

миопатии.ppt
- Количество слайдов: 35
 Миопатии
Миопатии
 n Миопатия – прогрессирующие мышечные дистрофии; относятся к наследственным заболеваниям. Миопатии могут передаваться по аутосомно-рецессивному, доминантному и сцепленному с полом типам. В основе развития миопатии лежат нарушение обмена в мышечных клетках, изменение синтеза нуклеиновых кислот, значительное преобладание ускоренного распада белков мышц над измененным их синтезом. Мышцы при миопатии истончены, часть волокон замещена жировой тканью. Основные признаки миопатии - нарастающая мышечная слабость, симметричная атрофия мышц, снижение сухожильных рефлексов, в поздних стадиях - деформация костей и суставов. Постоянно выражены вегетативнотрофические расстройства.
n Миопатия – прогрессирующие мышечные дистрофии; относятся к наследственным заболеваниям. Миопатии могут передаваться по аутосомно-рецессивному, доминантному и сцепленному с полом типам. В основе развития миопатии лежат нарушение обмена в мышечных клетках, изменение синтеза нуклеиновых кислот, значительное преобладание ускоренного распада белков мышц над измененным их синтезом. Мышцы при миопатии истончены, часть волокон замещена жировой тканью. Основные признаки миопатии - нарастающая мышечная слабость, симметричная атрофия мышц, снижение сухожильных рефлексов, в поздних стадиях - деформация костей и суставов. Постоянно выражены вегетативнотрофические расстройства.
 n Ландузи-Дежерина (плече-лопаточнолицевая") Наиболее часто встречаются следующие формы миопатий n Дюшенна n Эрба (тазо-плечевая) n Ландузи-Дежерина (плече-лопаточнолицевая миопатия).
Наиболее часто встречаются следующие формы миопатий n Дюшенна n Эрба (тазо-плечевая) n Ландузи-Дежерина (плече-лопаточнолицевая миопатия).
 Миопатия Дюшенна n n n Наследуется по рецессивному типу, связанному с Х-хромосмой. В подавляющем большинстве случаев болеют мальчики. Первые признаки заболевания появляются в первые 1 -3 года жизни ребенка. На первом году жизни дети начинают отставать в моторном развитии. С задержкой начинают сидеть, ходить, вставать. Движения неловкие, они часто спотыкаются и падают. В 2 -3 летнем возрасте выявляется патологическая утомляемость мышц утиная походка вставание из положения сидя на корточках происходит постепенно с активным использование рук( «взбирание по лесенке» или «взбирание по самому себе» ) симметричные атрофии мышц проксимальных отделов конечностей(сначала нижних, затем – верхних). Атрофии подвергаются мышцы тазового пояса, бедер, через 1 -3 года присоединяются атрофии мышц плечевого пояса, мышцы спины.
Миопатия Дюшенна n n n Наследуется по рецессивному типу, связанному с Х-хромосмой. В подавляющем большинстве случаев болеют мальчики. Первые признаки заболевания появляются в первые 1 -3 года жизни ребенка. На первом году жизни дети начинают отставать в моторном развитии. С задержкой начинают сидеть, ходить, вставать. Движения неловкие, они часто спотыкаются и падают. В 2 -3 летнем возрасте выявляется патологическая утомляемость мышц утиная походка вставание из положения сидя на корточках происходит постепенно с активным использование рук( «взбирание по лесенке» или «взбирание по самому себе» ) симметричные атрофии мышц проксимальных отделов конечностей(сначала нижних, затем – верхних). Атрофии подвергаются мышцы тазового пояса, бедер, через 1 -3 года присоединяются атрофии мышц плечевого пояса, мышцы спины.
