Молекулярная эволюция-лекция 5-Афонников.ppt
- Количество слайдов: 83
 Модели эволюции последовательностей • Описывают временные изменения в последовательностях генетических макромолекул • Для одиночных замен используются Марковские модели с конечным числом состояний • Развитие от простого к сложному
Модели эволюции последовательностей • Описывают временные изменения в последовательностях генетических макромолекул • Для одиночных замен используются Марковские модели с конечным числом состояний • Развитие от простого к сложному
 Эволюционное расстояние d между последовательностями – доля замен которая произошла в ходе эволюции AGTTC…A AGCAG…A AGCTA…A t • Задача: связать эволюционное расстояние и долю наблюдаемых различий p • В простейшем приближении можно использовать расстояние, основанное на частотах совпадений аминокислот (модель Пуассона). • Зная время и d можно установить скорость замен и наоборот, зная и d можно установить t. • Недостаток – не учитываются повторные и обратные замены, применимо на небольших временах дивергенции
Эволюционное расстояние d между последовательностями – доля замен которая произошла в ходе эволюции AGTTC…A AGCAG…A AGCTA…A t • Задача: связать эволюционное расстояние и долю наблюдаемых различий p • В простейшем приближении можно использовать расстояние, основанное на частотах совпадений аминокислот (модель Пуассона). • Зная время и d можно установить скорость замен и наоборот, зная и d можно установить t. • Недостаток – не учитываются повторные и обратные замены, применимо на небольших временах дивергенции
 Модель Джукса-Kантора A G C Замены случайны, независимы о других позиций, равновероятны с вероятностью Дискретное приближение T Непрерывное приближение
Модель Джукса-Kантора A G C Замены случайны, независимы о других позиций, равновероятны с вероятностью Дискретное приближение T Непрерывное приближение
=1 ) ( PA(0)=0 ) Особенности: PA(") Зависимость частоты нуклеотида ‘A’ от времени ( PA(0)=1 ) ( PA(0)=0 ) Особенности: PA( )= PT( )= PG( )= PC( )=1/4
Зависимость частоты нуклеотида ‘A’ от времени ( PA(0)=1 ) ( PA(0)=0 ) Особенности: PA( )= PT( )= PG( )= PC( )=1/4
 Для") Модель Kимуры A G C Учет разных частот Для транзиций – (T-C, A-G) Для трансверсий (остальные) - T Однако Pij различаются по парам замен (транзиции Y, трансверсии Z):
Модель Kимуры A G C Учет разных частот Для транзиций – (T-C, A-G) Для трансверсий (остальные) - T Однако Pij различаются по парам замен (транзиции Y, трансверсии Z):
 Свойства: • Сумма по столбцам равна") Матрица замен и ее свойства (матрица скоростей мутаций) Свойства: • Сумма по столбцам равна 1 • Эволюция за n шагов эквивалентна умножению на M n. • Если M=const, то существуют равновесные частоты, которые находятся из уравнения p=Mp
Матрица замен и ее свойства (матрица скоростей мутаций) Свойства: • Сумма по столбцам равна 1 • Эволюция за n шагов эквивалентна умножению на M n. • Если M=const, то существуют равновесные частоты, которые находятся из уравнения p=Mp
 Матрица замен: непрерывное время
Матрица замен: непрерывное время
 Различные меры расстояний последовательностей ДНК Определение расстояния зависит от типа модели. Примеры: Модель Джукса-Кантора: Модель Кимуры (P-частота транзиций, Q-частота трансверсий): Общий случай матрицы замен:
Различные меры расстояний последовательностей ДНК Определение расстояния зависит от типа модели. Примеры: Модель Джукса-Кантора: Модель Кимуры (P-частота транзиций, Q-частота трансверсий): Общий случай матрицы замен:
 Основная цепь (одинаковы) Пептидная связь • Относительно короткие цепи") Структура белков Боковая группа (различны) Основная цепь (одинаковы) Пептидная связь • Относительно короткие цепи (~100 – 1000 а. к. ) • 20 мономеров (различаются боковыми группами) • Пространственная структура нелинейна • Вторичная структура: , , • Третичная структура (глобула) : дальние контакты • Гетерогенность пространственной структуры
Структура белков Боковая группа (различны) Основная цепь (одинаковы) Пептидная связь • Относительно короткие цепи (~100 – 1000 а. к. ) • 20 мономеров (различаются боковыми группами) • Пространственная структура нелинейна • Вторичная структура: , , • Третичная структура (глобула) : дальние контакты • Гетерогенность пространственной структуры
 Уровни структурной организации белков Первичная Вторичная Третичная Четвертичная Последо. Локально Укладка всей вательность упорядоченцепи, за счет аминокислот ная осн. цепь дальних взаимод. Рисунок адаптирован из TUTORIAL ON PEPTIDE AND PROTEIN STRUCTURE ©J. E. Wampler, 1996 http: //bmbiris. bmb. uga. edu/wampler/tutorial/prot 0. html Укладка нескольких цепей (мономеров)
Уровни структурной организации белков Первичная Вторичная Третичная Четвертичная Последо. Локально Укладка всей вательность упорядоченцепи, за счет аминокислот ная осн. цепь дальних взаимод. Рисунок адаптирован из TUTORIAL ON PEPTIDE AND PROTEIN STRUCTURE ©J. E. Wampler, 1996 http: //bmbiris. bmb. uga. edu/wampler/tutorial/prot 0. html Укладка нескольких цепей (мономеров)
 Типы взаимодействий в белках и их вклад в стабильность глобулы Гидрофобные, ~1. 2 ккал/моль на одну группу -CH 2 - Водородные связи, 1. 6 1 ккал/моль на одну водородную связь Солевые мостики, 1 ккал/моль на поверхности, 3 -5 ккал/моль внутри В среднем 1. 7 ккал/моль на остаток. Для сравнения: k. T=0. 6 ккал. моль
Типы взаимодействий в белках и их вклад в стабильность глобулы Гидрофобные, ~1. 2 ккал/моль на одну группу -CH 2 - Водородные связи, 1. 6 1 ккал/моль на одну водородную связь Солевые мостики, 1 ккал/моль на поверхности, 3 -5 ккал/моль внутри В среднем 1. 7 ккал/моль на остаток. Для сравнения: k. T=0. 6 ккал. моль
 Влияние аминокислотных замен на стабильность и функцию белка. По фенотипическому проявлению мутации можно разделить на следующие классы: 1) нейтральные; 2) снижающие эффективность функционирования; 3) повышающие эффективность функционирования; 4) приводящие к возникновению новой функции. По молекулярному механизму действия замены остатков можно разделить на три основных группы: 1) изменяющие конформационную стабильность белков; 2) влияющие на процесс укладки (самоорганизации) полипептидной цепи в нативную глобулу; 3) изменяющие функцию белков за счет нарушения локальной конформации функциональных центров;
Влияние аминокислотных замен на стабильность и функцию белка. По фенотипическому проявлению мутации можно разделить на следующие классы: 1) нейтральные; 2) снижающие эффективность функционирования; 3) повышающие эффективность функционирования; 4) приводящие к возникновению новой функции. По молекулярному механизму действия замены остатков можно разделить на три основных группы: 1) изменяющие конформационную стабильность белков; 2) влияющие на процесс укладки (самоорганизации) полипептидной цепи в нативную глобулу; 3) изменяющие функцию белков за счет нарушения локальной конформации функциональных центров;
 Влияние аминокислотных замен на стабильность и функцию белка. Эффект замены существенно зависит от структурного контекста. Менее всего влияют на стабильность замены на поверхности белка и замены в неструктурных участках белка (высокое значение температурного фактора). Дестабилизирующие замены – такие, что увеличение свободной энергии в результате замены происходит более чем на 1 ккал/моль. Дестабилизирующие замены наблюдаются в гидрофобном ядре, затрагивают формирование солевых мостиков и водородных связей, влияют на конформацию основной цепи (замены Pro, Gly и наоборот).
