Митохондриальные болезни.pptx
- Количество слайдов: 39



Митохондриальные болезни Выполнил: Гапешин Р. А. 606 группа
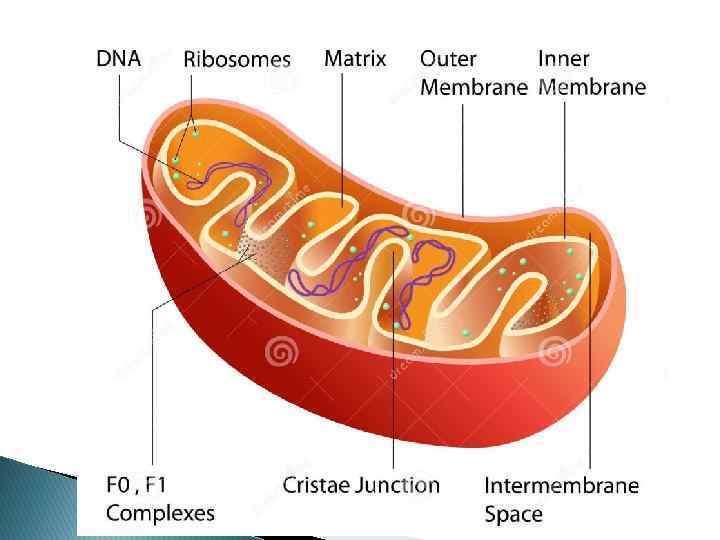
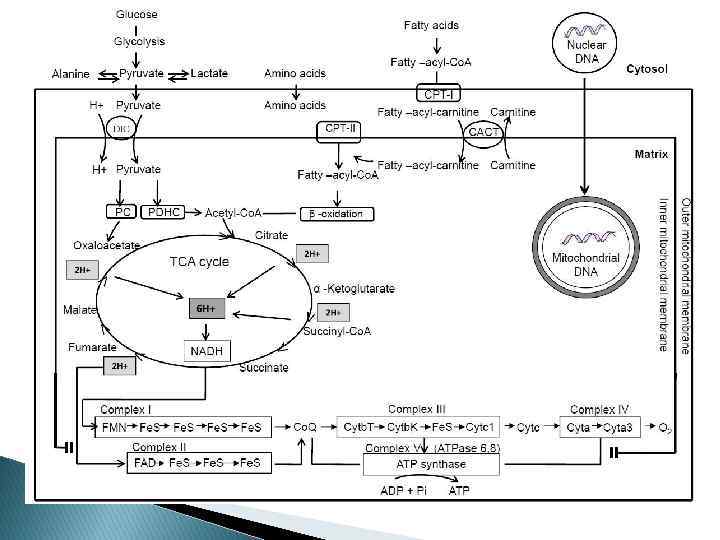
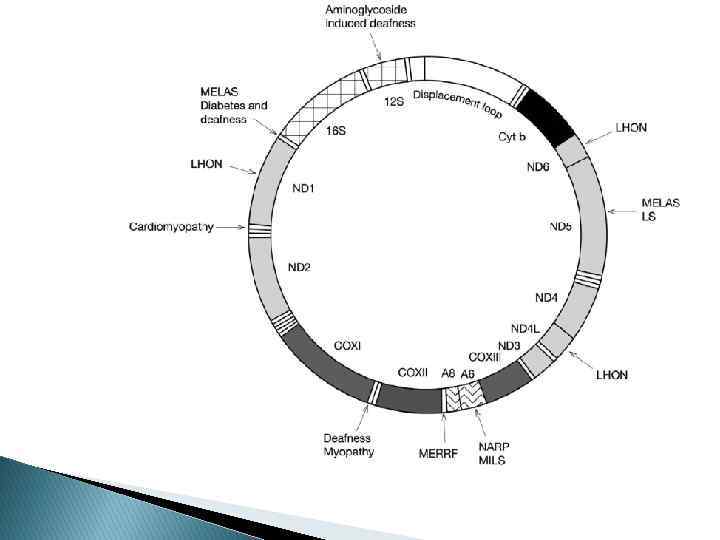