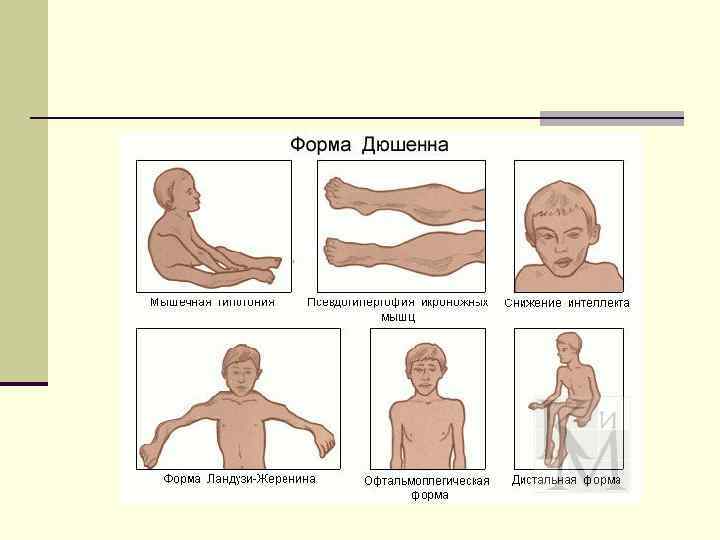
 Характерные для миопатий симптомы: осиная талия крыловидные лопатки, отошедшие от грудной клетки гиперлордоз в поясничном отделе позвоночника псевдогипертрофия икроножных мышц Со временем в пораженных мышцах развиваются контрактуры и ретракции. n Коленные рефлексы исчезают раньше всех, затем снижаются и исчезают рефлексы с сухожилий бицепса и трицепса. Ахилловы рефлексы остаются сохранными в течение длительного времени. n дистрофические изменения в костно-суставной системе. Характерны деформации стоп, позвоночника. n n n
Характерные для миопатий симптомы: осиная талия крыловидные лопатки, отошедшие от грудной клетки гиперлордоз в поясничном отделе позвоночника псевдогипертрофия икроножных мышц Со временем в пораженных мышцах развиваются контрактуры и ретракции. n Коленные рефлексы исчезают раньше всех, затем снижаются и исчезают рефлексы с сухожилий бицепса и трицепса. Ахилловы рефлексы остаются сохранными в течение длительного времени. n дистрофические изменения в костно-суставной системе. Характерны деформации стоп, позвоночника. n n n

 Миопатия Эрба-Рота n симптомы осиной талии n n n утиной походки крыловидных лопаток гиперлордоза в поясничном отделе позвоночника характерно появление контрактур и псевдогипертрофий снижаются сухожильные рефлексы Течение болезни чаще всего медленно-прогрессирующее.
Миопатия Эрба-Рота n симптомы осиной талии n n n утиной походки крыловидных лопаток гиперлордоза в поясничном отделе позвоночника характерно появление контрактур и псевдогипертрофий снижаются сухожильные рефлексы Течение болезни чаще всего медленно-прогрессирующее.
") Миопатия Эрба-Рота (тазо-плечевая форма)
Миопатия Эрба-Рота (тазо-плечевая форма)
 Миопатия Ландузи-Дежерина
Миопатия Ландузи-Дежерина
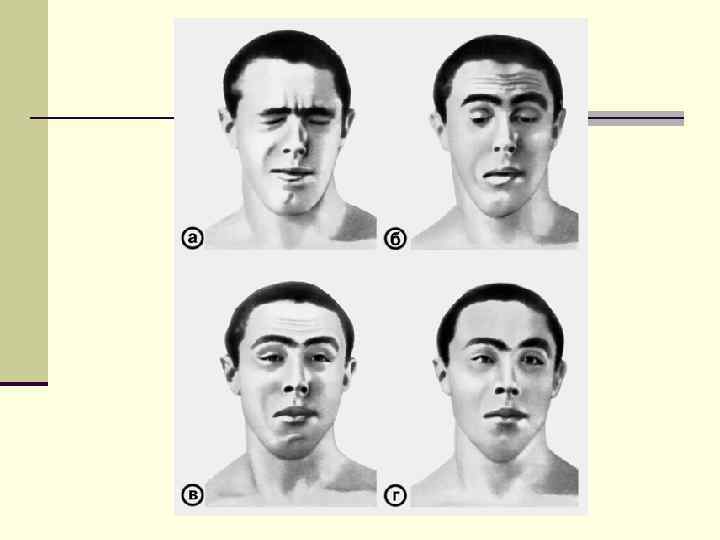
 n Поражаются мышцы плечевого пояса, лица и лопаток. n Вовлечение в процесс мимических мышц приводит к n n n появлению таких характерных симптомов, как «поперечная» улыбка Джоконды «полированный» лоб лагофтальм или «заячий» глаз (не закрывающийся) феномен «губы Тапира» за счет подвыворота губ и появления наружу слизистой оболочки лицо малоподвижное, мимики почти нет(лицо Сфинкса)
n Поражаются мышцы плечевого пояса, лица и лопаток. n Вовлечение в процесс мимических мышц приводит к n n n появлению таких характерных симптомов, как «поперечная» улыбка Джоконды «полированный» лоб лагофтальм или «заячий» глаз (не закрывающийся) феномен «губы Тапира» за счет подвыворота губ и появления наружу слизистой оболочки лицо малоподвижное, мимики почти нет(лицо Сфинкса)
 Миопатия Ландузи-Дежерина n Атрофии подвергаются двуглавая, трехглавая, большая грудная, передняя зубчатая и трапециевидная мышцы. Лопатки отходят от средней линии, становятся крыловидными, появляется симптом «свободных надплечий» . В некоторых случаях атрофии мышц распространяются на другие группы мышц. В икроножных и дельтовидных мышцах возможно развитие псевдогипертрофий. Сухожильные рефлексы снижаются с бицепса и трицепса. В пораженных мышцах снижен тонус.