Влияние аминокислотных замен на стабильность и функцию белка. Эффект замены существенно зависит от структурного контекста. Менее всего влияют на стабильность замены на поверхности белка и замены в неструктурных участках белка (высокое значение температурного фактора). Дестабилизирующие замены – такие, что увеличение свободной энергии в результате замены происходит более чем на 1 ккал/моль. Дестабилизирующие замены наблюдаются в гидрофобном ядре, затрагивают формирование солевых мостиков и водородных связей, влияют на конформацию основной цепи (замены Pro, Gly и наоборот).
 Исследование мутаций на примере Arc репрессора (Sauer et al. , 1996, Milla et al. , 1994) Проведены одиночные замены остатков на аланин в 51 -ой из 53 -х позиций белка. 25 мутаций – нейтральные (<1 ккал/моль) Повышают стабильность – единицы. Около половины – дестабилизируют белок.
Исследование мутаций на примере Arc репрессора (Sauer et al. , 1996, Milla et al. , 1994) Проведены одиночные замены остатков на аланин в 51 -ой из 53 -х позиций белка. 25 мутаций – нейтральные (<1 ккал/моль) Повышают стабильность – единицы. Около половины – дестабилизируют белок.
. Trp 14 Pro 8 > Leu, Ala") Пример мутации, повышающей стабильность белка (Arc репрессор). Trp 14 Pro 8 > Leu, Ala Замена Pro 8 на Leu или Ala увеличивает стабильность димера на 2. 5 ккал/моль. Это происходит в результате образования водородной связи атомами основной цепи с Trp 14 другого мономера. Такие мутации чрезвычайно редки. В стафилококковой нуклеазе из 290 замен только 33 привели к относительно небольшому увеличению стабильности.
Пример мутации, повышающей стабильность белка (Arc репрессор). Trp 14 Pro 8 > Leu, Ala Замена Pro 8 на Leu или Ala увеличивает стабильность димера на 2. 5 ккал/моль. Это происходит в результате образования водородной связи атомами основной цепи с Trp 14 другого мономера. Такие мутации чрезвычайно редки. В стафилококковой нуклеазе из 290 замен только 33 привели к относительно небольшому увеличению стабильности.
.") Влияние мутаций на активность белка (тирозил т. РНК синтетаза, (Fersht, 1987).
Влияние мутаций на активность белка (тирозил т. РНК синтетаза, (Fersht, 1987).
 Модель замен в белках 20 аминокислот. Замены в позициях независимы и определяются матрицей одинаковой для всех белков и всех позиций M(20 х20). Матрица замен M была определена эмпирически на основе анализа нескольких семейств гомологичных белков Дайхофф и сотр. (1978). Свойства матрицы Дайхофф: • Равновесные частоты равны частотам встречаемости аминокислот в последовательностях белков. • Наиболее часты замены аминокислот на аминокислоты, сходные по физико-химическим свойствам. • Исходная матрица нормирована на время, эквивалентное 1 замене на 100 позиций (1 PAM). • Для оценки вероятности замен через время t=n надо матрицу 1 PAM возвести в степень n.
Модель замен в белках 20 аминокислот. Замены в позициях независимы и определяются матрицей одинаковой для всех белков и всех позиций M(20 х20). Матрица замен M была определена эмпирически на основе анализа нескольких семейств гомологичных белков Дайхофф и сотр. (1978). Свойства матрицы Дайхофф: • Равновесные частоты равны частотам встречаемости аминокислот в последовательностях белков. • Наиболее часты замены аминокислот на аминокислоты, сходные по физико-химическим свойствам. • Исходная матрица нормирована на время, эквивалентное 1 замене на 100 позиций (1 PAM). • Для оценки вероятности замен через время t=n надо матрицу 1 PAM возвести в степень n.
 Как оценить матрицу замен? Матрица замен может быть оценена эмпирически. Матрица замен отражает скорость мутации, т. е. вероятность мутации за бесконечно малый промежуток времени. Поэтому для ее оценки Дайхофф взяли близкородственные последовательности нескольких десятков белковых семейств. Методом максимальной парсимонии построены предковые последовательности. Два основных фактора, определяющих параметры замен: Мутабильность остатка – как часто аминокислота мутирует? Специфика замен – на какие другие типы аминокислот замены более предпочтительны?
Как оценить матрицу замен? Матрица замен может быть оценена эмпирически. Матрица замен отражает скорость мутации, т. е. вероятность мутации за бесконечно малый промежуток времени. Поэтому для ее оценки Дайхофф взяли близкородственные последовательности нескольких десятков белковых семейств. Методом максимальной парсимонии построены предковые последовательности. Два основных фактора, определяющих параметры замен: Мутабильность остатка – как часто аминокислота мутирует? Специфика замен – на какие другие типы аминокислот замены более предпочтительны?
 Сравнение последовательностей вдоль ребра 7 -6 дает нам,") Пример Матрицы накопленных Пар замен (Aij) Сравнение последовательностей вдоль ребра 7 -6 дает нам, таким образом, 1 пару F A, 1 пару A F. Для ребра 6 -5 замены не наблюдаются. Ребро 5 -1 дает F A и A F. Ребро 5 -2 дает 0 замен, 6 -3 дает замены D A и A D, ребро 7 -4 также дает замены D A и A D. В результате получается матрица количества накопленных замен A, приведенная на рис. 2 (поскольку замены и неразличимы, матрица симметрическая).
Пример Матрицы накопленных Пар замен (Aij) Сравнение последовательностей вдоль ребра 7 -6 дает нам, таким образом, 1 пару F A, 1 пару A F. Для ребра 6 -5 замены не наблюдаются. Ребро 5 -1 дает F A и A F. Ребро 5 -2 дает 0 замен, 6 -3 дает замены D A и A D, ребро 7 -4 также дает замены D A и A D. В результате получается матрица количества накопленных замен A, приведенная на рис. 2 (поскольку замены и неразличимы, матрица симметрическая).
 из работы Дайхофф (х10) Наиболее часто встречающейся замена D") Матрицы накопленных Пар замен (Aij) из работы Дайхофф (х10) Наиболее часто встречающейся замена D и E (83 замены). Другими часто заменяющимися парами так же являются аминокислоты со сходными физико-химическими свойствами: A-S (77 пар) и A-T (59 пар) малые боковые группы; T-S (69 пар), малые аминокислоты с полярными боковыми группами; V-I (66 пар), гидрофобные. Для 35 пар аминокислот из 190 замен не наблюдалось. Отсутствие замен характерно, например, для триптофана (W), имеющего специфические свойства боковой группы (большой объем, сложную химическую структуру).