Митохондриальная ДНК 16, 569 bp Состоит из тяжелой и легкой цепей Имеет в составе 13 генов, кодирующих субъединицы для комплексов дыхательной цепи I (НАД-ДГ), III (цитохром b), IV (цитохром c оксидаза), V (АТФаза 6, АТФаза 8), 22 гена, кодирующих т. РНК и 2 гена, отвечающих за р. РНК

Особенности мт. ДНК наследуется по материнской линии Митохондрии полиплоидны, имеют 2 -10 копий мт. ДНК в каждой, тогда как каждая клетка содержит сотни митохондрий В нормальной клетке все копии мт. ДНК идентичны (гомоплазмия) Эффект «генетического горлышка бутылки» Эффект порога

Особенности мт. ДНК Все кодирующие последовательности состыкованы друг с другом мт. ДНК в норме не метилирована Экспрессия митохондриальных генов зависит не только от транскрипции, но и от взаимоотношений между транскрипцией ядерных генов и факторами трансляции с митохондриальными т. РНК и р. РНК Проявления заболевания зависят от патогенности мутации, пораженного гена и зависимости органа от обеспечения митохондрией энергии Генетический код мт. ДНК немного отличается от универсального генетического кода
 новорожденных Пути передачи: по материнской")
Митохондриальные болезни Частота более 1 на 7500 (1: 5000) новорожденных Пути передачи: по материнской линии, аутосомно-рецессивный, Х-сцепленный Впервые была описана шведскими учеными Luft et al, описавшими молодую женщину с гиперметаболизмом нетиреоидного происхождения в 1962 году В 1963 году Engel и Cunningham ненормальную пролиферацию митохондрий в мышце, окрашенной трихромом по Гомори, описанную как «шероховатые красные волокна» (ragged red fibers)

Классификация По клиническим проявлениям ◦ Синдромальные: MELAS, MNGIE, NARP, MERRF, MTS, CPEO, KSS, LBSL и др. ◦ Несиндромальные: поражение ЦНС, глаукома, ретинит, гипотироидизм, гипогонадизм, рвота, диаррея, поражение почек/печени, псевдообструкция ЖКТ, аритмии и др.

Классификация По типу нарушенного механизма ◦ Комплексы I, III, IV дыхательной цепи или коэнзим Q ◦ Процесс β-окисления ◦ Цикл Кребса ◦ Комплекс пируват-дегидрогиназы

Классификация По локализации дефекта ◦ Митохондриальная ДНК Точечная мутация Делеция Удвоение Деплеция ◦ Ядерная ДНК Точечная мутация Делеция Удвоение
 Подострая некротизирующая энцефаломиелопатия Чаще всего результат дефекта в мт. ДНК, кодирующей")
Синдром Лея (Leigh) Подострая некротизирующая энцефаломиелопатия Чаще всего результат дефекта в мт. ДНК, кодирующей цитохром с оксидазу (комплекс IV) или мутация в гене SURF 1 ядерной ДНК Частота 1: 40000 Манифестирует чаще всего в течение первых 2 лет жизни, но может проявиться и в 14 -43 года

Клиника Часто осложенное течение беременности Замедление психомоторного развития Слабость Эпилепсия Замедление роста Рвота и тошнота Атаксия Гипотония В крови повышенный уровень лактата и пирувата Лицевой дисморфизм и гипертрихоз (SURF 1)

Поражение черного вещества, красного ядра, субталамического ядра, скорлупы и ствола мозга