Миопатия Ландузи-Дежерина n Атрофии подвергаются двуглавая, трехглавая, большая грудная, передняя зубчатая и трапециевидная мышцы. Лопатки отходят от средней линии, становятся крыловидными, появляется симптом «свободных надплечий» . В некоторых случаях атрофии мышц распространяются на другие группы мышц. В икроножных и дельтовидных мышцах возможно развитие псевдогипертрофий. Сухожильные рефлексы снижаются с бицепса и трицепса. В пораженных мышцах снижен тонус.
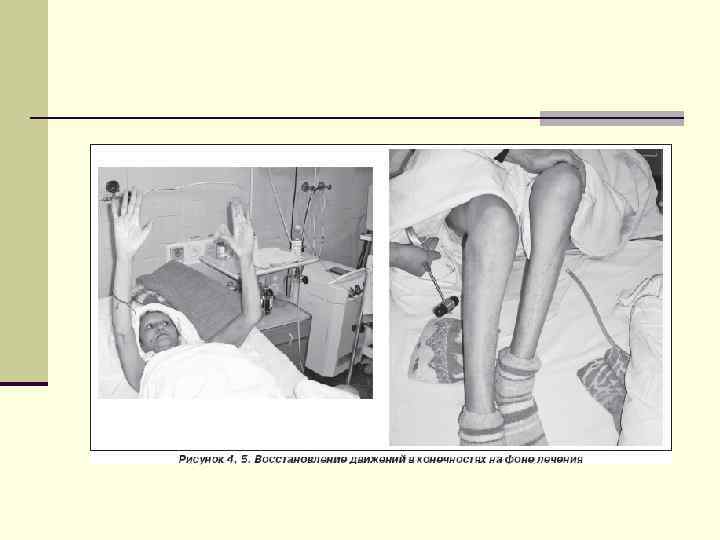
 Лечение миопатий n Направлено на сохранение и поддержание двигательной активности больных в течение как можно более длительного периода времени. Особое значение в этом процессе играет ЛФК. Она позволяет отсрочить обездвиженность больных. Занятия ЛФК предотвращают развитие контрактур и деформаций. Проводятся упражнения на объем движений, коррекция положения тела в кровати, кресле, частая смена положения и позы. Практикуется раннее применение шин. n Рекомендуется следить за весом больных. Избыточный вес ухудшает двигательные функции. n Из медикаментозных препаратов назначают АТФ, витамины группы В, витамин Е, анаболические стероиды (ретаболил, неробол).
Лечение миопатий n Направлено на сохранение и поддержание двигательной активности больных в течение как можно более длительного периода времени. Особое значение в этом процессе играет ЛФК. Она позволяет отсрочить обездвиженность больных. Занятия ЛФК предотвращают развитие контрактур и деформаций. Проводятся упражнения на объем движений, коррекция положения тела в кровати, кресле, частая смена положения и позы. Практикуется раннее применение шин. n Рекомендуется следить за весом больных. Избыточный вес ухудшает двигательные функции. n Из медикаментозных препаратов назначают АТФ, витамины группы В, витамин Е, анаболические стероиды (ретаболил, неробол).
 Миотонии n Миотония — наследственное заболевание, относящееся к каналопатиям (заболевания, связанные с патологией ионных каналов). Проявляется замедленным расслаблением мышц. n Характерные признаки миотонии — миотонические разряды, выявляемые игольчатой ЭМГ, и миотонические феномены, которые выявляются при клиническом обследовании.
