnemann, d-sarcoglycan (Nigro, 1996) telethonin (Moreira, 2000) TRIM 32 (Frosk, 2002) FKRP (Brockington, 2001) titin (Udd, 2002) POMT 1 (Balci, 2005)
 are generally milder n represent less than 10% of") autosomal dominant forms (LGMD 1) are generally milder n represent less than 10% of all LGMD n marked heterogeneity for LGMD 1, one gene per one single family n
autosomal dominant forms (LGMD 1) are generally milder n represent less than 10% of all LGMD n marked heterogeneity for LGMD 1, one gene per one single family n
 have an average prevalence of") autosomal recessive n n autosomal recessive forms (LGMD 2) have an average prevalence of 1: 14, 000 -1: 20, 000 at birth frequency differences among countries this depends on higher carrier frequencies of single mutations, as 550 del. A for calpain 3 in Croatia, L 276 I for FKRP in Northern Europe, 521 del. T for gamma-sarcoglycan in Northern Africa At least 25% of families are excluded from any known locus and 40% of typical LGMD cases have no mutation in any known gene
autosomal recessive n n autosomal recessive forms (LGMD 2) have an average prevalence of 1: 14, 000 -1: 20, 000 at birth frequency differences among countries this depends on higher carrier frequencies of single mutations, as 550 del. A for calpain 3 in Croatia, L 276 I for FKRP in Northern Europe, 521 del. T for gamma-sarcoglycan in Northern Africa At least 25% of families are excluded from any known locus and 40% of typical LGMD cases have no mutation in any known gene
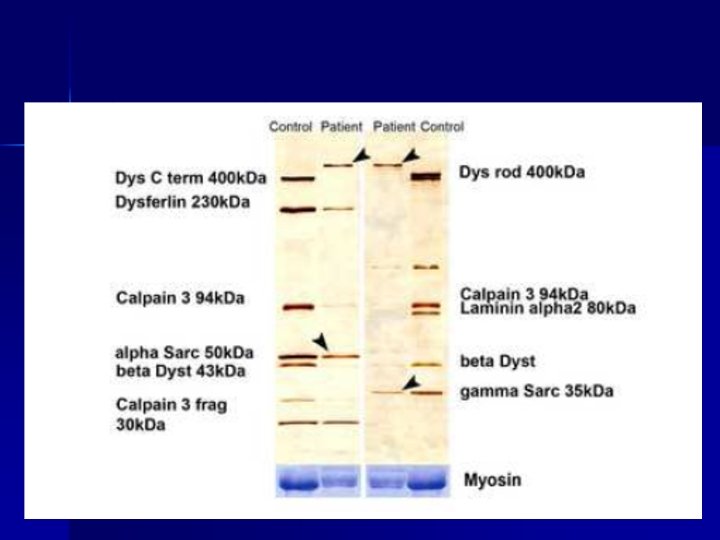
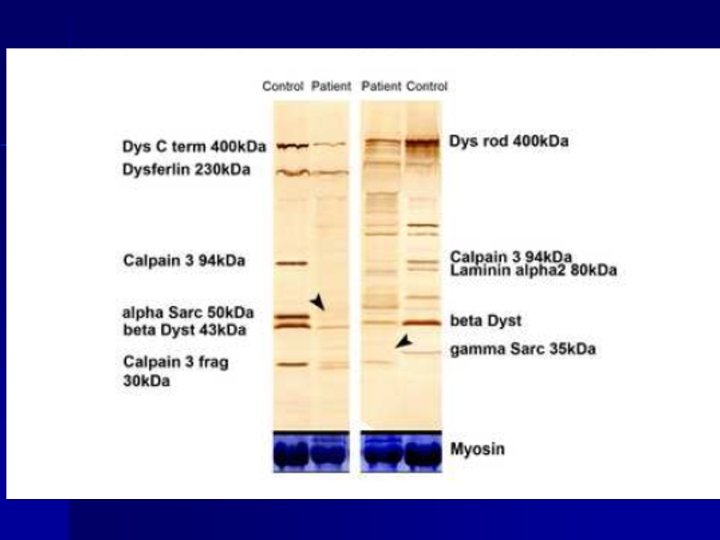
 n WB analysis n") Tools to address the diagnosis of LGMD Clinical presentation (MRI) n WB analysis n Segregation study n Mutation detection in patients n Mutation detection in normal subjects n Homogeneous collection of mutations and polymorphisms n
Tools to address the diagnosis of LGMD Clinical presentation (MRI) n WB analysis n Segregation study n Mutation detection in patients n Mutation detection in normal subjects n Homogeneous collection of mutations and polymorphisms n
 Segregation analysis n n Analysis of 30 polymorphic markers linked to LGMD 2 A, 2 B, 2 C-2 F, 2 I in sib pairs To find homozigosity…
Segregation analysis n n Analysis of 30 polymorphic markers linked to LGMD 2 A, 2 B, 2 C-2 F, 2 I in sib pairs To find homozigosity…
 Calpain 3 24 exons Myotilin dysferlin 55 exons Lamin A/C 13 exons a-sarcoglycan 10 exons Caveolin 3 2 exons (3) b-sarcoglycan 6 ex (7) g-sarcoglycan 8 ex (10) d-sarcoglyican 9 exons Telethonin 2 exons (3) TRIM 32 1 exons (7) FKRP 4 esons (8) Titin 363 ex (35) 9 exons