Поражение стриатума, левой radiatio optica
 Наследуется по материнской линии Частота 1: 12000 Точечная мутация")
NARP (Neuropathy, Ataxia, Retinitis Pigmentosa) Наследуется по материнской линии Частота 1: 12000 Точечная мутация Т на Г в нуклеотиде 8993 в митохондриальном гене MTATP 6, кодирующий субъединицу АТФазы 6 Манифестирует в молодом возрасте
 Слабость в проксимальных группах мышц")
Клиника Пигментная ретинопатия Ригидный зрачок Нистагм Слепота (сначала ночная) Слабость в проксимальных группах мышц Отставание в развитии Снижение слуха Судороги Атрофия кортикоспинального тракта Деменция Сенсорная нейропатия Мозжечковая атаксия

Поражение хвостатого ядра и скорлупы
 Манифестация чаще до 20 лет (5 месяцев – 50")
MNGIE (Mitochondrial Neuro. Gastro-Intestinal Encephalomyopathy) Манифестация чаще до 20 лет (5 месяцев – 50 лет) Мутация в гене тимидин-фосфорилазы (ECGF 1) ядерной ДНК, хромосома 22 q 13. 32 Наследуется по аутосомно-рецессивному типу Прогноз зависит от степени поражения ЖКТ и нарушения нутритивного статуса

Клиника Диарея, рвота Кишечная псевдообструкция Парез взора, птоз Прогрессирующая мышечная слабость, как в проксимальных так и в дистальных группах мышц Сенсомоторная периферическая нейропатия В меньшей степени страдают когнитивные функции Сенсоневральная тугоухость Лейкоэнцефалопатия Кахексия Кардиомиопатия

A, B – поражение височных долей, трапециевидного тела, мозжечка, ствола мозга C, D – поражение полушарий, моста
 Чаще всего в результате точечной мутации в")
MELAS (Mitochondrial Encephalomyopathy, Lact. Acidosis, Stroke-like episodes) Чаще всего в результате точечной мутации в гене мт. ДНК, кодирующей т. РНК, где А заменяется на Г в 3243 нуклеотиде Частота мутации 1: 6000 (Финляндия), 1: 424 (Австралия) Может быть результатом и мутации в ядерной ДНК Манифестация заболевания до 40 лет, чаще в 6 -10 лет Наследуется по материнской линии

Клиника Гемианопсия, чаще гомонимная Слепота Тошнота, рвота Инсультоподобные эпизоды, проявляющиеся головной болью, головокружением, очаговой неврологической симптоматикой (парезы, параличи) Судороги, фокальные и генерализованные

Гипогонадизм Деменция Почечная тубулопатия Гипертрофическая кардиомиопатия Бесплодие Сенсоневральная тугоухость Сахарный диабет Кровь: повышение уровня лактата Биопсия мышц: при электронной микроскопии под сарколеммой выявляются скопления митохондрий гигантских размеров, неправильной формы; на поперечном срезе окрашенные трихромом по Гомори мышечные волокна имеют вид красного ободка с неровными контурами (так называемые «шероховатые красные волокна» , составляющие до 20– 25% всех мышечных волокон на срезе ИГХ мышечных волокон: дефицит цитохром с оксидазы

A – поражение левой постцентральн ой извилины B – поражение правой постцентральн ой извилины C– билатеральное поражение затылочных долей D– усугубление поражения правой затылочной доли

Диагностика поражения ЦНС Клинические проявления: судороги, инсультоподобные эпизоды, слабость, нарушения походки, атаксия, мигрень, психозы, когнитивные нарушения, деменция Морфология: атрофия, кальцификаты, кровоизлияния, кисты, лакуны, очаги гиперинтенсивности СМЖ: повышение уровня лактата, пирувата, белка, кетоновых тел, плейоцитоз

Диагностика поражения ЦНС Функциональные исследования: ◦ SPECT: локальная гипоперфузия ◦ ПЭТ: локальные зоны сниженного метаболизма ◦ МР-спектроскопия: повышение уровня лактата, снижения уровня холина, креатина, N-ацетил аспартата ◦ ЭЭГ: локальное или диффузное замедление ритма, пароксизмы ◦ Индуцированные вызванные потенциалы: удлинение латентного периода, снижение амплитуды