Миотонии n Миотония — наследственное заболевание, относящееся к каналопатиям (заболевания, связанные с патологией ионных каналов). Проявляется замедленным расслаблением мышц. n Характерные признаки миотонии — миотонические разряды, выявляемые игольчатой ЭМГ, и миотонические феномены, которые выявляются при клиническом обследовании.
 n хондродистрофическая n конгенитальная аутосомно-доминантная n конгенитальная аутосомно-рецессивная n") Классификация n дистрофическая (двух типов) n хондродистрофическая n конгенитальная аутосомно-доминантная n конгенитальная аутосомно-рецессивная n парамиотония Эйленбурга n нейромиотония
Классификация n дистрофическая (двух типов) n хондродистрофическая n конгенитальная аутосомно-доминантная n конгенитальная аутосомно-рецессивная n парамиотония Эйленбурга n нейромиотония

 Клиническая картина Симптом «кулака» — основной клинический тест на выявление миотонии: пациент не может быстро разжать кулак, для этого ему нужно время и определенные усилия. При повторных попытках такой миотонический феномен угасает за исключением миотонии Эйленбурга, когда скованность, наоборот, усиливается с каждой повторной попыткой. n Скованность также наблюдается при разжимании сжатых челюстей, быстро открыть зажмуренные глаза, быстро встать со стула. n На игольчатой электромиографии выявляют один из самых характерных для миотонии феноменов — миотонические разряды, сопровождающиеся звуком «пикирующего бомбардировщика» , возникающие при введении и перемещении игольчатого электрода. n Отличительной клинической особенностью врожденной миотонии является гипертрофия отдельных мышечных групп, которая создает впечатление об атлетическом телосложении пациента. n
Клиническая картина Симптом «кулака» — основной клинический тест на выявление миотонии: пациент не может быстро разжать кулак, для этого ему нужно время и определенные усилия. При повторных попытках такой миотонический феномен угасает за исключением миотонии Эйленбурга, когда скованность, наоборот, усиливается с каждой повторной попыткой. n Скованность также наблюдается при разжимании сжатых челюстей, быстро открыть зажмуренные глаза, быстро встать со стула. n На игольчатой электромиографии выявляют один из самых характерных для миотонии феноменов — миотонические разряды, сопровождающиеся звуком «пикирующего бомбардировщика» , возникающие при введении и перемещении игольчатого электрода. n Отличительной клинической особенностью врожденной миотонии является гипертрофия отдельных мышечных групп, которая создает впечатление об атлетическом телосложении пациента. n
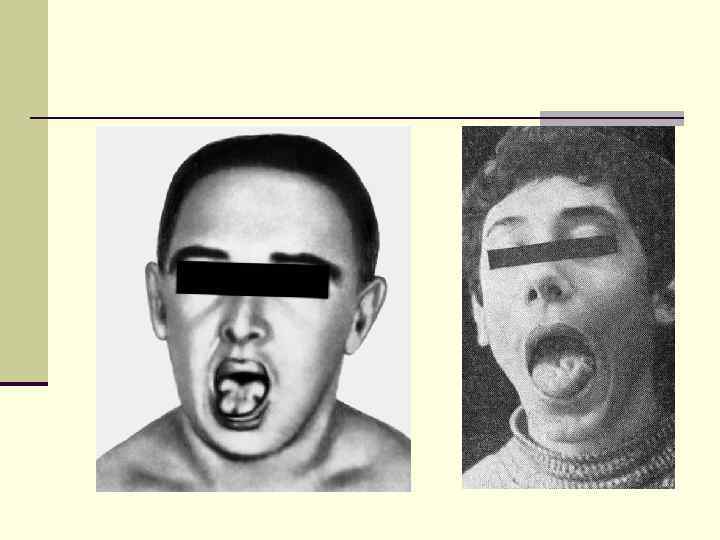
 Клиническая картина n В большинстве случаев мышечная сила сохранена, но иногда бывает снижена в дистальных мышцах рук. n Дистрофическая миотония — мультисистемное заболевание. В большинстве случаев неврологические симптомы сочетаются с сердечной патологией (гипертрофия левого желудочка, аритмия), церебральными симптомами (гиперсомния, сниженный уровень интеллекта), эндокринными расстройствами (нарушение менструального цикла у женщин; гипогонадизм и импотенция у мужчин). n Для парамиотонии типична т. н. «холодовая миотония» — возникновение мышечного спазма и пареза на холоде; такие приступы могут длиться от нескольких минут до нескольких часов. n Клиническими проявлениями нейромиотоний являются мышечная скованность, спазмы (безболезненные) и постоянная мышечная активность на ЭМГ.