Calpain 3 24 exons Myotilin dysferlin 55 exons Lamin A/C 13 exons a-sarcoglycan 10 exons Caveolin 3 2 exons (3) b-sarcoglycan 6 ex (7) g-sarcoglycan 8 ex (10) d-sarcoglyican 9 exons Telethonin 2 exons (3) TRIM 32 1 exons (7) FKRP 4 esons (8) Titin 363 ex (35) 9 exons
 Case 1 n n n The gene is known It is composed of five small size exons There are 10 patients, sons of consanguineous parents Expected mutations are homozygous Mutations have never been identified in this gene There is no other member of the same gene families (or pseudogenes) in the genome
Case 1 n n n The gene is known It is composed of five small size exons There are 10 patients, sons of consanguineous parents Expected mutations are homozygous Mutations have never been identified in this gene There is no other member of the same gene families (or pseudogenes) in the genome
 Case 2 n n n The gene is known The putative function of the gene product is to serve as a transcription factor Expected mutations are dominant Mutations have never been identified in this gene There are other members of the same gene families (or pseudogenes) in the genome
Case 2 n n n The gene is known The putative function of the gene product is to serve as a transcription factor Expected mutations are dominant Mutations have never been identified in this gene There are other members of the same gene families (or pseudogenes) in the genome
, sequencing") Sequencing n With the ongoing reduction of costs (today about 2 -4 €/run), sequencing of PCR products is applied for mutation detection n Sequencing is often thought of as the 'gold standard' for mutation detection. n This perception is distorted due to the fact that this is the only method of mutation identification, but this does not mean it is the best for mutation detection
Sequencing n With the ongoing reduction of costs (today about 2 -4 €/run), sequencing of PCR products is applied for mutation detection n Sequencing is often thought of as the 'gold standard' for mutation detection. n This perception is distorted due to the fact that this is the only method of mutation identification, but this does not mean it is the best for mutation detection
 –when searching for heterozygous DNA differences there a number") Sequencing artifacts FALSE POSITIVE (specificity) –when searching for heterozygous DNA differences there a number of potential mutations, together with sequence artifacts, compressions and differences in peak intensities that must be rechecked with additional primers and costs FALSE NEGATIVE (sensitivity) –loss of information farther away or closer to the primer –does not detect a minority of mutant molecules in a wild-type environment