Диагностика Кровь: повышение уровней креатин-киназы, лактата, пирувата, соотношения гидроксибутират/аминоацетат Моча: повышение уровня лактата, органические кислоты Электронная микроскопия: аномальные митохондрии, паракристаллиновые включения Биопсия и гистология мышц: шероховатые красные мышечные волокна >2%, включения гликогена, липидов
, цитохрома b (III), цитохром с оксидазы (IV), сукцинатдегидрогиназы")
Диагностика ИГХ: снижение активности НАДНдегидрогиназы (I), цитохрома b (III), цитохром с оксидазы (IV), сукцинатдегидрогиназы Генетическое тестирование ◦ ◦ Southern blot ПЦР ДНК-микрочипы Секвенирование мт. ДНК
 Клиническая картина (мах 4 балла) ◦")
Диагностические критерии митохондриальных болезней (Wolf et al, 2007) Клиническая картина (мах 4 балла) ◦ Мышечные симптомы (мах 2 балла) ◦ Нарушения ЦНС (мах 2 балла) ◦ Мультисистемное поражение (мах 3 балла) Метаболические нарушения и данные нейровизуализации (мах 4 балла) Данные гистологического исследования (мах 4 балла)
 8 -12 - МБ 5 -7")
Диагностические критерии митохондриальных болезней (Wolf et al, 2007) 8 -12 - МБ 5 -7 – МБ вероятна 2 -4 – МБ возможна 1 – МБ маловероятна
 Ингибиторы холинэстеразы Седативные средства Нейролептики (арипипразол)")
Специфическая симптоматическая терапия Противоэпилептические средства (кроме вальпроевой кислоты) Ингибиторы холинэстеразы Седативные средства Нейролептики (арипипразол) Антидепрессанты Ингибиторы ДОФА и Д-миметики Противоспастические средства (Баклофен, тизанидин, ботуллотоксин) β-блокаторы (при глаукоме) Гормональная терапия и др.
 Витамин")
Средства, удаляющие токсические метаболиты Антиоксиданты ◦ ◦ ◦ ◦ Квиноны (Co. Q, идебенон) Витамин Е Липоевая кислота ГКС Витамин С Глутатион Другие средства (эдаравон, ингибиторы АТ-II, мито. Q) Средства, снижающие уровень лактата ◦ Бикарбонаты ◦ Дихлоруксусная кислота

Доноры и акцепторы электронов Рибофлавин Сукцинат Альтернативные источники энергии Креатин-моногидрат

Кофакторы L-аргинин L-карнитин Аспартат Тиамин Фолиевая кислота

Другие средства и методы Медь Инфузии глюкозы Переливание крови и ее компонентов Гемодиализ Инвазивные методы лечения (постановка кардиостимулятора) Хирургические методы лечения Диета Физиотерапия Образ жизни

Экспериментальная этиотропная терапия Использование донорских соматических клеток Генная терапия ◦ Аллотопическая и ксенотопическая экспрессия ◦ Внедрение «здоровых» копий мт. ДНК в митохондрию ◦ Шифт генов Использование зародышевых клеток
 Угнетающие репликацию мт. ДНК и вызывающие")
Избегание употребления ЛС Вызывающие мутации мт. ДНК (карбоплатин) Угнетающие репликацию мт. ДНК и вызывающие деплецию мт. ДНК или снижающие активность дыхательной цепи (аналоги нуклеозидов) Нарушающие транскрипцию мт. ДНК (IF) Блокаторы дыхательной цепи (карведилол, бупивакаин, фенотиазины) Неконкурентные ингибиторы АТФазы (β-АБ) Снижающие уровень эндогенного Co. Q (статины) Ингибиторы β-окисления (тетрациклины, амиодарон) Тормозящие синтез митохондриальных белков (барбитураты, хлорамфеникол) Связывающие карнитин и снижающие активность дыхательной цепи (вальпроаты, доксирубицин)
")
Пренатальная диагностика Биопсия ворсин хориона Амниоцентез Пункция пупочной вены (не применяется)
Митохондриальные болезни.pptx