Клиническая картина n В большинстве случаев мышечная сила сохранена, но иногда бывает снижена в дистальных мышцах рук. n Дистрофическая миотония — мультисистемное заболевание. В большинстве случаев неврологические симптомы сочетаются с сердечной патологией (гипертрофия левого желудочка, аритмия), церебральными симптомами (гиперсомния, сниженный уровень интеллекта), эндокринными расстройствами (нарушение менструального цикла у женщин; гипогонадизм и импотенция у мужчин). n Для парамиотонии типична т. н. «холодовая миотония» — возникновение мышечного спазма и пареза на холоде; такие приступы могут длиться от нескольких минут до нескольких часов. n Клиническими проявлениями нейромиотоний являются мышечная скованность, спазмы (безболезненные) и постоянная мышечная активность на ЭМГ.
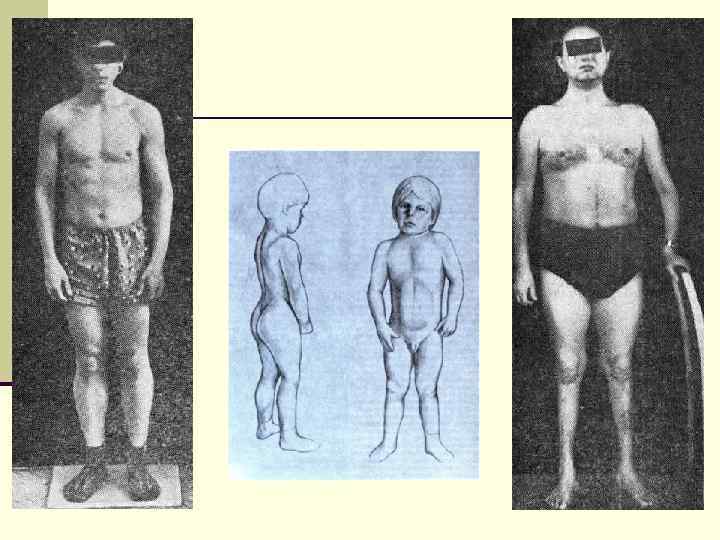
 Диагностика n n n С помощью лабораторных исследований при нейромиотонии выявляют антитела к потенциал-зависимым калиевым каналам, а дистрофическая миотония отличается незначительным повышением активности КФК в крови. Основными инструментальными методом диагностики миотонии является игольчатая электромиография (ЭМГ), на которой определяют миотонические разряды — патогномоничный признак миотонии. Проводится исследование при помощи вызванных потенциалов и электронейрография. При парамиотонии на ЭМГ регистрируют нормальные ПДЕ и редкие миотонические разряды. Проводят холодовую пробу: незначительное охлаждение вызывает миотонические разряды, при дальнейшем охлаждении наступает «биоэлектрическое молчание» (исчезают как миотонические феномены, так и ПДЕ). ДНК-диагностика дистрофической миотонии основана на выявлении повышенного количества CTG-повторов в гене DMPK.
Диагностика n n n С помощью лабораторных исследований при нейромиотонии выявляют антитела к потенциал-зависимым калиевым каналам, а дистрофическая миотония отличается незначительным повышением активности КФК в крови. Основными инструментальными методом диагностики миотонии является игольчатая электромиография (ЭМГ), на которой определяют миотонические разряды — патогномоничный признак миотонии. Проводится исследование при помощи вызванных потенциалов и электронейрография. При парамиотонии на ЭМГ регистрируют нормальные ПДЕ и редкие миотонические разряды. Проводят холодовую пробу: незначительное охлаждение вызывает миотонические разряды, при дальнейшем охлаждении наступает «биоэлектрическое молчание» (исчезают как миотонические феномены, так и ПДЕ). ДНК-диагностика дистрофической миотонии основана на выявлении повышенного количества CTG-повторов в гене DMPK.