Sequencing artifacts FALSE POSITIVE (specificity) –when searching for heterozygous DNA differences there a number of potential mutations, together with sequence artifacts, compressions and differences in peak intensities that must be rechecked with additional primers and costs FALSE NEGATIVE (sensitivity) –loss of information farther away or closer to the primer –does not detect a minority of mutant molecules in a wild-type environment
") Current mutation scanning techniques n n n n n SSCP (single strand conformation polymorphism) HA (heteroduplex analysis) CCM (chemical cleavage of mismatch) CSGE (conformation sensitive gel electrophoresis) DGGE (denaturing gradient gel electrophoresis) DHPLC (denaturing HPLC) PTT (protein truncation test) DGCE (denaturing gradient capillary electrophoresis) direct sequencing
Current mutation scanning techniques n n n n n SSCP (single strand conformation polymorphism) HA (heteroduplex analysis) CCM (chemical cleavage of mismatch) CSGE (conformation sensitive gel electrophoresis) DGGE (denaturing gradient gel electrophoresis) DHPLC (denaturing HPLC) PTT (protein truncation test) DGCE (denaturing gradient capillary electrophoresis) direct sequencing
 SSCP
SSCP
 Mutation detection by heteroduplex analysis: the mutant DNA must first be hybridized with the wild-type DNA to form a mixture of two homoduplexes and two h e t e r o d u p l e xe s
Mutation detection by heteroduplex analysis: the mutant DNA must first be hybridized with the wild-type DNA to form a mixture of two homoduplexes and two h e t e r o d u p l e xe s
 Heteroduplex analysis
Heteroduplex analysis
 DHPLC denaturing HPLC from Transgenomic
DHPLC denaturing HPLC from Transgenomic
 DHPLC analysis at different temperatures of the column
DHPLC analysis at different temperatures of the column
 Analysis of dystrophin exon 59 Homoduplex DNA: PCR fragments are identicals Heteroduplex DNA: PCR fragments are different
Analysis of dystrophin exon 59 Homoduplex DNA: PCR fragments are identicals Heteroduplex DNA: PCR fragments are different
 UV 2 0 1: 2") DHPLC analysis of the CAPN 3 gene (exon 11) UV 2 0 1: 2 FLUO 100 0 1: 4 1: 6 1: 8 1: 10
DHPLC analysis of the CAPN 3 gene (exon 11) UV 2 0 1: 2 FLUO 100 0 1: 4 1: 6 1: 8 1: 10
 PLATE B PLATE A POOLED PLATES A+B DHPLC analysis
PLATE B PLATE A POOLED PLATES A+B DHPLC analysis
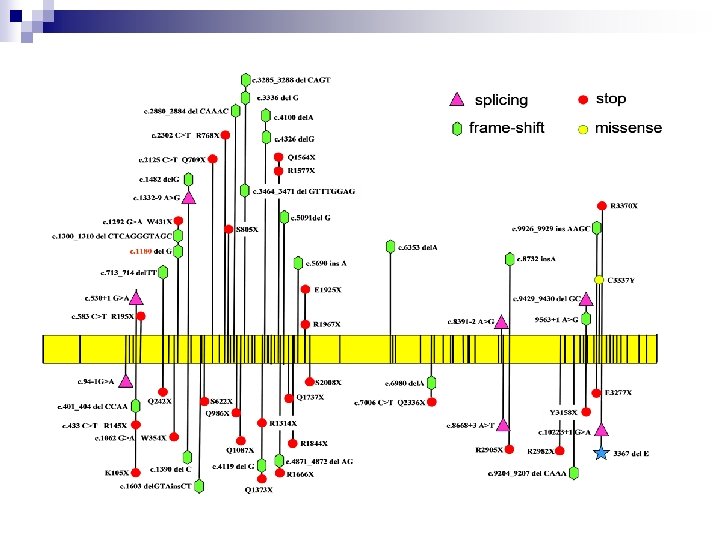