Матрицы накопленных Пар замен (Aij) из работы Дайхофф (х10) Наиболее часто встречающейся замена D и E (83 замены). Другими часто заменяющимися парами так же являются аминокислоты со сходными физико-химическими свойствами: A-S (77 пар) и A-T (59 пар) малые боковые группы; T-S (69 пар), малые аминокислоты с полярными боковыми группами; V-I (66 пар), гидрофобные. Для 35 пар аминокислот из 190 замен не наблюдалось. Отсутствие замен характерно, например, для триптофана (W), имеющего специфические свойства боковой группы (большой объем, сложную химическую структуру).
 Относительная мутабильность аминокислот. Seq 1: ADA Выровненные Seq 2: ADB последовательности Мутабильность характеризует свойство аминокислоты мутировать. mi=(число замен аминокислоты i)/(число аминокислот i) mi= i j. Aij/pi Накопленная статистика: Тип аминокислоты A B D Число замен 1 1 0 Число встречаемости 3 1 2 Частота встречаемости(fi) 0. 5 0. 17 0. 33 Относительная мутабильность(mi) 0. 33 1 0
Относительная мутабильность аминокислот. Seq 1: ADA Выровненные Seq 2: ADB последовательности Мутабильность характеризует свойство аминокислоты мутировать. mi=(число замен аминокислоты i)/(число аминокислот i) mi= i j. Aij/pi Накопленная статистика: Тип аминокислоты A B D Число замен 1 1 0 Число встречаемости 3 1 2 Частота встречаемости(fi) 0. 5 0. 17 0. 33 Относительная мутабильность(mi) 0. 33 1 0
 Для примера из рисунка получаем p. A=13/28=0. 47,") Пример Матрицы накопленных Пар замен (Aij) Для примера из рисунка получаем p. A=13/28=0. 47, p. F=3/28=0. 1, p. C=7/28=0. 25, p. D=5/28=0. 18. Мутабильности будут пропорциональны величинам m. A=8/0. 47=17, m. F=4/0. 1=40, m. C=0, m. D=4/0. 18=22. Видно, что хотя число наблюдаемых замен для аминокислоты A больше остальных, ее мутабильность относительно F и D меньше, в силу того, что число аминокислот А в выравниваниях существенно больше.
Пример Матрицы накопленных Пар замен (Aij) Для примера из рисунка получаем p. A=13/28=0. 47, p. F=3/28=0. 1, p. C=7/28=0. 25, p. D=5/28=0. 18. Мутабильности будут пропорциональны величинам m. A=8/0. 47=17, m. F=4/0. 1=40, m. C=0, m. D=4/0. 18=22. Видно, что хотя число наблюдаемых замен для аминокислоты A больше остальных, ее мутабильность относительно F и D меньше, в силу того, что число аминокислот А в выравниваниях существенно больше.
. Делеция интерпретируется как") Относительная мутабильность разных аминокислот. Относительная мутабильность аминокислот по отношению к ALA(100). Делеция интерпретируется как 21 тип остатка Ser 149 Asp 90 Gly 48 Met 122 Thr 90 Phe 45 Asn 111 Gap 84 Arg 44 lle 110 Val 80 Leu 38 Glu 102 Lys 57 Tyr 34 Ala 100 Pro 56 Cys 27 Gln 98 His 50 Trp 22 Вариабельные: Ser, Met, Asn, Ile, Gly. Консервативные: Trp, Cys, Tyr, Leu, Arg.
Относительная мутабильность разных аминокислот. Относительная мутабильность аминокислот по отношению к ALA(100). Делеция интерпретируется как 21 тип остатка Ser 149 Asp 90 Gly 48 Met 122 Thr 90 Phe 45 Asn 111 Gap 84 Arg 44 lle 110 Val 80 Leu 38 Glu 102 Lys 57 Tyr 34 Ala 100 Pro 56 Cys 27 Gln 98 His 50 Trp 22 Вариабельные: Ser, Met, Asn, Ile, Gly. Консервативные: Trp, Cys, Tyr, Leu, Arg.
 Частота встречаемости аминокислот. Ala Lys Val Ser Glu . 096. 085. 078. 057. 053 Asn lle Arg Tyr Met . 042. 035. 034. 030. 012 Gly Leu Thr Asp Phe . 090. 084. 062. 053. 045 Pro His Gln Cys Trp Этот параметр отражает относительную представленность частот в банке данных, или Равновесные частоты аминокислот. . 041. 034. 032. 025. 012
Частота встречаемости аминокислот. Ala Lys Val Ser Glu . 096. 085. 078. 057. 053 Asn lle Arg Tyr Met . 042. 035. 034. 030. 012 Gly Leu Thr Asp Phe . 090. 084. 062. 053. 045 Pro His Gln Cys Trp Этот параметр отражает относительную представленность частот в банке данных, или Равновесные частоты аминокислот. . 041. 034. 032. 025. 012
 Оценка матрицы замен. Mij – замена аминокислоты j на i Aij – элемент матрицы парных сумм. mi – относительная мутабильность аминокислоты - константа, котороая определяется средней скоростью замен. Диагональные элементы вычисляются как Среднее число замен можно вычислить по диагональным элементам матрицы : Константу выбирают из условия чтобы среднее число замен равнялось 1 мутация на 100 позиций белка – 1 PAM:
Оценка матрицы замен. Mij – замена аминокислоты j на i Aij – элемент матрицы парных сумм. mi – относительная мутабильность аминокислоты - константа, котороая определяется средней скоростью замен. Диагональные элементы вычисляются как Среднее число замен можно вычислить по диагональным элементам матрицы : Константу выбирают из условия чтобы среднее число замен равнялось 1 мутация на 100 позиций белка – 1 PAM:
 Аминокислота после замены (i) PAM 1: Вероятность не измениться (ALA на") Исходная аминокислота (j) Аминокислота после замены (i) PAM 1: Вероятность не измениться (ALA на ALA): 0. 9867; измениться: 0. 0133 (1. 33%) Вероятность замены ALA на GLU равна 10/10000=0. 1% Вероятность замены ALA на SER равна 28/10000=0. 28% Вероятность замены ALA на ARG равна 1/10000=0. 01%
Исходная аминокислота (j) Аминокислота после замены (i) PAM 1: Вероятность не измениться (ALA на ALA): 0. 9867; измениться: 0. 0133 (1. 33%) Вероятность замены ALA на GLU равна 10/10000=0. 1% Вероятность замены ALA на SER равна 28/10000=0. 28% Вероятность замены ALA на ARG равна 1/10000=0. 01%
 Оценка матрицы вероятности замен на больших разных эволюционных расстояниях.
Оценка матрицы вероятности замен на больших разных эволюционных расстояниях.