 Лечение n Целью лечения нейромиотонии является устранение постоянной мышечной активности и достижение возможной ремиссии, целью лечения миотонии — снижение выраженности миотонических проявлений. n Немедикаментозное лечение миотонии состоит из диеты с ограничением солей калия, ЛФК, массажа, электромиостимуляции, а также предупреждения переохлаждений, так как при холоде усиливаются все миотонические реакции. n Радикального медикаментозного лечения миотонии не существует, поэтому в целях уменьшении выраженности миотонических проявлений применяют фенитоин (200 -400 мг/сутки), а для снижения уровня калия — диуретики. В некоторых случаях удается достичь ремиссии с помощью иммуносупрессивной терапии: внутривенное введение иммуноглобулина человека (400 мг/кг), преднизолон (1 мг/кг/сутки), циклофосфамид.
Лечение n Целью лечения нейромиотонии является устранение постоянной мышечной активности и достижение возможной ремиссии, целью лечения миотонии — снижение выраженности миотонических проявлений. n Немедикаментозное лечение миотонии состоит из диеты с ограничением солей калия, ЛФК, массажа, электромиостимуляции, а также предупреждения переохлаждений, так как при холоде усиливаются все миотонические реакции. n Радикального медикаментозного лечения миотонии не существует, поэтому в целях уменьшении выраженности миотонических проявлений применяют фенитоин (200 -400 мг/сутки), а для снижения уровня калия — диуретики. В некоторых случаях удается достичь ремиссии с помощью иммуносупрессивной терапии: внутривенное введение иммуноглобулина человека (400 мг/кг), преднизолон (1 мг/кг/сутки), циклофосфамид.
 Миоплегии n Пароксизмальная миоплегия, или периодический паралич, - редкое наследственное заболевание, характеризующееся приступами вялого паралича скелетных мышц.
Миоплегии n Пароксизмальная миоплегия, или периодический паралич, - редкое наследственное заболевание, характеризующееся приступами вялого паралича скелетных мышц.
 n гиперкалиемическая или семейная эпизодическая адинамия (болезнь Гамсторп)") Классификация n Гипокалиемическая форма (болезнь Вестфаля-Шахновича) n гиперкалиемическая или семейная эпизодическая адинамия (болезнь Гамсторп) n нормокалиемическая форма.
Классификация n Гипокалиемическая форма (болезнь Вестфаля-Шахновича) n гиперкалиемическая или семейная эпизодическая адинамия (болезнь Гамсторп) n нормокалиемическая форма.
 n Передается по аутосомно-доминантному типу с неполной пенетрантностью. Мужчины болеют") Гипокалиемическая форма (болезнь Вестфаля-Шахновича) n Передается по аутосомно-доминантному типу с неполной пенетрантностью. Мужчины болеют значительно чаще женщин. n Начало заболевания относится к детскому или юношескому возрасту, в основном от 10 до 18 лет, хотя известны случаи возникновения заболевания и в возрасте после 30 лет. n Приступы возникают, как правило, ночью или под утро, больные просыпаются с явлениями паралича конечностей, мышц туловища и шеи. В тяжелых случаях слабость может распространяться на мышцы лица, дыхательную мускулатуру. Резко снижается тонус, исчезают сухожильные рефлексы.
Гипокалиемическая форма (болезнь Вестфаля-Шахновича) n Передается по аутосомно-доминантному типу с неполной пенетрантностью. Мужчины болеют значительно чаще женщин. n Начало заболевания относится к детскому или юношескому возрасту, в основном от 10 до 18 лет, хотя известны случаи возникновения заболевания и в возрасте после 30 лет. n Приступы возникают, как правило, ночью или под утро, больные просыпаются с явлениями паралича конечностей, мышц туловища и шеи. В тяжелых случаях слабость может распространяться на мышцы лица, дыхательную мускулатуру. Резко снижается тонус, исчезают сухожильные рефлексы.
 n Наблюдаются вегетативные симптомы n гиперемия лица, n потливость, n") Гипокалиемическая форма (болезнь Вестфаля-Шахновича) n Наблюдаются вегетативные симптомы n гиперемия лица, n потливость, n изменение частоты пульса, дыхания, n повышенная жажда, n в редких случаях тошнота.
Гипокалиемическая форма (болезнь Вестфаля-Шахновича) n Наблюдаются вегетативные симптомы n гиперемия лица, n потливость, n изменение частоты пульса, дыхания, n повышенная жажда, n в редких случаях тошнота.