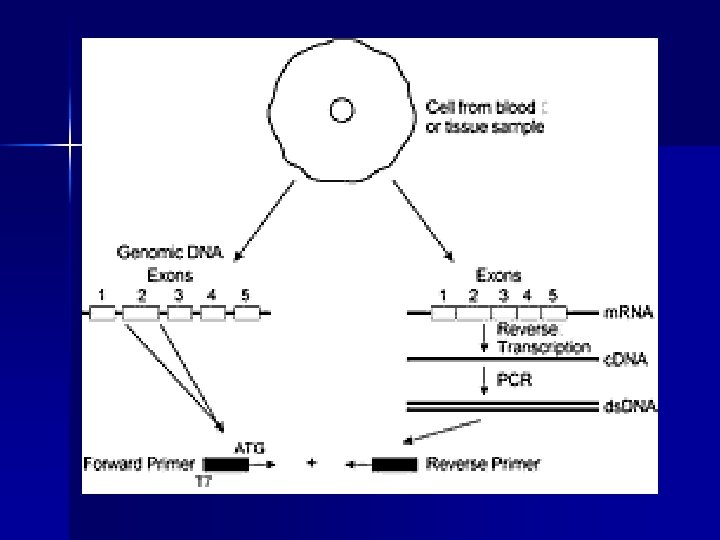
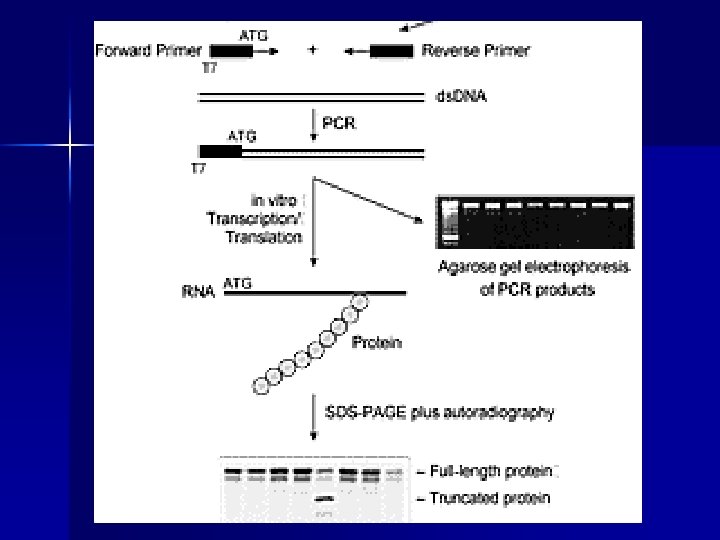
 PTT protein truncation test n n n Sensitivity 1000 -bp fragment > 85% Detects only nonsense mutations Post PCR time: 48 -72 hours (translation/trascription, gel preparation, loading and run, analysis of results) Use of 35 S radioactivity No special equipment required m. RNA as starting template
PTT protein truncation test n n n Sensitivity 1000 -bp fragment > 85% Detects only nonsense mutations Post PCR time: 48 -72 hours (translation/trascription, gel preparation, loading and run, analysis of results) Use of 35 S radioactivity No special equipment required m. RNA as starting template
 n Polycystic Kidney Disease PKD 1 95%") Applications of PTT (% of truncating mutations) n Polycystic Kidney Disease PKD 1 95% n Familial Adenomatous Polyposis APC 95% n Ataxia telangiectasia ATM 90% n Hereditary breast and ovarian cancer BRCA 1 -2 90% n Duchenne Muscular Dystrophy DMD 90%? n Fanconi anemia FAA 80% n Hereditary non-polyposis colorectal cancer h. MSH 1 -2 70%80% n Neurofibromatosis type 2 NF 2 65% n Hunter Syndrome IDS 50% n Neurofibromatosis type 1 NF 1 50% n Cystic Fibrosis CFTR 15%
Applications of PTT (% of truncating mutations) n Polycystic Kidney Disease PKD 1 95% n Familial Adenomatous Polyposis APC 95% n Ataxia telangiectasia ATM 90% n Hereditary breast and ovarian cancer BRCA 1 -2 90% n Duchenne Muscular Dystrophy DMD 90%? n Fanconi anemia FAA 80% n Hereditary non-polyposis colorectal cancer h. MSH 1 -2 70%80% n Neurofibromatosis type 2 NF 2 65% n Hunter Syndrome IDS 50% n Neurofibromatosis type 1 NF 1 50% n Cystic Fibrosis CFTR 15%
 genotyping • MIP genotyping uses circularizable probes with 5′ and") Molecular inversion probe (MIP) genotyping • MIP genotyping uses circularizable probes with 5′ and 3′ ends that anneal upstream and downstream of the SNP site leaving a 1 bp gap • Polymerase extension with d. NTPs and a non-stranddisplacing polymerase is used to fill in the gap
Molecular inversion probe (MIP) genotyping • MIP genotyping uses circularizable probes with 5′ and 3′ ends that anneal upstream and downstream of the SNP site leaving a 1 bp gap • Polymerase extension with d. NTPs and a non-stranddisplacing polymerase is used to fill in the gap
 • Ligation seals the nick, and exonuclease I is used to remove excess unannealed and unligated circular probes • The resultant product is PCR-amplified and the orientation of the primers ensures that only circularized probes will be amplified • The resultant product is hybridized and read out on an array of universal-capture probes