 Исходная аминокислота По мере увеличения времени эволюции элементы матрицы Mij по столбцам будут стремиться к величинам fi – долям аминокислот в банке данных Аминокислота после замены PAM 250. Вероятность не измениться (ALA на ALA): 0. 13; измениться: 0. 87 (87%) Вероятность замены ALA на GLU равна 5/100=5% (PAM 1: 0. 1%) Вероятность замены ALA на SER равна 9/100=9% (PAM 1: 0. 28%) Вероятность замены ALA на ARG равна 3/100=3% (PAM 1: 0. 01%)
Исходная аминокислота По мере увеличения времени эволюции элементы матрицы Mij по столбцам будут стремиться к величинам fi – долям аминокислот в банке данных Аминокислота после замены PAM 250. Вероятность не измениться (ALA на ALA): 0. 13; измениться: 0. 87 (87%) Вероятность замены ALA на GLU равна 5/100=5% (PAM 1: 0. 1%) Вероятность замены ALA на SER равна 9/100=9% (PAM 1: 0. 28%) Вероятность замены ALA на ARG равна 3/100=3% (PAM 1: 0. 01%)
 Среднее число наблюдаемых замен p Расстояния белковых последовательностей • Матрицы серии PAM-N отражают вероятность Среднее число замены наблюдаемых замен аминокислот p: ai aj за время эволюции, эквивалентное N PAM единиц. Расстояние PAM (d) (среднее число произошедших замен на 100 позиций белка)
Среднее число наблюдаемых замен p Расстояния белковых последовательностей • Матрицы серии PAM-N отражают вероятность Среднее число замены наблюдаемых замен аминокислот p: ai aj за время эволюции, эквивалентное N PAM единиц. Расстояние PAM (d) (среднее число произошедших замен на 100 позиций белка)
 Оценка матрицы замен на более современных данных: матрица JTT. В 1992 году Jones, Taylor and Thornton оценили матрицу замен, аналогичную Дайхофф, но по большему числу последовательностей. Реконструкция предковых последовательностей не использовалась. Полученная модель замен названа JTT. Она считается лучше модели Дайхофф. Jones DT, Taylor WR & Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Computer Applications in the Biosciences 8: 275 -282.
Оценка матрицы замен на более современных данных: матрица JTT. В 1992 году Jones, Taylor and Thornton оценили матрицу замен, аналогичную Дайхофф, но по большему числу последовательностей. Реконструкция предковых последовательностей не использовалась. Полученная модель замен названа JTT. Она считается лучше модели Дайхофф. Jones DT, Taylor WR & Thornton JM (1992) The rapid generation of mutation data matrices from protein sequences. Computer Applications in the Biosciences 8: 275 -282.
 Сравниваются две аминокислотные последовательности. S 1=ARNLLSAA S 2=AKQLLSLA") Матрицы сходства аминокислот (используются для выравнивания) Сравниваются две аминокислотные последовательности. S 1=ARNLLSAA S 2=AKQLLSLA • • Проверка двух гипотез: H 1, две сравниваемые последовательности являются гомологичными, т. е. в ходе процесса молекулярной эволюции кодируемые ими белки произошли от одного общего предка и, следовательно, обладают сходной структурой и функцией. Вероятность наблюдать пару выравненных аминокислот - p(αi, • βi) H 0, альтернативная гипотеза, две сравниваемые последовательности не являются гомологичными, белки имеют разные структуры и выполняют различные функции. Вероятность наблюдать пару выровненных аминокислот p(αi) p(βi), где p(α)=f(a) – равновесная частота встречаемости аминокислоты α Нужно сравнить: p(αi, βi) и p(αi) p(βi): s=log(p(αi, βi)/ p(αi) p(βi)). Если s>0 , то последовательности гомологичные
Матрицы сходства аминокислот (используются для выравнивания) Сравниваются две аминокислотные последовательности. S 1=ARNLLSAA S 2=AKQLLSLA • • Проверка двух гипотез: H 1, две сравниваемые последовательности являются гомологичными, т. е. в ходе процесса молекулярной эволюции кодируемые ими белки произошли от одного общего предка и, следовательно, обладают сходной структурой и функцией. Вероятность наблюдать пару выравненных аминокислот - p(αi, • βi) H 0, альтернативная гипотеза, две сравниваемые последовательности не являются гомологичными, белки имеют разные структуры и выполняют различные функции. Вероятность наблюдать пару выровненных аминокислот p(αi) p(βi), где p(α)=f(a) – равновесная частота встречаемости аминокислоты α Нужно сравнить: p(αi, βi) и p(αi) p(βi): s=log(p(αi, βi)/ p(αi) p(βi)). Если s>0 , то последовательности гомологичные
 Вес выравнивания Сравниваются две аминокислотные последовательности. S 1=ARNLLSAA S 2=AKQLLSLA Гипотеза 1: две последовательности произошли от общего предка (гомологи) в результате процесса эволюции по Дайхофф с матрицей М (t=150), что означает, что вероятность замены аминокислоты i->j Prob(i->j)=wij. (W(150)=M 150) Гипотеза 2 : две последовательности являются случайными, что означает, что вероятность замены аминокислоты i->j Prob(i->j)=fj – равна частоте встречаемости аминокислоты j. . L 1/L 2 =П(l)[wij / fj] ln(L 1/L 2)=ln(П(l)wij/ fj)= (l)[ln(wij/ fj)]= (l)sij; S=sij – матрица весов сравнения аминокислот.
Вес выравнивания Сравниваются две аминокислотные последовательности. S 1=ARNLLSAA S 2=AKQLLSLA Гипотеза 1: две последовательности произошли от общего предка (гомологи) в результате процесса эволюции по Дайхофф с матрицей М (t=150), что означает, что вероятность замены аминокислоты i->j Prob(i->j)=wij. (W(150)=M 150) Гипотеза 2 : две последовательности являются случайными, что означает, что вероятность замены аминокислоты i->j Prob(i->j)=fj – равна частоте встречаемости аминокислоты j. . L 1/L 2 =П(l)[wij / fj] ln(L 1/L 2)=ln(П(l)wij/ fj)= (l)[ln(wij/ fj)]= (l)sij; S=sij – матрица весов сравнения аминокислот.
 в") Матрицы сходства аминокислот S 1: ………αi………… S 2: ………βi………… Как вычислить p(αi, βi) в модели Дайхофф? Вероятность наблюдать α в позиции i: f(α) Вероятность наблюдать замену α-> β за эволюционное время N PAM : M β α(N), где M(N)=(M(1))N - матрица вероятностей замен Тогда p(αi, βi)= f(α) M β α(N) и sαβ=log(p(αi, βi)/ p(α) p(β))= log(f(α) M β α(N) / f(α) f(β)) = log(M β α(N) / f(β)) S – используется в выравнивании (матрица сравнения аминокислот) и определяют вес выравнивания
Матрицы сходства аминокислот S 1: ………αi………… S 2: ………βi………… Как вычислить p(αi, βi) в модели Дайхофф? Вероятность наблюдать α в позиции i: f(α) Вероятность наблюдать замену α-> β за эволюционное время N PAM : M β α(N), где M(N)=(M(1))N - матрица вероятностей замен Тогда p(αi, βi)= f(α) M β α(N) и sαβ=log(p(αi, βi)/ p(α) p(β))= log(f(α) M β α(N) / f(α) f(β)) = log(M β α(N) / f(β)) S – используется в выравнивании (матрица сравнения аминокислот) и определяют вес выравнивания
 Матрица сравнения аминокислот PAM 250
Матрица сравнения аминокислот PAM 250
 Практический вопрос 1 Требуется построить выравнивание двух последовательностей с ожидаемым уровнем сходства 40%. Какое выравнивание будет лучше: построенное по матрице 50 PAM, 100 PAM или 250 PAM?