 Приступ может длиться от 1 ч до нескольких суток, большей") Гипокалиемическая форма (болезнь Вестфаля-Шахновича) Приступ может длиться от 1 ч до нескольких суток, большей частью 2 -4 ч. Постепенно слабость исчезает, появляются движения сначала в дистальных, затем в проксимальных отделах конечностей, в этот период больные стремятся активно двигаться, что способствует ликвидации приступа. n У некоторых больных приступы наблюдаются ежедневно, когда имеется как бы status myoplegicus, или они проявляются в виде постоянной умеренной слабости, выраженной больше в утренние часы. В таких случаях у больных может развиться миопатический синдром с умеренной гипотрофией мышц, особенно в проксимальных отделах конечностей, снижение сухожильных рефлексов. n Характерны провоцирующие факторы - употребление большого количества углеводов в пище, переохлаждение, физические перегрузки, злоупотребление поваренной солью, алкоголем. n У женщин приступы чаще развиваются за 1 -2 дня или в 1 -й день менструаций. n n
Гипокалиемическая форма (болезнь Вестфаля-Шахновича) Приступ может длиться от 1 ч до нескольких суток, большей частью 2 -4 ч. Постепенно слабость исчезает, появляются движения сначала в дистальных, затем в проксимальных отделах конечностей, в этот период больные стремятся активно двигаться, что способствует ликвидации приступа. n У некоторых больных приступы наблюдаются ежедневно, когда имеется как бы status myoplegicus, или они проявляются в виде постоянной умеренной слабости, выраженной больше в утренние часы. В таких случаях у больных может развиться миопатический синдром с умеренной гипотрофией мышц, особенно в проксимальных отделах конечностей, снижение сухожильных рефлексов. n Характерны провоцирующие факторы - употребление большого количества углеводов в пище, переохлаждение, физические перегрузки, злоупотребление поваренной солью, алкоголем. n У женщин приступы чаще развиваются за 1 -2 дня или в 1 -й день менструаций. n n
 Гиперкалиемическая форма, или семейная эпизодическая адинамия n Заболевание передается по аутосомно- доминантному типу с высокой пенетрантностью. Мужчины и женщины страдают почти одинаково часто. Эта форма миоплегии наблюдается намного реже, чем гипокалиемический вариант. Начало заболевания в большинстве случаев в возрасте до 10 лет.
Гиперкалиемическая форма, или семейная эпизодическая адинамия n Заболевание передается по аутосомно- доминантному типу с высокой пенетрантностью. Мужчины и женщины страдают почти одинаково часто. Эта форма миоплегии наблюдается намного реже, чем гипокалиемический вариант. Начало заболевания в большинстве случаев в возрасте до 10 лет.
 Гиперкалиемическая форма, или семейная эпизодическая адинамия n Приступ развивается обычно днем, провоцируется состоянием голода или состоянием покоя, которому предшествовала физическая работа. Первыми симптомами развивающегося пароксизма являются парестезии в области лица, в руках и ногах, слабость начинается с дистальных отделов конечностей и быстро генерализуется, включая мышцы лица. Развивается гипотония, сухожильная арефлексия. Длительность приступов, как правило, меньше чем при гипокалиемической форме и составляет в среднем 1 -2 ч. Характерна массивность вегетативных расстройств - профузный пот, сильная жажда, сердцебиение, повышение АД. Иногда отмечается повышение механической возбудимости мышц.
Гиперкалиемическая форма, или семейная эпизодическая адинамия n Приступ развивается обычно днем, провоцируется состоянием голода или состоянием покоя, которому предшествовала физическая работа. Первыми симптомами развивающегося пароксизма являются парестезии в области лица, в руках и ногах, слабость начинается с дистальных отделов конечностей и быстро генерализуется, включая мышцы лица. Развивается гипотония, сухожильная арефлексия. Длительность приступов, как правило, меньше чем при гипокалиемической форме и составляет в среднем 1 -2 ч. Характерна массивность вегетативных расстройств - профузный пот, сильная жажда, сердцебиение, повышение АД. Иногда отмечается повышение механической возбудимости мышц.
 Нормокалиемическая форма n Заболевание, так же как и другие формы, - семейное, передается по аутосомнодоминантному типу. Эта форма встречается крайне редко, до настоящего времени описано всего 4 семьи, одна из них - в отечественной литературе [Ильина И. А. и др. , 1977]. Первые приступы развиваются в возрасте до 10 лет.