• Ligation seals the nick, and exonuclease I is used to remove excess unannealed and unligated circular probes • The resultant product is PCR-amplified and the orientation of the primers ensures that only circularized probes will be amplified • The resultant product is hybridized and read out on an array of universal-capture probes
 Golden. Gate genotyping assay n n n Golden. Gate uses extension ligation between annealed locus-specific oligos (LSOs) and allelespecific oligos (ASOs) An allele-specific primer extension step is used to preferentially extend the correctly matched ASO (at the 3′ end) up to the 5′ end of the LSO primer Ligation then closes the nick
Golden. Gate genotyping assay n n n Golden. Gate uses extension ligation between annealed locus-specific oligos (LSOs) and allelespecific oligos (ASOs) An allele-specific primer extension step is used to preferentially extend the correctly matched ASO (at the 3′ end) up to the 5′ end of the LSO primer Ligation then closes the nick
 Golden. Gate genotyping assay n n A subsequent PCR amplification step is used to amplify the appropriate product using common primers to ‘built-in’ universal PCR sites in the ASO and LSO sequences The resultant PCR products are hybridized and read out on an array of universal-capture probes
Golden. Gate genotyping assay n n A subsequent PCR amplification step is used to amplify the appropriate product using common primers to ‘built-in’ universal PCR sites in the ASO and LSO sequences The resultant PCR products are hybridized and read out on an array of universal-capture probes
 454 technology: DNA fragmentation and adaptor ligation
454 technology: DNA fragmentation and adaptor ligation
 454 technology: a water-in-oil emulsion is created: a single molecule of DNA with a single bead
454 technology: a water-in-oil emulsion is created: a single molecule of DNA with a single bead
 454 technology: Beads with clones are selected and assembled onto a planar substrate
454 technology: Beads with clones are selected and assembled onto a planar substrate
 454 technology: Sequencing by synthesis pyrosequencing Up to 100 Million bp in 8 hours can be read Ambiguities arise for homopolymeric tracts
454 technology: Sequencing by synthesis pyrosequencing Up to 100 Million bp in 8 hours can be read Ambiguities arise for homopolymeric tracts
 7. 4 x coverage 234 runs 24. 5 billions bp
7. 4 x coverage 234 runs 24. 5 billions bp
 11 genetic diseases !!
11 genetic diseases !!
 Nimble. Gen sequence capture
Nimble. Gen sequence capture