Практический вопрос 1 Требуется построить выравнивание двух последовательностей с ожидаемым уровнем сходства 40%. Какое выравнивание будет лучше: построенное по матрице 50 PAM, 100 PAM или 250 PAM?
 Среднее число замен d Ответ Расстояние PAM Более точное выравнивание будет достигнуто при выборе матрице PAM, соответствующей ожидаемому эволюционному расстоянию
Среднее число замен d Ответ Расстояние PAM Более точное выравнивание будет достигнуто при выборе матрице PAM, соответствующей ожидаемому эволюционному расстоянию
 Практический вопрос 2 Требуется произвести поиск по возможности более близкородственных белков в базе данных SWISSPROT с помощью программы BLAST. Какую матрицу выбрать? Ответ: для более качественного поиска близкородственных белков необходимо выбирать матрицу, соответствующую ожидаемому уровню их сходства. Эволюционное расстояние (доля замен) Высокое сходство низкое сходство Номер матрицы PAM 30 50 150 Номер матрицы BLOSUM (отражает процент сходства посл-тей) 80 60 45
Практический вопрос 2 Требуется произвести поиск по возможности более близкородственных белков в базе данных SWISSPROT с помощью программы BLAST. Какую матрицу выбрать? Ответ: для более качественного поиска близкородственных белков необходимо выбирать матрицу, соответствующую ожидаемому уровню их сходства. Эволюционное расстояние (доля замен) Высокое сходство низкое сходство Номер матрицы PAM 30 50 150 Номер матрицы BLOSUM (отражает процент сходства посл-тей) 80 60 45
 Имеются n наблюдений случайной величины x – вектор наблюдений x=(x 1,") Функция правдоподобия (ФП) Имеются n наблюдений случайной величины x – вектор наблюдений x=(x 1, x 2, …xn); Вероятность наблюдать значение x зависит от некоторого параметра : p(x| ). Тогда вероятность наблюдать n значений x=(x 1, x 2, …xn) равна L(x| )=p(x 1| ) p(x 2| ) …. p(xn| ) L(X| ) называют функцией правдоподобия. Ее удобно использовать при оценке параметров распределений p(x| ). Идея: выбрать такой параметр, который максимизирует вероятность наблюдать набор значений x=(x 1, x 2, …xn).
Функция правдоподобия (ФП) Имеются n наблюдений случайной величины x – вектор наблюдений x=(x 1, x 2, …xn); Вероятность наблюдать значение x зависит от некоторого параметра : p(x| ). Тогда вероятность наблюдать n значений x=(x 1, x 2, …xn) равна L(x| )=p(x 1| ) p(x 2| ) …. p(xn| ) L(X| ) называют функцией правдоподобия. Ее удобно использовать при оценке параметров распределений p(x| ). Идея: выбрать такой параметр, который максимизирует вероятность наблюдать набор значений x=(x 1, x 2, …xn).
 – p, вероятность решки (Р)") Пример : бросание монетки Бросаем монету, вероятность орла (O) – p, вероятность решки (Р) – 1 -p. В данном случае параметр, от которого зависит вероятность наблюдать событие O – p. Наблюдаем 11 бросаний монет: ООРОРРО Функция правдоподобия: L=pp(1 − p)(1 − p)p(1 − p)p L=p 5(1 − p)6 L(p)
Пример : бросание монетки Бросаем монету, вероятность орла (O) – p, вероятность решки (Р) – 1 -p. В данном случае параметр, от которого зависит вероятность наблюдать событие O – p. Наблюдаем 11 бросаний монет: ООРОРРО Функция правдоподобия: L=pp(1 − p)(1 − p)p(1 − p)p L=p 5(1 − p)6 L(p)
") Оценка параметра p Обычно используют логарифм Ф. П. (логарифмирование не меняет положение максимума)
Оценка параметра p Обычно используют логарифм Ф. П. (логарифмирование не меняет положение максимума)
 Если много параметров В случае нескольких параметров определяется поверхность правдоподобия ОМП контур
Если много параметров В случае нескольких параметров определяется поверхность правдоподобия ОМП контур
 Функция правдоподобия и проверка гипотез Данные Гипотеза 1 Гипотеза 2 Условная вероятность Выбирается гипотеза, при которой вероятность наблюдать набор данных выше.
Функция правдоподобия и проверка гипотез Данные Гипотеза 1 Гипотеза 2 Условная вероятность Выбирается гипотеза, при которой вероятность наблюдать набор данных выше.
 Пример: тест на модель частот нуклеотидов Пусть имеется последовательность нуклеотидов, в которых частоты их встречаемости A C G T Гипотеза Н 1: частоты встречаемости A C G T равны их оценкам в последовательности Гипотеза Н 2: частоты встречаемости A = C= G = T =1/4 (модель Джукса-Кантора)
Пример: тест на модель частот нуклеотидов Пусть имеется последовательность нуклеотидов, в которых частоты их встречаемости A C G T Гипотеза Н 1: частоты встречаемости A C G T равны их оценкам в последовательности Гипотеза Н 2: частоты встречаемости A = C= G = T =1/4 (модель Джукса-Кантора)
 Сравнение значений ФП Гипотеза 1 Гипотеза 2 Принять гипотезу 1 выгоднее, первая модель более вероятна.
Сравнение значений ФП Гипотеза 1 Гипотеза 2 Принять гипотезу 1 выгоднее, первая модель более вероятна.
 Пример сравнения двух последовательностей Сравниваются две последовательности, эволюционировавшие в течении времени t со скоростью замен t Простейший случай – 2 нуклеотида, модель Джукса-Кантора
Пример сравнения двух последовательностей Сравниваются две последовательности, эволюционировавшие в течении времени t со скоростью замен t Простейший случай – 2 нуклеотида, модель Джукса-Кантора
 Филогенетические деревья Корень Время Порядок ветвления – точки на шкале времени • Необходимы для восстановления эволюционной истории • Описывают эволюционные отношения для набора из нескольких последовательностей
Филогенетические деревья Корень Время Порядок ветвления – точки на шкале времени • Необходимы для восстановления эволюционной истории • Описывают эволюционные отношения для набора из нескольких последовательностей
, соответствуют таксономическим") Филогенетические деревья Конечные узлы Внутренние узлы Корень A-E – конечные узлы (листья), соответствуют таксономическим единицам (OTU); F-I внутренние узлы (предковые) Таксономические единицы: виды, популяции, особи, гены, белки. Потомки эволюционируют независимо. Топология – порядок ветвления узлов дерева.
Филогенетические деревья Конечные узлы Внутренние узлы Корень A-E – конечные узлы (листья), соответствуют таксономическим единицам (OTU); F-I внутренние узлы (предковые) Таксономические единицы: виды, популяции, особи, гены, белки. Потомки эволюционируют независимо. Топология – порядок ветвления узлов дерева.