Нормокалиемическая форма n Заболевание, так же как и другие формы, - семейное, передается по аутосомнодоминантному типу. Эта форма встречается крайне редко, до настоящего времени описано всего 4 семьи, одна из них - в отечественной литературе [Ильина И. А. и др. , 1977]. Первые приступы развиваются в возрасте до 10 лет.
 Нормокалиемическая форма n Тяжесть пароксизмов варьирует от полного паралича всех конечностей, туловища, а также мускулатуры лица, до умеренной мышечной слабости. Длительность их обычно значительно большая, чем при других формах, и составляет от нескольких дней до 2 -3 нед. Мышечная слабость медленно нарастает и еще более медленно уменьшается. Имеются указания о снижении чувствительности по периферическому типу в период пароксизма. Резко снижаются сухожильные рефлексы и мышечный тонус. У некоторых больных отмечается гипертрофия отдельных мышц, атлетическое телосложение.
Нормокалиемическая форма n Тяжесть пароксизмов варьирует от полного паралича всех конечностей, туловища, а также мускулатуры лица, до умеренной мышечной слабости. Длительность их обычно значительно большая, чем при других формах, и составляет от нескольких дней до 2 -3 нед. Мышечная слабость медленно нарастает и еще более медленно уменьшается. Имеются указания о снижении чувствительности по периферическому типу в период пароксизма. Резко снижаются сухожильные рефлексы и мышечный тонус. У некоторых больных отмечается гипертрофия отдельных мышц, атлетическое телосложение.
 Диагностика и дифференциальная диагностика n Заболевание диагностируется на основании результатов генеалогического анализа, особенностей клинической картины (темп нарастания мышечной слабости, провоцирующие факторы), результатов лабораторного и биохимического обследования (нормальное содержание калия в сыворотке крови, электровозбудимость мышц). n Заболевание следует отличать от миоплегий, развивающихся в результате эндокринных заболеваний: тиреотоксикоза, болезни Конна (первичный гиперальдостеронизм), болезни Аддисона и др.
Диагностика и дифференциальная диагностика n Заболевание диагностируется на основании результатов генеалогического анализа, особенностей клинической картины (темп нарастания мышечной слабости, провоцирующие факторы), результатов лабораторного и биохимического обследования (нормальное содержание калия в сыворотке крови, электровозбудимость мышц). n Заболевание следует отличать от миоплегий, развивающихся в результате эндокринных заболеваний: тиреотоксикоза, болезни Конна (первичный гиперальдостеронизм), болезни Аддисона и др.
 Лечение n При гипокалиемическом параличе в момент приступа назначают 10% раствор хлорида калия по 1 -2 столовой ложке через каждые 12 ч, панангин внутрь или внутривенно по 1 мл на изотоническом растворе хлорида натрия, что ускоряет -окончание приступа. n Для профилактики приступов рекомендуются диуретики - дихлотиазид (гипотиазид) по 2, 5 мг 5 раз в неделю, диакарб по 0, 25 г 2 -3 раза в день. Следует избегать пищи, богатой калием, увеличить в суточном рационе количество углеводов и поваренной соли. Рекомендуется дробное питание с укороченными интервалами между приемами пищи. При лечении больных нормокалиемическим периодическим параличом рекомендуется дополнительный прием 8 -10 г поваренной соли ежедневно. Некоторое улучшение наблюдается приеме диакарба в дозе 0, 25 г 2 раза в день.
Лечение n При гипокалиемическом параличе в момент приступа назначают 10% раствор хлорида калия по 1 -2 столовой ложке через каждые 12 ч, панангин внутрь или внутривенно по 1 мл на изотоническом растворе хлорида натрия, что ускоряет -окончание приступа. n Для профилактики приступов рекомендуются диуретики - дихлотиазид (гипотиазид) по 2, 5 мг 5 раз в неделю, диакарб по 0, 25 г 2 -3 раза в день. Следует избегать пищи, богатой калием, увеличить в суточном рационе количество углеводов и поваренной соли. Рекомендуется дробное питание с укороченными интервалами между приемами пищи. При лечении больных нормокалиемическим периодическим параличом рекомендуется дополнительный прием 8 -10 г поваренной соли ежедневно. Некоторое улучшение наблюдается приеме диакарба в дозе 0, 25 г 2 раза в день.