 Не все деревья имеют корень i, j, - внутренние узлы, последовательности для которых неизвестны • Не все методы построения деревьев могут давать положение корня
Не все деревья имеют корень i, j, - внутренние узлы, последовательности для которых неизвестны • Не все методы построения деревьев могут давать положение корня
 Основанные на наблюдаемых") Группы методов построения деревьев Основанные на эволюционных расстояниях (UPGMA, ближайшего соседа) Основанные на наблюдаемых признаках нуклеотидах, аминокислотах (метод максимальной экономии, максимального правдоподобия).
Группы методов построения деревьев Основанные на эволюционных расстояниях (UPGMA, ближайшего соседа) Основанные на наблюдаемых признаках нуклеотидах, аминокислотах (метод максимальной экономии, максимального правдоподобия).
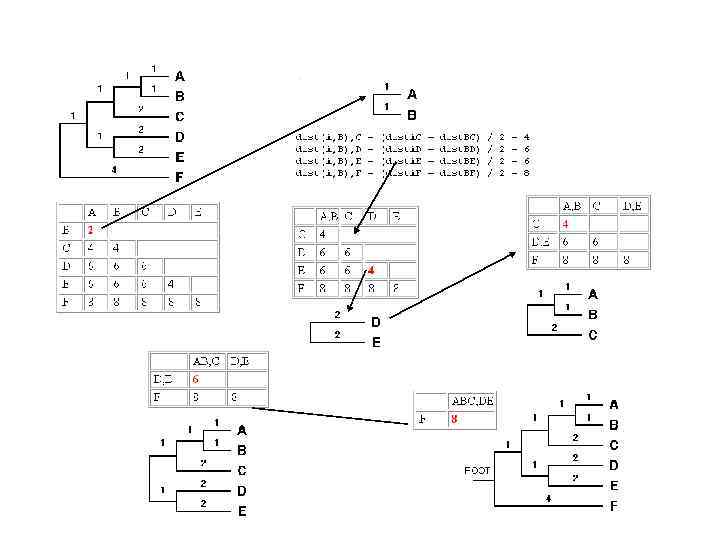
 Методы построения деревьев: UPGMA • Расстояние между кластером X и кластером Y равно среднему от парных расстояний между последовательностя ми этих кластеров • Предполагает равномерность замен (молек. часы) во всех таксонах • Расстояние=2*длину ветви • Дает всегда дерево с корнем
Методы построения деревьев: UPGMA • Расстояние между кластером X и кластером Y равно среднему от парных расстояний между последовательностя ми этих кластеров • Предполагает равномерность замен (молек. часы) во всех таксонах • Расстояние=2*длину ветви • Дает всегда дерево с корнем
 UPGMA ошибается если скорости замен различны
UPGMA ошибается если скорости замен различны
 Метод ближайшего соседа В этом подходе расстояния нормируются на среднюю удаленность ri OTU от других OTU При построении дерева два ближайших узла i, j заменяются новым узлом n; расстояния пересчитываются по следующим правилам: Для каждого к На следующем шаге выбирается пара i, j, для которых Dij минимально ( Dij=dij-ri-rj )
Метод ближайшего соседа В этом подходе расстояния нормируются на среднюю удаленность ri OTU от других OTU При построении дерева два ближайших узла i, j заменяются новым узлом n; расстояния пересчитываются по следующим правилам: Для каждого к На следующем шаге выбирается пара i, j, для которых Dij минимально ( Dij=dij-ri-rj )
 Для пары A, B Средняя удаленность Первый шаг – стартуем с звездного дерева Второй шаг - A, B образуют новую единицу, U U
Для пары A, B Средняя удаленность Первый шаг – стартуем с звездного дерева Второй шаг - A, B образуют новую единицу, U U
, • Clustal.") Программы реализующие данных подход • Neighbor в пакете Phylip (Felsentein, Univ. Washington), • Clustal. W (D. Higgins), • Distnj в пакете Protml (Adachi and Hasegawa, Univ. Tokyo) Учитывает неравномерность скоростей эволюции на ветвях Быстрый, может использоваться для больших семейств Иногда могут встречаться отрицательные расстояния.
Программы реализующие данных подход • Neighbor в пакете Phylip (Felsentein, Univ. Washington), • Clustal. W (D. Higgins), • Distnj в пакете Protml (Adachi and Hasegawa, Univ. Tokyo) Учитывает неравномерность скоростей эволюции на ветвях Быстрый, может использоваться для больших семейств Иногда могут встречаться отрицательные расстояния.
 Дерево можно перестраивать и оценивать Перестановка соседних узлов Число различных топологий дерева Без корня С корнем Сокращение и перестройка поддеревьев • Каждому дереву можно присвоить числовую характеристику и сравнивать их
Дерево можно перестраивать и оценивать Перестановка соседних узлов Число различных топологий дерева Без корня С корнем Сокращение и перестройка поддеревьев • Каждому дереву можно присвоить числовую характеристику и сравнивать их
 Метод парсимонии Пример построения дерева для набора из четырех последовательностей, с двузнаковым алфавитом (0, 1) 1) В первом дереве изменения происходят только один раз (+) 2) Во втором дереве 1 появляется (+) и теряется (*) 3) В третьем дереве 1 появляется независимо два раза (+) Дерево (1) содержит минимальное число эволюционных событий – его и выбираем.
Метод парсимонии Пример построения дерева для набора из четырех последовательностей, с двузнаковым алфавитом (0, 1) 1) В первом дереве изменения происходят только один раз (+) 2) Во втором дереве 1 появляется (+) и теряется (*) 3) В третьем дереве 1 появляется независимо два раза (+) Дерево (1) содержит минимальное число эволюционных событий – его и выбираем.
 • Paup (David Swofford) Приемлем") Программы реализующие данных подход • Protpars (Felsentein, пакет Phylip) • Paup (David Swofford) Приемлем для последовательностей с высокой гомологией. Нельзя использовать для сильно дивергировавших последовательностей!
Программы реализующие данных подход • Protpars (Felsentein, пакет Phylip) • Paup (David Swofford) Приемлем для последовательностей с высокой гомологией. Нельзя использовать для сильно дивергировавших последовательностей!
, при условии, что эволюция происходила") Методы максимального правдоподобия • Вычисляется вероятность наблюдения данных (D), при условии, что эволюция происходила по данной топологии (T) – функция правдоподобия L(D|T) • Вычисляется рекурсивно от листьев к вершине • Выбирается дерево, которое дает max(L) • Решение зависит от выбора модели замен
Методы максимального правдоподобия • Вычисляется вероятность наблюдения данных (D), при условии, что эволюция происходила по данной топологии (T) – функция правдоподобия L(D|T) • Вычисляется рекурсивно от листьев к вершине • Выбирается дерево, которое дает max(L) • Решение зависит от выбора модели замен
; Задана топология дерева (порядок") Как оценить правдоподобие дерева? Задана модель эволюции (матрица скоростей замен); Задана топология дерева (порядок ветвления) и длины ветвей (ti=скорость замен * время)
Как оценить правдоподобие дерева? Задана модель эволюции (матрица скоростей замен); Задана топология дерева (порядок ветвления) и длины ветвей (ti=скорость замен * время)
 Если бы внутренние узлы были известны
Если бы внутренние узлы были известны
") Но они неизвестны Необходимо просуммировать по всем нуклеотидам во внутренних узлах дерева (усреднить)
Но они неизвестны Необходимо просуммировать по всем нуклеотидам во внутренних узлах дерева (усреднить)
 – Вероятность наблюдать нуклеотид типа sj в") Подсчет методом сокращения Felsenstein, 1981 Prob(sj|s, vj) – Вероятность наблюдать нуклеотид типа sj в дочернем узле j при условии, что в родительском узле i находится символ s и время эволюции составило vj.
Подсчет методом сокращения Felsenstein, 1981 Prob(sj|s, vj) – Вероятность наблюдать нуклеотид типа sj в дочернем узле j при условии, что в родительском узле i находится символ s и время эволюции составило vj.
 общей предковой последовательности L 0(i) Итоговое значение") Итоговое значение усредняется по частотам нуклеотидов (аминокислот) общей предковой последовательности L 0(i) Итоговое значение Ф. П. : перемножаются для всех позиций (независимость мутаций в позициях)
Итоговое значение усредняется по частотам нуклеотидов (аминокислот) общей предковой последовательности L 0(i) Итоговое значение Ф. П. : перемножаются для всех позиций (независимость мутаций в позициях)
 • Fast. DNAML (ДНК)") Программы реализующие данных подход • DNAML (пакет Phylip, ДНК ) • Fast. DNAML (ДНК) • Prot. ML (ДНК и белки, Adachi and Hasegawa) • Puzzle (Днк и белки, Strimmer and von Haeseler) • Phyml (ДНК, белки ; Guindon, Gasquel) • Ra. XML (ДНК, белки (если очень много последовательностей – более 1000, быстрый эффективный), Stamatakis)
Программы реализующие данных подход • DNAML (пакет Phylip, ДНК ) • Fast. DNAML (ДНК) • Prot. ML (ДНК и белки, Adachi and Hasegawa) • Puzzle (Днк и белки, Strimmer and von Haeseler) • Phyml (ДНК, белки ; Guindon, Gasquel) • Ra. XML (ДНК, белки (если очень много последовательностей – более 1000, быстрый эффективный), Stamatakis)
 Методы максимального правдоподобия • С помощью методов МП можно оценивать и другие параметры: так как и матрица замен, и скорости замен могут быть такими параметрами. L=L(T, M, t…. . ). • Можно усложнять модель, добавляя новые параметры. • Метод имеет статистическое обоснование • Но требует большого количества вычислений Матрица замен, оцененная методом МП – WAG [Whealan and Goldman] (лучше Dayhoff, JTT).
Методы максимального правдоподобия • С помощью методов МП можно оценивать и другие параметры: так как и матрица замен, и скорости замен могут быть такими параметрами. L=L(T, M, t…. . ). • Можно усложнять модель, добавляя новые параметры. • Метод имеет статистическое обоснование • Но требует большого количества вычислений Матрица замен, оцененная методом МП – WAG [Whealan and Goldman] (лучше Dayhoff, JTT).
 Вопросы к экзамену Что такое эволюционное расстояние? В чем отличие модели нуклеотидных замен Кимуры от модели Джукса-Кантора? Классификация аминокислотных замен в белках. Особенности модели эволюции аминокислотных последовательностей Дайхоф. Что такое мутабильность аминокислоты? Каким образом матрицы сходства аминокислот (матрица весов сравнения аминокислот) связаны с матрицами скоростей замен? Что такое филогенетическое дерево? Какие существуют методы построения филогенетических деревьев? Метод расстояний на примере UPGMA и его недостатки.
Вопросы к экзамену Что такое эволюционное расстояние? В чем отличие модели нуклеотидных замен Кимуры от модели Джукса-Кантора? Классификация аминокислотных замен в белках. Особенности модели эволюции аминокислотных последовательностей Дайхоф. Что такое мутабильность аминокислоты? Каким образом матрицы сходства аминокислот (матрица весов сравнения аминокислот) связаны с матрицами скоростей замен? Что такое филогенетическое дерево? Какие существуют методы построения филогенетических деревьев? Метод расстояний на примере UPGMA и его недостатки.

 Модели эволюции белковых последовательностей • Белки – гетерогенные объекты. • Содержат различные типы вторичных структур • Различные участки доступности растворителю • Содержат функциональные сайты • Давление отбора для разных участков белка различно.
Модели эволюции белковых последовательностей • Белки – гетерогенные объекты. • Содержат различные типы вторичных структур • Различные участки доступности растворителю • Содержат функциональные сайты • Давление отбора для разных участков белка различно.
 Особенности эволюции белков скорости фиксации замен для различных ветвей эволюционного дерева могут быть различными, что не соответствует предположению о "молекулярных часах" (Ayala et al. , 1997; Ayala, 1997); скорости фиксации замен аминокислот могут различаться для различных позиций белка вдоль его последовательности (Uzzel and Corbin, 1971; Morozov et al. , 2000); вероятности замен аминокислот (определенные как элементы матрицы аминокислотных замен) для различных участков глобулы или даже для отдельных позиций белковой последовательности могут быть различными (Overington, 1992). параметры эволюционного процесса (в частности элементы матрицы M ) могут зависеть от времени (Benner et al. , 1994). мутации в различных позициях белка могут фиксироваться зависимым образом.
Особенности эволюции белков скорости фиксации замен для различных ветвей эволюционного дерева могут быть различными, что не соответствует предположению о "молекулярных часах" (Ayala et al. , 1997; Ayala, 1997); скорости фиксации замен аминокислот могут различаться для различных позиций белка вдоль его последовательности (Uzzel and Corbin, 1971; Morozov et al. , 2000); вероятности замен аминокислот (определенные как элементы матрицы аминокислотных замен) для различных участков глобулы или даже для отдельных позиций белковой последовательности могут быть различными (Overington, 1992). параметры эволюционного процесса (в частности элементы матрицы M ) могут зависеть от времени (Benner et al. , 1994). мутации в различных позициях белка могут фиксироваться зависимым образом.
 Гетерогенность скоростей замен в белках. В одном белке различные домены могут иметь разные скорости замен Проинсулин Инсулин
Гетерогенность скоростей замен в белках. В одном белке различные домены могут иметь разные скорости замен Проинсулин Инсулин
 Особенности эволюции белков скорости фиксации замен для различных ветвей эволюционного дерева могут быть различными, что не соответствует предположению о "молекулярных часах" (Ayala et al. , 1997; Ayala, 1997); скорости фиксации замен аминокислот могут различаться для различных позиций белка вдоль его последовательности (Uzzel and Corbin, 1971; Morozov et al. , 2000); вероятности замен аминокислот (определенные как элементы матрицы аминокислотных замен) для различных участков глобулы или даже для отдельных позиций белковой последовательности могут быть различными (Overington, 1992). параметры эволюционного процесса (в частности элементы матрицы M ) могут зависеть от времени (Benner et al. , 1994). мутации в различных позициях белка могут фиксироваться зависимым образом.
Особенности эволюции белков скорости фиксации замен для различных ветвей эволюционного дерева могут быть различными, что не соответствует предположению о "молекулярных часах" (Ayala et al. , 1997; Ayala, 1997); скорости фиксации замен аминокислот могут различаться для различных позиций белка вдоль его последовательности (Uzzel and Corbin, 1971; Morozov et al. , 2000); вероятности замен аминокислот (определенные как элементы матрицы аминокислотных замен) для различных участков глобулы или даже для отдельных позиций белковой последовательности могут быть различными (Overington, 1992). параметры эволюционного процесса (в частности элементы матрицы M ) могут зависеть от времени (Benner et al. , 1994). мутации в различных позициях белка могут фиксироваться зависимым образом.
") Гетерогенность скоростей замен в позициях белка Относительные темпы замен для Ig-k (Morozov, 2000)
Гетерогенность скоростей замен в позициях белка Относительные темпы замен для Ig-k (Morozov, 2000)
 Сравнение матриц замен для разных участков белка Режимы эволюции для разных участков глобулы различны! Петли на поверхности Погруженная спираль Рисунок адаптирован из лекции R. Goldstein, Modeling Evolution at the Amino Acid Level , http: //online. itp. ucsb. edu/online/infobio 01/goldstein 1/
Сравнение матриц замен для разных участков белка Режимы эволюции для разных участков глобулы различны! Петли на поверхности Погруженная спираль Рисунок адаптирован из лекции R. Goldstein, Modeling Evolution at the Amino Acid Level , http: //online. itp. ucsb. edu/online/infobio 01/goldstein 1/
 Матрицы замен, специфичные для позиций • Основная идея – описывать замены для разных структурных классов остатков разными матрицами замен (Koshi & Goldstein; Wako & Blundell; Overington).
Матрицы замен, специфичные для позиций • Основная идея – описывать замены для разных структурных классов остатков разными матрицами замен (Koshi & Goldstein; Wako & Blundell; Overington).
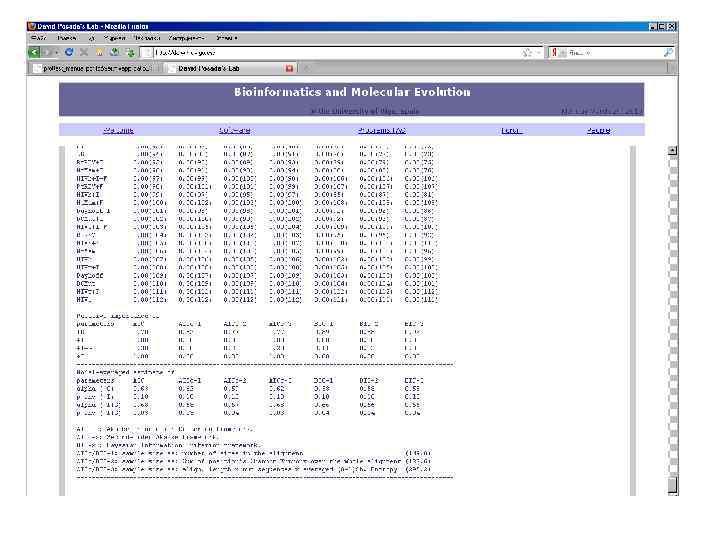
 Prot. Test- подбор модели замен аминокислот, оптимальной для белкового семейства http: //darwin. uvigo. es/
Prot. Test- подбор модели замен аминокислот, оптимальной для белкового семейства http: //darwin. uvigo. es/
 • • • Dayhoff (Dayhoff et al. 1978) JTT (Jones") Тестируемые модели (типы матриц) • • • Dayhoff (Dayhoff et al. 1978) JTT (Jones et al. 1992) WAG (Whelan and Goldman 2001) mt. REV (Adachi and Hasegawa 1996) Mt. Mam (Cao et al. 1998), VT (Muller and Vingron 2000) Cp. REV (Adachi et al. 2000) Rt. REV (Dimmic et al. 2002) Mt. Art (Abascal et al. 2007) HIVb/HIVw (Nickle et al. 2007) LG (Le and Gascuel 2008) Blosum 62 (Henikoff and Henikoff 1992)
Тестируемые модели (типы матриц) • • • Dayhoff (Dayhoff et al. 1978) JTT (Jones et al. 1992) WAG (Whelan and Goldman 2001) mt. REV (Adachi and Hasegawa 1996) Mt. Mam (Cao et al. 1998), VT (Muller and Vingron 2000) Cp. REV (Adachi et al. 2000) Rt. REV (Dimmic et al. 2002) Mt. Art (Abascal et al. 2007) HIVb/HIVw (Nickle et al. 2007) LG (Le and Gascuel 2008) Blosum 62 (Henikoff and Henikoff 1992)
 Дополнительные особенности моделей • +I – учет доли сайтов в которых замены не происходит (invariable sites) • +G – учет различных категорий сайтов по вариабельности • +F – учет частот встречаемости аминокислот в выравнивании
Дополнительные особенности моделей • +I – учет доли сайтов в которых замены не происходит (invariable sites) • +G – учет различных категорий сайтов по вариабельности • +F – учет частот встречаемости аминокислот в выравнивании
 Для оценки качества модели используется критерий Акаике • L – значение функции правдоподобия модели (условная вероятность получить наблюдаемые данные при заданных параметрах модели) • K – число параметров модели Меньшие значения соотвествуют лучшей модели. Для нескольких моделей можно сравнить с лучшей:
Для оценки качества модели используется критерий Акаике • L – значение функции правдоподобия модели (условная вероятность получить наблюдаемые данные при заданных параметрах модели) • K – число параметров модели Меньшие значения соотвествуют лучшей модели. Для нескольких моделей можно сравнить с лучшей:
 Пример анализа:
Пример анализа:
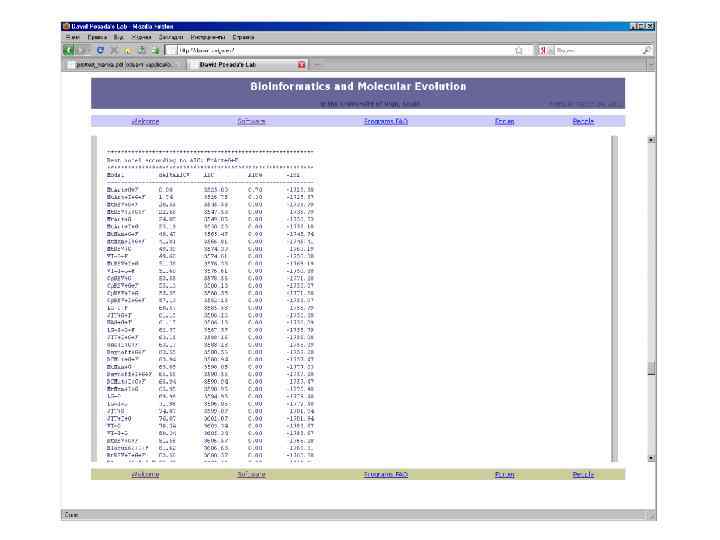
 Результаты сравнения моделей
Результаты сравнения моделей